
Abstract
The epithelial derived Harderian gland consists of 2 types of secretory cells. The more numerous type A cells are responsible for the secretion of lipid droplets, while type B cells produce dark granules of multilamellar bodies. The process of autophagy is constitutively active in the Harderian gland, as confirmed by our analysis of LC3 processing in GFP-LC3 transgenic mice. This process is compromised by epithelial deletion of Atg7. Morphologically, the Atg7 mutant glands are hypotrophic and degenerated, with highly vacuolated cells and pyknotic nuclei. The mutant glands accumulate lipid droplets coated with PLIN2 (perilipin 2) and contain deposits of cholesterol, ubiquitinated proteins, SQSTM1/p62 (sequestosome 1) positive aggregates and other metabolic products such as porphyrin. Immunofluorescence stainings show that distinct cells strongly aggregate both proteins and lipids. Electron microscopy of the Harderian glands reveals that its organized structure is compromised, and the presence of large intracellular lipid droplets and heterologous aggregates. We attribute the occurrence of large vacuoles to a malfunction in the formation of multilamellar bodies found in the less abundant type B Harderian gland cells. This defect causes the formation of large tertiary lysosomes of heterologous content and is accompanied by the generation of tight lamellar stacks of endoplasmic reticulum in a pseudo-crystalline form. To test the hypothesis that lipid and protein accumulation is the cause for the degeneration in autophagy-deficient Harderian glands, epithelial cells were treated with a combination of the proteasome inhibitor and free fatty acids, to induce aggregation of misfolded proteins and lipid accumulation, respectively. The results show that lipid accumulation indeed enhanced the toxicity of misfolded proteins and that this was even more pronounced in autophagy-deficient cells. Thus, we conclude autophagy controls protein and lipid catabolism and anabolism to facilitate bulk production of secretory vesicles of the Harderian gland.
Abbreviations:
- Atg7, autophagy related 7
- Atg12, autophagy related 12
- BCA, bicinchoninic acid assay
- BSA, bovine serum albumin
- BODIPY, boron-dipyrromethene fluorescent dye
- Cre, Cre recombinase
- DMSO, dimethyl sulfoxide
- ELISA, enzyme-linked immunosorbent assay
- ER, edoplasmic reticulum
- f, floxed
- FC, free cholesterol
- GFP, green fluorescent protein
- HaGl, Harderian gland
- KLICK, keratosis lineariz with ichthyosis congenita and sclerosing keratoderma
- KRT14, Keratin 14
- LD, Lipid drops
- LSM, laser scanning microscope
- MAP1LC3A/B (LC3), microtubule-associated protein 1 light chain 3 α/β
- MG312, synthetic peptide Z-Leu-Leu-Leu-al
- ORO, oil red O
- PARP, poly (ADP-ribose) polymerase
- PCR, polymerase chain reaction
- PLIN2, perilipin 2
- RFU, relative fluorecent units
- SQSTM1, sequestosome 1/p62
- TBS-T, Tris buffered saline with Tween 20
- TLC, thin layer chromatography
- UV, ultraviolet
Introduction
Maintenance of metabolic homeostasis is crucial for proper cellular function, whereas an imbalance leads to accumulation of unwanted (e.g., misfolded proteins) or toxic products, which are hallmarks of degenerative diseases and aging.Citation1,2 To preserve a tolerable equilibrium a multitude of coping mechanisms have evolved in biological systems.
In proliferating cells such a coping mechanism for toxic metabolites is the rejuvenation of cells by asymmetric division. This helps to clear one of the 2 daughter cells, generally the replicating cell, from aggregates and potentially damaging proteins.Citation3 Another coping mechanism observed in the multilayered epidermis is shedding.Citation4 Keratinocytes, arising from basally located stem cells, accumulate proteins during their differentiation until they reach the most differentiated apical layer, the stratum corneum.Citation5 These cells are eventually removed by shedding from the apical surface. Alternatively, protein aggregates may even induce proliferation, as recently reported for a mutant form of α-1-antitrypsin, which induced hepatocellular carcinoma.Citation6
In resting cells the default pathway of clearing misfolded proteins is proteasomal and/or lysosomal degradation, or even secretion. Any impairment of clearance causes the formation of vacuoles and aggregates.Citation7 The aggresome is a specialized structure, accumulating ubiquitinated and misfolded proteins, particularly in amyloid diseases.Citation8 In Alzheimer disease, one major neurodegenerative disorder, misfolded proteins are deposited inside the neurons as neurofibrillary tangles composed of the Tau protein, and extracellularly and in blood vessels as amyloid plaques, composed of Amyloid-β peptides.Citation9 Protein deposits in a crystalline or crystalline like form are readily observed during neurodegeneration, as they are in chronic liver diseases and in myopathies.Citation10,11 A beneficial role of aggregation has been demonstrated in a model for Huntington disease, another neurodegenerative disease associated with trinucleotide repeats and intracellular protein aggregates, in which the unbound specimens are even more toxic than the aggregates.Citation12
Another efficient way for the intracellular clearance and renewing is autophagic degradation.Citation13 Macroautophagy (hereafter autophagy) is an especially versatile clearance system capable of turning over cellular organelles, proteins and lipids, thereby recycling their constituents.Citation14,15 Autophagy is initiated by a cascade of modification steps termed the ATG12 and LC3/Atg8-conjugation systems, leading to the formation of autophagosomes.Citation16 A crucial step in this cascade is the conversion of microtubule-associated protein light chain 3 (LC3) from its unlipidated form (LC3-I) to a lipid conjugated form (LC3-II), which is incorporated into the autophagosome membrane. The autophagosome engulfs cytoplasmic constituents and organelles and subjects them to lysosomal degradation. A GFP-conjugated form of LC3 and the conversion of LC3-I to LC3-II are widely used as markers to monitor autophagy.Citation17 Although autophagy has generally been considered as a nonselective process induced during starvation, other forms of autophagy function as specific degradation of mitochondria (mitophagy), peroxisomes (pexophagy) or the edoplasmic reticulum (reticulophagy).Citation18,19,20 For misfolded protein aggregates, the proteins SQSTM1 (sequestosome 1) and NBR1 (neighbor of BRCA1 gene 1), which both contain ubiquitin and LC3 binding domains, serve as selective adaptors to guide ubiquitinated, misfolded proteins to phagophores.Citation13 Consequently, inhibition of autophagy causes accumulation of polyubiquitinated proteins/SQSTM1 aggregates.Citation21 Physiologically, defects in autophagy have been directly linked to developmental defects, aging, autoimmunity and atherosclerosis.Citation22–25
The Harderian gland is, like other exocrine glands such as the lacrimal, the sebaceous or the mammary gland, an epithelial lineage cell derived organ. It is located retro-orbitally and present in most vertebrates, forming a tubuloalveolar network with a layer of luminal columnar epithelial cells surrounded by basal myoepithelial cells. In mice, the glandular epithelium consists of 2 types of secretory cells, the more numerous type A cells being responsible for the secretion of lipid droplets, while the type B cells contain dark granules of multilamellar bodies.Citation26
Harderian ductal cells are highly metabolically active and primarily secrete the triglyceride analog 1-alkyl-2,3-diacylglycerol, porphyrin, and multilamellar vesicles.Citation27,28,29 The main route of secretion is exocytosis, but apocrine and holocrine secretion is also observed. Secreted lipids participate in protection and lubrication of the cornea and in water repulsion and thermoregulation of the fur.Citation30 The function of multilamellar bodies could be reminiscent of those found in pneumocytes type 2 to stabilize the surface.Citation31 Porphyrin, is released primarily by holocrine secretion and can be found as intraluminal accretions mostly in heterologous aggregates with lipids and proteins.Citation32 Due to the large amounts of porphyrins, which accumulate, the Harderian gland is highly exposed to oxidative stress.Citation33,34 The occurrence of autophagosomes in the Harderian gland was observed in electron micrographs and further demonstrated by immunofluorescence in GFP-LC3 reporter mice.Citation23,35
Whereas defective clearance mechanisms and accumulation of aberrant metabolic products have been widely investigated, the consequences of the perturbation of metabolic pathways in cells of the epithelial lineage are not yet understood. We have thus investigated the role of autophagy in the Harderian gland using a Krt14-Cre-driven Atg7-deficient mouse model.
Results
Generation of epithelial-specific ATG7-deficient mice
The Harderian gland is an epithelial derived organ with a layer of luminal columnar epithelial cells surrounded by basal myoepithelial cells.Citation29 In young mice myoepithelial cells express smooth muscle differentiation markers in addition to those of basal keratinocytes and are progenitors of luminal cells. In adulthood myoepithelial cells differentiate to myofibroblasts thereby loosing basal epithelial markers. Thus we investigated the expression of the epithelial KRT14 (keratin 14) in the Harderian gland of young mice. As seen in the immunohistochemistry of 3-wk-old mice, basal myoepithelial cells, morphologically characterized by an elongated nucleus parallel to the basement membrane, stain positive for KRT14 (). Hence a transgenic line expressing Cre recombinase under control of a Krt14 promoter can be used as a deletion-trigger in the Harderian gland.
Figure 1. Characterization of autophagy-deficient atg7−/− HaGls. (A) KRT14 expression in myoepithelial cells of the HaGl visualized by immunohistochemistry against KRT14 in young mice (3-wk-old). These basal layer cells in the ducts are formed by myoepithelial cells, which are mitotically active. Ductal cells with large and round nuclei form the inner surface of the ductal lumen. The lumen is partially filled with an amorphous substance from these secretory cells. Myoepithelial cell (→), secretory cell (*), ductal lumen (L). Size bar = 20 μm. (B) Genotyping PCR showing loss of floxed allele in HaGls of Atg7f/f Krt14-cre (atg7−/−) mice. (C) Immunoblot for ATG7, LC3 and GAPDH on Atg7f/f and atg7−/− HaGl tissue homogenates showing almost complete absence of ATG7 (upper band, lower band represents unspecific signal) and of the processed LC3-II form of LC3-I. The lack of the LC3-II band in atg7−/− samples demonstrates abrogation of autophagy. Note: The presence of residual ATG7 protein likely results from other cell types present in the HaGl, such as blood cells. (D) Sections of GFP-LC3 transgenic Atg7f/f and atg7−/− mice showing the presence of a discrete number of ductal cells that display GFP puncta reminiscent of autophagosomes in Atg7f/f. Note: The cell in the center of the Atg7f/f section is binucleated, a feature of the HaGl. In contrast, and in agreement with the increase in LC3-I in the atg7−/− background, GFP-LC3 is diffusely distributed and accumulates in these samples, indicative of free form of GFP-LC3 not incorporated into autophagic vesicles. Note: a few GFP spots can still be detected in atg7−/− cells possibly resulting from GFP aggregates. *, vacuoles completely excluding GFP; arrow, small spots also excluding GFP found in both genotypes; L, ductal lumen. Nuclei were visualized with Hoechst. Size bar = 10 μm.
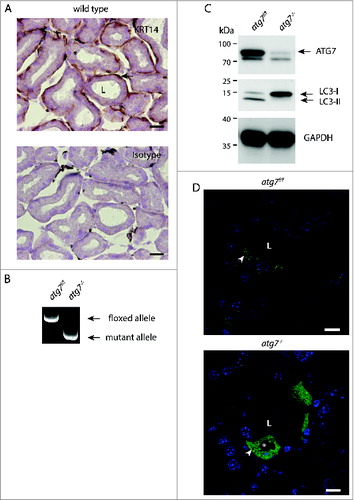
Atg7-floxed mice (Atg7f/f) were crossed with a transgenic line expressing Cre recombinase under the control of a Krt14 promoter to generate epithelial atg7-deficient mice, hereafter referred to as atg7−/−.Citation22,36 Deletion of the floxed sequence in genomic DNA from Harderian gland was confirmed by PCR analysis (). Accordingly, ATG7 protein was almost undetectable in Harderian gland homogenates (). Functional inhibition of autophagy in the gland was confirmed by immunoblotting, showing accumulation of unlipidated LC3-I form and diminution of the processed lipidated LC3-II form (). This result was confirmed by the analysis of LC3-I processing in 3 additional independent Harderian gland samples of each genotype (Fig. S1A).
The occurrence of autophagosomes and their cellular distribution in Harderian glands was analyzed in sections from GFP-LC3 transgenic animals by fluorescence microscopy. In the Harderian gland of GFP-LC3 transgenic control mice about 25% (n = 3) of ductal cells, identified by their large round nuclei, show numerous GFP-”puncta” ( and S1B, S2B). At high magnification it became obvious that many of the puncta in Atg7f/f animals, were not perfectly circular, but often displayed a crescent-like appearance (Fig. S1B). In contrast, in sections of the Harderian gland of Atg7-deficient mice a diffuse cellular GFP staining, indicative of free GFP-LC3 was observed. A few residual GFP-spots were still visible in atg7−/− animals, potentially resulting from intracellular GFP-aggregates, because GFP is prone to accumulation when overexpressed (, lower picture).Citation37 Furthermore, in ductal cells of Atg7-deficient Harderian glands, large intracellular circular areas excluding GFP completely were observed (asterisk in ). These areas originate from large intracellular vacuoles present in Atg7-deficient mice, which can be clearly seen histologically ( lower panel). It is of note that only approximately 5% of the atg7−/−, GFP-LC3 accumulating cells contain large vacuoles ( and S2B). These large vacuoles differ in size from the more abundant tiny spots found in both genotypes, which also exclude GFP. Massive vacuolization was described in cells under endoplasmic reticulum (ER) stress caused by a defect in protein degradation induced with proteasome and/or autophagy inhibitors.Citation38 From this set of experiments we conclude that the Harderian gland can be used as a model to investigate the role of autophagy by conditional gene deletion.
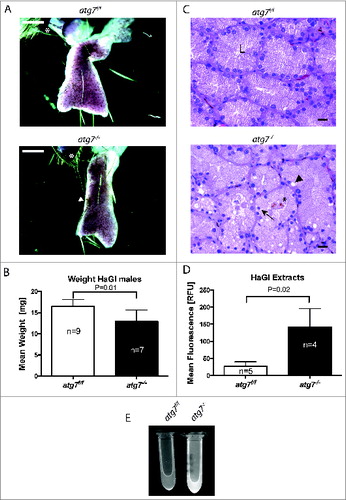
Figure 2. Pathology of autophagy-deficient HaGls. (A) Appearance of carmine red stained whole mount HaGl. Two to 3 lobes are visible with needle shaped pigmented deposits originating from crystalline porphyrin. The preparations contain part of the eyelid (*) for orientation. The atg7−/− glands appear smaller and contain numerous areas of accumulated crystalline porphyrin. White arrowhead corresponds to crystalline pigmented deposits. Size bars = 1 mm. (B) Bar graph quantifying organ weight revealing reduced weight of atg7−/− glands compared to Atg7f/f controls. P = 0.013. (C) Haematoxylin and eosin stained 5 μm sections of Atg7f/f and atg7−/− Harderian glands. In atg7−/− glands the following pathological characteristics were observed: loss of the organized appearance of the ducts including large vacuoles and deposits of pigmented material (porphyrin). Nuclei were often condensed or variable in size and located away from the basement toward the glandular lumen. L, glandular lumen; *, pigmented material; black arrowhead, large vacuoles; black arrow, suprabasal nucleus. Size bars = 20 μm. (D) Quantification of porphyrin levels within the HaGl under UV bright field fluorescence. Statistical analysis indicated a significantly higher fluorescence of atg7−/− extracts, P = 0.02. (E) Markedly increased porphyrin content in the atg7−/− HaGl extracts. The content of porphyrins is visualized by UV bright field fluorescence.
Autophagy inhibition induces hypotrophy, porphyria and tissue degeneration in the Harderian gland
As previously reported, Krt14-Cre conditionally deleted atg7−/− animals were indistinguishable from control animals with regard to body weight, fertility and immunoprofile.Citation39 Harderian gland preparations from adult mice, however, revealed a reduced organ size that correlated with a weight reduction of the gland: 12.91 ± 2.7 mg in atg7−/− versus 16.46 ± 1.7 mg in age-matched control Atg7f/f mice (P = 0.013) (). Furthermore, histological analysis of the glands revealed degenerative changes, including loss of the organized ductal morphology, highly vacuolated cells and small/shrunken darkly stained (pyknotic) nuclei of irregular shape (). Occasionally nuclei were shifted from the basement toward the lumen of the duct. Tissue degeneration was substantiated by detection of cell death by (TdT-mediated dUTP-biotin Nick End Labeling) staining in atg7−/− ducts (Fig. S2C). Further, these morphological alterations were clearly visible in ultrathin sections of Harderian glands (Fig. S3). Most prominently, nuclei were observed in the lumen of atg7−/− ducts and some cells displayed deformed nuclei in the cell center surrounded by liposomes. Large heterologous aggregates were identified juxtanuclear in atg7−/− cells (Fig. S3B).
The observed degeneration of the Harderian gland was accompanied by luminal deposition of porphyrin, a pigmented material ().Citation28 Since this compound absorbs UV light with an absorbency maximum near 400 nm and emits light in the visible red, the porphyrin content of Harderian gland extracts was verified by spectral analysis and quantified under UV bright fluorescence (, and S4).Citation30 Statistical analysis indicated a significantly higher fluorescence of atg7−/− (142.2 ± 54) vs. Atg7f/f extracts (26.9 ± 13 RFU; P = 0.02). Taken together this phenotype is characteristic for a degeneration of the Harderian gland.
Degeneration is accompanied by an accumulation of neutral lipids and misfolded proteins
The Harderian gland secrets large amounts of lipids. Since autophagy has been shown to directly regulate lipid- and protein-homeostasis, we decided to investigate molecular markers for both misfolded proteins and lipid accumulation.Citation15,40
PLIN2/adipophilin (perilipin 2) is a peripherally associated membrane protein of lipid droplets and in atherosclerosis it concentrates in cholesterol deposits of foam cells.Citation41,42 Immunoblots from homogenates of the Harderian gland consistently showed an increase of PLIN2 (). Similarly, thin layer chromatography (TLC) displayed differential accumulation of free cholesterol (FC) and showed an overall lipid spectrum similar to the epidermis ().Citation43 In relation to the gland weight the FC was elevated (P = 0.002, n = 4) (). Qualitative and quantitative analysis of FC, free fatty acids and triglycerides was performed by gas chromatography, revealing that only FC was significantly elevated by 60%, (P = 0.06, n = 3) (). This accumulation was further substantiated by fluorescent staining of cryo sections with the free cholesterol dye filipin. Cholesterol accumulating cells were only identified in atg7−/− ducts (). Occasionally, filipin positive spots were also observed in the ductal lumen (Fig. S5A). Other neutral lipid dyes such as BODIPY and ORO could not quantitatively discriminate between the genotypes, however morphologically lipid loaded cells could be identified in sections of atg7−/− glands (Fig. S5B). Hence, we clearly observe a difference in cholesterol levels in atg7−/− glands, and possibly also an accumulation of neutral lipids given the marked differences in PLIN2.
Figure 3 (See previous page). Molecular phenotype of lipid and misfolded protein response. (A) Immunoblot of Harderian gland samples for the lipid droplet marker PLIN2, the protein aggregate marker SQSTM1, and ubiquitin as a marker for misfolded protein response. Consistently in the absence of autophagy, atg7−/− gland samples accumulated PLIN2, SQSTM1 protein, and SQSTM1 aggregates (higher molecular weight species of SQSTM1, which is not resolved on the SDS gel), as well as ubiquitin in comparison to the Atg7f/f control samples. (B) Determination of lipid composition by thin layer chromatography. HaGl extracts, normalized to their wet weight. Four different animals of each genotype were separated by thin layer chromatography with solvents for a broad lipid spectrum. In extracts of atg7−/− glands the amount of free cholesterol (FC) is increased. A prominent band corresponds to the triglycerides analog (TG) 1-Alkyl-2,3-diacylglycerol, which according to the literature is the most abundant class of lipids of the Harderian gland. Wax esters and cholesterol esters (CE), free fatty acids (FFA), Ceramides (Cer), polar lipids (mostly phospholipids, PL). (C) Quantification of free cholesterol from the TLC. Analysis of the band intensity showed a 30% increase in of free cholesterol compared relative to the gland weight (P = 0.0017). (D) Immunohistochemistry for the protein aggregate marker SQSTM1. While no SQSTM1 is detected in sections of the Atg7f/f control, the signals for SQSTM1 have a granular and circular appearance and were sometimes localized in a juxtanuclear position or at the base of the duct in sections of atg7−/− glands. Here the size of the SQSTM1 granule indicates large inclusions and deposits, which were found in a mosaic like pattern affecting cells unequally. In addition, SQSTM1 is weak and diffusely distributed in the cytoplasm and in the secretions within the ductal lumen. The diffuse SQSTM1 distribution may reflect a more immature state of potentially not further aggregated cellular forms of misfolded proteins. Black arrowhead corresponds to large vacuoles found in atg7−/− cells that did not stain for SQSTM1. L, ductal lumen. Size bar = 20 μm. (E) Ubiquitin accumulation in atg7−/− glands. Ubiquitinated proteins are highly abundant in the cytoplasm of atg7−/− ductal cells compared to controls. Ubiquitin staining appears partly granular indicating aggregates. Cell debris containing ubiquitin can be detected in the ductal lumen (L). Size bar = 20 μm. (F) Cholesterol aggregating cells of atg7−/− glands. Filipin III binds to diffuse and aggregated cholesterol in atg7−/− glands. Such cells highly accumulating cholesterol (white arrow) were absent in control sections. Some vacuoles exclude cholesterol (white arrowhead). Size bar = 20 μm. (G) Coimmuno-fluorescence of SQSTM1 protein aggregates and PLIN2 lipid droplets. Sections of atg7−/− HaGls reveal an overall higher abundance of PLIN2-positive lipids droplets. SQSTM1 staining is granular and diffuse. Interestingly, discrete cells accumulate both PLIN2-positive lipid droplets and SQSTM1, while this is not observed in cells of control animals. PLIN2 is stained in red, SQSTM1 in green, nuclei are stained in blue by Hoechst. Size bar 20 μm. (H) Single cell laser scanning image of a SQSTM1 and PLIN2 double-positive cell. PLIN2 staining has a circular, droplet like appearance, while SQSTM1 stains large granules and diffusely in the cytoplasmic. SQSTM1 staining is close to the droplets but does not entirely cover the PLIN2 positive structures. Largely these markers appear separate. Note that in this particular cell the nucleus is heavily deformed. PLIN2 stains in red, SQSTM1 in green, nuclear stain Hoechst in blue. Size bar = 5 μm. (I) Gas chromatographic analysis of neutral lipids of the HaGl. Free cholesterol is markedly increased in lipid extracts (n = 3, P = 0.06), whereas triglycerides and free fatty acids do not differ.
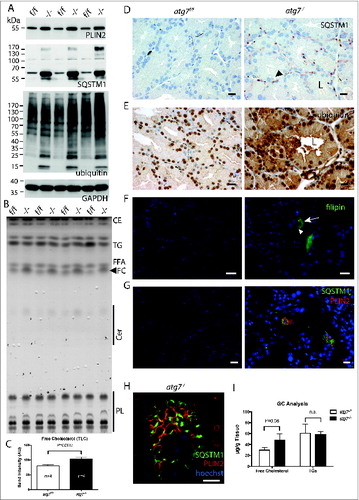
In addition, we analyzed SQSTM1 expression, a multifunctional adaptor protein, which directly interacts with ubiquitin. On the one hand, SQSTM1 is a cargo marker for protein and protein aggregate removal by autophagy. On the other hand, it forms a shell-like structure around protein aggregates potentially shielding them and thereby protecting cells from misfolded protein induced cell death.Citation21 Accumulation of SQSTM1 in ubiquitin-positive protein inclusions of hepatocytes and neurons upon conditional Atg7 deletion has been previously reported.Citation40 Likewise, strong accumulation of ubiquitin and SQSTM1 was observed by immunoblotting of atg7−/− Harderian gland homogenates compared to controls (). Accordingly, a pronounced signal for ubiquitin was also observed by immunohistochemistry on sections of atg7−/− glands (). In the SQSTM1 immunoblots, we noted the presence of a high molecular weight smear of SQSTM1 aggregates not resolved by electrophoresis (asterisk in ). Such a high molecular weight smear is indicative for aggregates of polyubiquitinated misfolded forms of proteins occurring during inhibition or saturation of the proteasome and under defective autophagy.Citation8,44 Immunohistochemistry on sections of atg7−/− Harderian glands showed an intense granular and circular SQSTM1 staining (). The size of the SQSTM1 positive spots and their circular appearance is in agreement with a previous report of SQSTM1 forming a shell around large aggregates of misfolded proteins.Citation21 Large juxtanuclear aggregates formed of membranous, multi vesicular structures and lipid droplets were observed in ultra thin sections (Fig. S3B). Nevertheless the large vacuoles observed in atg7−/− glands did not stain for SQSTM1 (arrowhead in ). Little to no SQSTM1 staining was detected in Harderian glands of control animals, demonstrating an effective clearance of SQSTM1 aggregates by autophagy under normal conditions (). As differential sexual regulation of the Harderian gland has been shown in another species, we examined these pathology markers in the females as well.Citation45 However, we found no striking difference of SQSTM1 and PLIN2 accumulation between both sexes in mice (Fig. S6A).
Because of the accumulation of SQSTM1, its circular staining, together with excessive lipid storage, we hypothesized that SQSTM1 directly interferes with lipid storage, for instance by targeting lipid droplets via SQSTM1 to autophagosomes. This is supported by the observation of a similar lipid accumulation phenotype in a SQSTM1-deficient mouse model.Citation46 We therefore used immunofluorescence to test whether SQSTM1 and PLIN2 colocalize. As expected an overall higher abundance of PLIN2 in Harderian glands of atg7−/− animals was observed by immunofluorescence (). Furthermore, in a discrete number of cells PLIN2 was highly abundant, which was not seen in the control. Peculiarly some of these lipid-overloaded cells were also positive for SQSTM1 ( and S6B). One of the SQSTM1 and PLIN2 double-positive cells was visualized in detail on a laser scanning microscope (LSM; and Fig. S7). Note that in this droplet loaded cell the nucleus became heavily deformed. The LSM revealed that while the granular SQSTM1 staining was close to the PLIN2-positive stained circular droplets it did not fully overlap with the PLIN2 positive structures. Rather, misfolded aggregated proteins may expose their hydrophobic surfaces, which could bring SQSTM1 in close proximity to lipid droplets (for single channel images see Fig. S7).Citation47 Thus we conclude that a direct involvement of SQSTM1 in the turnover of lipid droplets seems unlikely. Although the occurrence of a misfolded protein response and defects in neutral lipid turnover are well established in the absence of autophagy, the co-occurrence of both pathologies has to our knowledge not yet been reported.
Deficiency in autophagy leads to the formation of tertiary lysosomes and lamellar stacks of the endoplasmic reticulum
One of the striking phenotypes in ATG7-deficient Harderian glands, is the formation of large intracellular vacuoles, found in 5% of total cells in both sexes. These vacuoles were not positive for SQSTM1 () or the lipid markers filipin and PLIN2 (), and stained only faintly with the neutral lipid dye BODIPY (Fig. S5B). Hence, we conclude that this represents a different phenotypic aspect of atg7−/− Harderian glands, potentially affecting type B cells.
In electron micrographs of Harderian glands of both genotypes we could identify type B cells secreting multilamellar bodies (Fig. S9).Citation48 These cells contain compartments required for a high anabolic rate namely a prominent ER and a large Golgi (). A detailed analysis of an Atg7f/f cell () revealed that the multilamellar bodies originate by fusion of lamellar bundles with lysosomes ( and S10A) and grow by the absorption of lipid droplets (dark stained) ().
Figure 4 (See previous page). Ultrastructure of ductal Harderian gland cell. (A) Type B HaGl cell containing multilamellar bodies (LB) with a woolly appearance in different stages of maturation. (B) Nascent lamellar body of an early stage. During the formation of these secretory vesicles, small primary lysosomes (white arrowhead) fuse with the nascent lamellar body containing a few lamellae (white arrow). (C) Nascent lamellar body of a later stage. Further lamellae (white arrow) are acquired by the incorporation of dark lipid drops (*) and the fusion of lysosomes (white arrowhead). (D) Tertiary lysosomes in atg7−/− HaGls. A tertiary lysosome (Ly) adjacent to the nucleus occupies a large area of the cytoplasm in an autophagy-deficient mouse. The content of this vacuole is not fully homogeneous, but has several pieces of membrane stacks and lipids (**). This 7-μm spanning compartment is separated from the cytoplasm by a unit membrane and has a further inclusion (E). In addition, tight lamella stacks, lipid clefts and smaller tertiary lysosomes are observed in the cytoplasm of this cell. (E) Inclusions of the tertiary lysosome. The enlarged area displays rough endoplasmic reticulum (rER), tight lamella stacks (double arrows), lipid inclusions (**) and electron dense areas potentially originating from disassembled lamellae. (F) High magnification of tight lamella stacks revealed that they are continuous with the rough endoplasmic reticulum (rER), where ribosomes are stripped off at a certain site (opposing arrows). (G) Further tight lamellar ER stacks (double arrows) with additional inclusions of rER and lipids (**). LB, multilamellar body; rER, rough endoplasmic reticulum; M, mitochondria; G, Golgi; arrowhead, primary lysosome; arrow, lamella; Ly, tertiary lysosome; LD, lipid droplet of an adjacent cell; N, nucleus; opposing arrows, demarcation line between rough endoplasmic reticulum and lamellar stacks; double arrows, tight lamella stacks; *, small electron dense/dark lipid drop; **, lipid inclusions. Size bars: 1 μm ((A)and D), 500 nm (B, C, (E)and G).
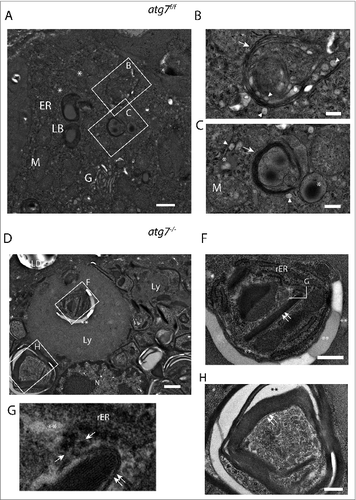
Multilamellar body formation involves autophagy in vitro in type 2 pneumocytes and hence could be impaired in atg7−/− Harderian glands, leading to an accumulation of their constituents.Citation49 In electron micrographs of atg7−/− Harderian glands we identified cells containing a large vacuole surpassing the size of the nucleus (). This vacuole differs from swollen lipid droplets and protein aggregates by its structure and size. Based on the morphology they are similar to tertiary lysosomes described for human diseases, typically containing inclusions of undegraded material.Citation50 The tertiary lysosomes seen in are separated from the cytoplasm, and from another inclusion (), by unit membranes. In addition, many tight lamellar stacks were observed within the lysosomal inclusion () and also in the cytoplasm (). As seen in , the lamellar stack in the center of the lysosome appears to be continuous with the rough ER. Close to the tip of the stack, the ER is opened giving rise to electron translucent area, possibly lipids. Further ribosomes are lost at this site. The tight lamellar ER stacks can be seen in various pseudo crystalline forms with additional inclusions of rough ER ().
We suggest that our observations demonstrate that in the absence of autophagy, lysosomes that are usually required for the formation of multilamellar bodies give rise to huge tertiary lysosomes with various inclusions, accompanied by an accumulation of tight lamellar stacks of the ER.
Combined effect of protein and lipid toxicity in autophagy-deficient cells
Protein and lipid accumulation are hallmarks of human degenerative disease.Citation51–54 However, the interplay of these factors and their effects on different cells are poorly understood.
Based on our observations we hypothesized that the combination of misfolded proteins and excess of lipids exerts a toxic effect on cells and that this effect is intensified if autophagy is compromised. As Harderian gland cells cannot be cultured, we turned to primary murine keratinocytes from Atg7-deficient and control animals as a source for our in vitro experiment examining the interplay of protein and lipid accumulation. We reasoned that keratinocytes may be a good alternative cell, as both Harderian gland cells and keratinocytes are of epithelial origin. Furthermore, dysfunction of the proteasome activates an unfolded protein response in primary keratinocytes and the epidermis displays a lipid composition analogous to the TLC spectrum of the Harderian gland.Citation43,55 Most importantly, lipids are also incorporated into lamellar bodies in this tissue.Citation56
For the in vitro experiment we cultivated the keratinocytes under conditions causing either a misfolded protein response, or lipid accumulation, or both. Thus, to provoke lipid accumulation we used easily incorporated free fatty acids, which are both esterified to triglycerides and cholesterol ester. Accumulation of misfolded proteins was induced by the addition of the proteasome inhibitor MG132 causing an accumulation of ubiquitinated proteins and the misfolded protein aggregate marker SQSTM1 ().Citation57 SQSTM1 accumulation was more pronounced in keratinocytes isolated from atg7−/− than from Atg7f/f animals. The enhanced intensity of the LC3-II band in Atg7f/f and LC3-I band in atg7−/− keratinocytes further indicates the induction of autophagy during MG132 mediated proteasome inhibition (). Likewise, palmitate supplementation caused an upregulation of the lipid droplet associated protein PLIN2, indicating accumulation of intracellular lipids (). Analogously to the protein aggregation phenotype, the lipid accumulation phenotype was more pronounced in atg7−/− keratinocytes. When adding both agents together, both, the increase in misfolded protein accumulation and lipid storage, were more pronounced than when adding the reagents separately, reflected by the higher abundance of the respective markers. Again as expected, this was more aggravated in the autophagy-deficient cells (). Similar results were observed at a later time point with respect to the lipid marker PLIN2 and the marker for the misfolded protein response, ubiquitin (Fig. S11). In conclusion, we can artificially induce the phenotype resembling that observed in the Harderian gland with respect to accumulation of misfolded proteins and lipids in cultivated keratinocytes.
Figure 5 (See previous page). Effect of misfolded protein stress and excess of lipids in Atg7f/f and atg7−/− keratinocytes. (A) Immunoblot of extracts from primary murine keratinocytes treated with MG132 (250 nM) or palmitate (62.5 and 125 μM) or a combination of both for 16 h. Controls were treated with DMSO and the lipid carrier bovine serum albumin. The addition of MG132 causes an accumulation of ubiquitinated proteins and the misfolded protein aggregate marker SQSTM1. The addition of palmitate causes an upregulation of the lipid droplet protein PLIN2. Both responses, the misfolded protein and lipid accumulation, are more pronounced in keratinocytes isolated from atg7−/− animals. Similarly, PARP cleavage is more pronounced in autophagy-deficient keratinocytes. Cell death was further quantified by histone ELISA in this experiment (lower panel). In all treatments atg7−/− keratinocytes are more sensitive than control cells. The strongest induction of cell death was measured when combining MG132 and palmitate. Experiments were done in triplicates. (B) Inhibition of autophagy with bafilomycin A1. C57Bl/6 derived keratinocytes were treated with or without bafilomycin A1 (50 nM), MG132 (250 nM) and/or palmitate or oleate (both 62.5 (lc) and 125 μM (hc)). The induction of cell death seen in bafilomycin A1-treated keratinocytes by palmitate or in combination of palmitate and MG132 is similar to those in atg7−/− cells. MG132 alone was a less potent inducer of cell death in this setting. The addition of oleate was tolerated by keratinocytes. Palmitate (PA), oleate (OA).
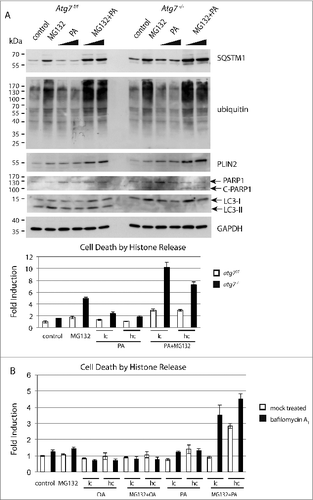
Because degenerating keratinocytes undergo cell death, apoptosis was measured in cultured cells using 2 independent methods. During apoptosis PARP1 (poly [ADP-ribose] polymerase 1) is cleaved from a full-length 116-kDa to an 89-kDa form (C-PARP1), which can be detected on an immunoblot.Citation58 When we assayed for PARP1 cleavage, we noted that control cells were able to cope to a certain extent with the stress induced either by misfolded protein accumulation induced by MG132 or an excess of lipids through addition of palmitate, when added separately (). While atg7−/− cells were more sensitive to the misfolded protein accumulation stress, they tolerated excess of lipid stress. However, under combined conditions aggravated cell death occurred particularly in atg7−/− cells. Histone release upon DNA fragmentation is another hallmark for cell death and can be easily quantified using an ELISA.Citation59 Consequently histone release was measured under the same conditions as in the PARP cleavage assay. Again, MG132 caused toxic stress to control and Atg7−/− cells, but cell death was only markedly elevated in the latter, thereby demonstrating a defect in handling misfolded proteins upon autophagy deficiency. When combining MG132 and palmitate a severe increase in toxicity was observed in atg7−/− cells, in contrast this treatment was mostly tolerated in atg7f/f cells (). Taken together, in all treatments atg7−/− cells were more sensitive to the induction of cell death than control cells and this was most obvious in a combined MG132 and palmitate treatment.
The isolated atg7−/− keratinocytes may have acquired irreversible defects before treatment, potentially influencing the experimental outcome. We thus used the lysosomal inhibitor bafilomycin A1 to abrogate autophagy in vitro in wild-type keratinocytes. In addition to palmitate, we introduced oleate, a less problematic free fatty acid to provoke lipid accumulation. The results assessing cell death by quantification of histone release in this setting were in agreement with those using atg7−/− cells (). As has been reported previously, oleate exhibits a less toxic effect than palmitate, even in combination with MG132.Citation60 The only marked difference was in the inhibition of the proteasome, which induced higher toxicity in atg7−/− keratinocytes (). This result further supports our finding that the use of a proteasome inhibitor in combination with palmitate has a higher toxicity than an individual exposure, which becomes pronounced when autophagy is compromised.
Discussion
Our results show that basal levels of autophagy are required for maintenance of the Harderian gland. Lack of autophagy in ductal cells causes a disturbance of the metabolic homeostasis, which leads to the formation of large tertiary lysosomes and provokes an accumulation of lipids and misfolded proteins, accompanied by tissue degeneration. A massive destruction of the Harderian gland associated with lymphocyte infiltration has been described in a nonobese diabetic mouse model.Citation61 In contrast, no difference in infiltrate was observed in our model, ruling out the possibility that tissue degeneration is caused by autoimmunity.Citation39 Our interpretation that the phenotype is caused by a disturbed metabolic homeostasis is supported by the results from the in vitro experiments.
Both secretory cell types were affected in autophagy-deficient Harderian glands; type A cells accumulated heterologous protein and lipid aggregates, while large vacuoles and tight lamellar stacks of ER were formed in type B cells. The underlying processes are obviously different, but have in common that in both cases lipids and proteins, which are secreted, accumulate.
The observation that either lipids or misfolded proteins accumulate has been described in autophagy-deficient models of adipocytes, neurons, myocytes, and other cells, but has been so far not reported to occur in combination as described here for the Harderian gland.Citation15,22,52,62 As such we would like to put forward the concept that a combination of both enhances cell death. This is supported by our in vitro keratinocyte experiments, where the combined stimulus of misfolded proteins together with an excess of lipids led to an enhanced cell death in wild-type cells. The effect on cell death was even more prominent in autophagy-deficient cells. In agreement with Singh and colleagues and Hara and colleagues, we conclude that autophagy serves as a mechanism for clearing lipids and proteins, and that it is cytoprotective in our model.Citation52,63 Conclusively, loss of white adipose tissue has been demonstrated in an Atg7-deficient adipocyte model.Citation63 Interestingly, the lipotoxic effect in our in vitro experiment, caused by the free fatty acid palmitate, was negligible in comparison to the toxicity of misfolded proteins. Palmitate seems to rather promote the toxicity of misfolded proteins, than causing toxicity on its own. Vice versa, if a cell is under misfolded protein stress, it will be especially sensitive to lipotoxicity. Consistent with our hypothesis an increase in toxicity when combining palmitate and MG132 was observed in wild-type keratinocytes treated with the autophagy inhibitor bafilomycin A1 (). In contrast, oleate had no effect. In this context the controversy of autophagy being cytoprotective or cytotoxic during free fatty acid induced cell death may be due to the presence or absence of costimuli, such as misfolded proteins, under the different culture conditions.Citation64,65 The cross talk between the misfolded protein accumulation response and lipotoxicity may take place in the ER. In the ER, misfolded proteins trigger stress and free fatty acids are synthesized into triglycerides.Citation66,67 In the absence of autophagy both stimuli together may overload the capacity of the ER and induce cell death synergistically. Under normal conditions, autophagy is activated in response to ER stress caused by misfolded proteins, thus counteracting cell death.Citation38 An involvement of the ER in our model is supported by the observation of large vacuoles in the absence of autophagy in sections of the Harderian gland. In vitro, cell vacuolization has been described as an indicator of ER-stress caused by misfolded proteins.Citation38 It should be noted that ER stress signaling is different in humans than in other mammals, as the functional executor CASP12/caspase 12 is lost in humans.Citation68
A striking morphological phenotype in ATG7-deficient Harderian glands is the formation of large tertiary lysosomes up to several μm in size. In lysosomal storage diseases, these compartments are pathologically enlarged and accumulate undegraded material, depending on the identity of the cell and on the nature of their defects.Citation50 It is the basic concept of autophagy that lysosomes fuse with autophagosomes, hence this explains the formation of aberrant lysosomes in the absence of autophagy. The high abundance of lysosomes in atg7−/− glands is consistent with the accumulation of free cholesterol as in atherosclerosis macrophages which can store excessive cholesterol in lysosomes.Citation69 Similarly, in Niemann-Pick disease type C intracellular cholesterol trafficking is disturbed, leading to neuronal dystrophy associated with an accumulation of lamellated inclusions in mouse models of the disease.Citation70 The ultrastructure of tertiary lysosomes observed in atg7−/− Harderian gland cells is heterologous and contains electron translucent patches as well as membrane stacks. The membrane stacks found in these cells are highly organized staples of smooth ER in a pseudo crystalline state. Adjacent to these sites some darker areas of fragmented membranes were seen. Possibly, these pseudo crystals of smooth ER undergo fragmentation to smaller pieces of membranes. It is also likely that the expansion of the ER serves to counterbalance unfolded protein response of secretory proteins as seen in yeast, and autophagy reduces excessive ER thereafter.Citation71
A subset of Harderian gland cells, the type B cells (Fig. S9), form multilamellar bodies by fusion of lipid droplets and primary lysosomes (). The lipid droplets and the growing multilamellar bodies are an interesting union. This probably depicts a fundamental biological process in which a fluid phase is organized into biological membranes (). The diameter of the structure is more than 1 μm, and some autophagosomes observed in the GFP-LC3 model appear to be similar in size ( and S1B), thus supporting the possibility of a direct involvement of autophagy. In agreement, an involvement of autophagy in the generation of multilamellar bodies in type 2 pneumocytes has been demonstrated previously in vitro.Citation49 As multilamellar bodies were seen in both control and autophagy-deficient Harderian glands, we assume that autophagy only participates in certain steps. Interestingly, autophagy has been shown to mediate protein secretion in the specific condition of oncogene-induced senescence, a state of continuous metabolic activity.Citation72 Additionally autophagy is directly involved in unconventional protein secretion in D. discoideum, P. pastoris and S. cerevisiae.Citation73,74 Alternatively autophagy might be responsible for clearing defective organelles involved in this process as lysosomes (autophagy-mediated lysosome turnover), or controlling the ER.Citation20,75
It is of note that in contrast to abolishing autophagy and challenging keratinocytes in vitro, inhibition of autophagy in vivo in the skin leads to only a very mild phenotype.Citation36 This may be explained by the observation that proteasomal activity is responsible for maintaining protein homeostasis in keratinocytes.Citation55 The crucial role of the proteasome in the skin was previously demonstrated as a gradual loss of the proteasome toward the granular layer of the skin caused the disease KLICK genodermatosis.Citation76 It is known that lamellar bodies, secretory vesicles of the skin, deliver cholesterol and proteins to the surface and could there for be affected by ablation of autophagy.Citation77 Hints of defective lamellar bodies formation were initially observed in the skin of ATG7-deficient mice, but could not be confirmed in a follow-up study.Citation36
Ultrastructural analysis of ductal cells showed aggregates of heterologous content to be concentrated in a juxtanuclear position, where they mostly compromise the nucleus ( and S3). This may be explained by the fact that misfolded proteins are shuttled by the microtubule network connected to the nucleus, which may be impaired in cells of the Harderian gland.Citation57
The observed shedding of atg7−/− Harderian gland cells into the lumen (Fig. S3A), may serve the purpose to either maintain secretory activity or to remove protein and/or lipid accumulating cells from the gland. This form of secretion (holocrine secretion) has previously been observed in the rat Harderian gland, and may be a consequence of a pathological condition.Citation32 In the Syrian hamster, enhanced oxidative stress in females is correlated with increase of holocrine secretion, and it is postulated that autophagy is involved in this form of secretion.Citation78,79 Holocrine secretion in the Harderian gland may also point to a defect in clearing cells from the ductal lumen in the absence of autophagy. In other glandular epithelial cells, cell detachment induces autophagy as a cytoprotective mechanism and in the Drosophila, the development of the salivary gland is delayed, when autophagy is inhibited.Citation80,81
Our experiments lead us to propose the following model to explain how autophagy facilitates secretion (see ): the lysosome converts protein and lipid vesicles dedicated for secretion. Protein excess may lead to their ubiquitination and subjection to proteolysis, while similarly, excess lipids may undergo lipolysis. Alternatively, larger protein aggregates will be SQSTM1-tagged, while problematic lipids will be concentrated in PLIN2-carrying vesicles.Citation13,15 Both structures are normally cleared by autophagy, subjected to lysosomal degradation and secretion. Hence the absence of autophagy causes a vast accumulation of both destabilizing the cell.
Figure 6. Model of autophagy facilitating secretion. The main mechanism of Harderian gland secretion is to directly target proteins and lipids to the lysosome (gray) to generate secretory vesicles (green, round). However, an oversupply of proteins and lipids generated by the high metabolic activity of these epithelial cells causes ubiquitination and SQSTM1 tagging of proteins and PLIN2 coating of lipid drops. These are cleared by autophagy (green) and thus targeted to the lysosome. The inhibition of autophagy (lower part) leads to aggregation, which may cause loss of cell attachment and cell death (apocrine secretion).
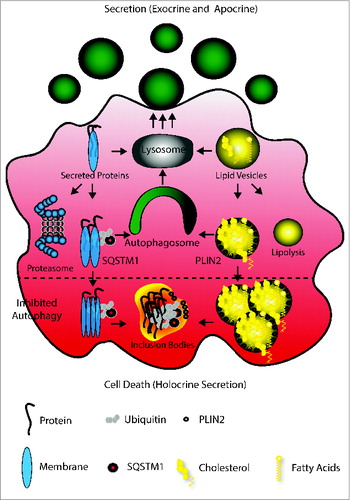
To balance cell death, coping mechanisms to maintain homeostasis may be induced. One of these homeostatic events, in the absence of autophagy, is the formation of inclusion bodies, lipid storages or the generation of heterologous aggregates. In that context, it has been previously proposed that the formation of inclusion bodies acts as a cell protective mechanism in Huntington disease and that triglyceride accumulation protects against free fatty acid-induced lipotoxicity.Citation12,60 Based on this model, we propose that PLIN2 may have a similar function for lipids as SQSTM1 for proteins.
Besides the obvious parallels to lysosomal storage diseases, other atg7−/− epithelial derived organs also displayed PLIN2 and/or SQSTM1 accumulation, but in a very weak manifestation. This includes the skin, the mammary gland and lacrimal gland (unpublished observations). In this respect the Harderian gland serves as a model for the role of autophagy in secretion.
Materials and Methods
Mouse husbandry
To obtain epithelial autophagy-deficient mice, mice carrying the Atg7-floxed allele (referred to as Atg7f/f) were crossed to Krt14-cre transgenic mice (strain Tg(Krt14-cre)1Amc/J) (Jackson Laboratory) to obtain epithelial Atg7-deficient mice (referred to atg7−/−).Citation22 For the generation of autophagosome reporter mice, the GFP-LC3 transgene was further crossed into the Atg7f/f Krt14-cre background.Citation82 The alleles were genotyped as previously described.Citation36 The animal experiments were approved by the Ethics Review Committee for Animal Experimentation of the Medical University of Vienna, Austria (approval number BMWF-66.009/0124-II/10b/2010).
Histology and fluorescence microscopy
For the in situ GFP-LC3 fluorescence analysis, mice were euthanized by carbon dioxide asphyxiation, then immediately perfused through the left ventricle first with cold phosphate-buffered saline (PBS), followed by 4% paraformaldehyde in PBS to fix the tissue. Harderian glands (HaGls) were collected and further fixed with the same fixative for 4 h, followed by treatment with 15% sucrose in PBS for 4 h at room temperature and then with 30% sucrose solution overnight at 4 °C. Tissue samples were embedded in optimal cutting temperature (OCT) medium (Tissue-Tek, 4583) and stored at −80 °C. The presence of GFP-LC3 puncta was investigated using a confocal laser microscope (Zeiss, LSM700, Jena, Germany). For the histological investigation, mice were sacrificed and HaGls were fixed with 7.5% formaldehyde in PBS for 24 h and embedded in paraffin. The following primary antibodies were used for the detection of specific antigens in a 4°C overnight incubation using the following dilutions: rabbit anti-KRT14 1:1000 (Covance, PRB-155P), rabbit polyclonal anti-SQSTM1 1:4000 (Enzo, PW9860), guinea pig anti-PLIN2/adipophilin 1:2000 (Acris, BP5012), rabbit anti-ubiquitin (DakoCytomation, Z0458) 1:500. Filipin III (Cayman, 70440) 0.05 mg/ml in PBS-10% FCS. Hoechst 33258 (Life Technologies, H3569) and DAPI (Sigma, D9542) was used to label the nuclei. Secondary antibodies against the respective species were Alexa Fluor (Life Technologies, A11074 and A11034) or anti-rabbit HRP-conjugated antibody (Dako, P0448). Images were acquired using fluorescence microscopy (Zeiss, LSM700, Jena, Germany; or Olympus, AX70, Shinjuku, Tokio, Japan).
Transmission electron microscopy
Animals were perfused as described above. Samples of approximately 1 mmCitation3 were dissected from HaGls and subsequently fixed with 2.5% glutaraldehyde (Sigma, G7526) in 0.1 mol/l phosphate buffer, postfixed with 2% osmium tetroxide in the same buffer, dehydrated in a graded series of ethanol, and embedded in Agar 100 epoxy resin (Agar Scientific, AGR1045). Ultrathin sections were cut at a nominal thickness of 70 nm, poststained with 2% uranyl acetate and lead citrate and inspected in a transmission electron microscope (Morgagni, FEI 268D TEM, Hillsboro, Oregon, USA) operated at 80 kV. Images were acquired using an 11-megapixel CCD camera from (Olympus-SIS, Shinjuku, Tokio, Japan).
Keratinocyte cultivation and treatment
Epidermal keratinocytes in single cell suspensions were prepared from tail and ear of adult mice, by protease digestion. The cells were suspended according to a modification of the method of Hager and colleagues in low-calcium growth medium KGM (Lonza, CC-3111) and plated at about 20% confluence in Costar 12-well plates coated with collagen (Collagen Corp., Vitrogen-100).Citation83 At confluence cells were incubated with MG132 (Sellek, S2619) dissolved in dimethyl sulfoxide (DMSO; Sigma, D8418) and bovine serum albumin (Roth, 00522), conjugated palmitate (Sigma, P9767), and neutralized oleic acid (Sigma, O1008) which was prepared as previously described.Citation15 For inhibition of autophagy, keratinocytes from C57Bl/6 mice were isolated as above and pretreated for 2 h with 100 nM bafilomycin A1 (Sigma, B1793) or its solvent DMSO. Thereafter the medium was exchanged to 50 nM bafilomycin A1 or DMSO in the presence of the other reagents or respective amounts of solvents or carriers and cultivated for 16 h. After incubation cells were harvested for immunoblotting in RIPA buffer containing protease inhibitor cocktail (Sigma, P8340). The induction of cell death was assessed by the quantitative detection of histone-associated DNA fragments in the cell death detection ELISA kit (Roche, 11 544 675 001) of adherent cells. Experiments were performed in triplicate.
Immunoblotting
Snap frozen whole HaGls were homogenized in RIPA buffer containing protease inhibitor cocktail (Sigma, P8340) in a Precellys bead tissue grinder (Peqlab, 91-PCS-CK14). The insoluble debris was removed by centrifugation and the protein concentration was measured by the BCA (Pierce, 23235) method. Ten to 20 μg of each sample was separated on precast polyacrylamide gels (Bio-Rad, 456–1094) and transferred to PVDF membranes (Bio-Rad, 162–0177). After incubation bands were visualized with SuperSignal chemiluminescent substrate Pierce (Thermo Scientific, 34079). The following first step antibodies were used for the detection of specific antigens in an overnight incubation in 4% nonfat dry milk (Bio-Rad, 170–6404) in TBS-T: mouse anti-GAPDH 1:4000 (Acris, BM439), rabbit polyclonal anti-SQSTM1 1:4000 (Enzo, PW9860), rabbit polyclonal anti-LC3 1:2000 (GeneTex, GTX82986), rabbit anti-ATG7 1:4000 (Sigma, A2856), rabbit anti-PARP1 1:1000 (Cell Signaling Technology, 9542), or incubated in 5% BSA (Sigma, A9647) TBS-T: guinea pig anti-PLIN2/adipophilin 1:4000 (Acris, BP5012), monoclonal mouse anti-ubiquitin clone FK2 1:2000 (Enzo, PW8810). Secondary antibodies against the respective species were diluted 1:10000. For reprobing, blots were inactivated in 30% H2O2 for 15 min at 37 °C.
Lipid analysis
Snap frozen HaGls were homogenized and total lipids extracted according to a modification of the method according to Bligh and Dyer in (chloroform/methanol/formic acid 2:1:0.5 v:v:v) and taken up in chloroform.Citation84 Total lipid extracts were separated by one-dimensional thin layer chromatography as previously described.Citation85 In brief, total lipids were separated on silica gel 60 TLC plates (Merck Millipore, 105721) and following solvent systems were used sequentially: chloroform/methanol/water 40:10:1 (v/v/v) to 10 cm, chloroform/methanol/acetic acid 190:9:1 (v/v/v) to 16 cm and hexane/diethylether/acetic acid 70:30:1 (v/v/v) to 20 cm. In all experiments, the plates were dried under air stream before they were developed with a new mobile phase. Lipids were visualized by exposing the plates to 10% copper sulfate in an 8.5% aqueous solution of ortho-phosphoric acid and subsequent drying and heating at 180 °C. Lipid classes were tentatively identified using standards for cholesterol esters (Sigma, C0289), Triolein (Sigma, T7140), palmitic acid (Sigma, P0500), cholesterol (Sigma, C8667), C18 ceramide (Avanti, 860518) and cholesteryl linoleate (Avanti, C0289). Extracts were normalized to the total tissue weight.
For gas chromatography HaGl extracts were prepared as described above then solubilized with 100 μl of isopropanol/n-heptane/acetonitrile 35:12:52 (v/v). Twenty μl of this solution was introduced into the HPLC device and cholesterol was analyzed as previously described.Citation86 Total triglycerides were measured by means of the CHOD-PAP method on an automated analyzer system (Advia 1800, Siemens Healthcare Diagnostics GmbH, Eschborn, Germany).Citation87
Porphyrin analysis
Harderian porphyrins were prepared as described.Citation30 Extracts were vacuum dried and dissolved in 50 μl chloroform. The total volume of each sample was brought to 1 ml with 0.25 N HCl. For measurement 250 μl sample was mixed with 250 μl 0.25 N HCl and placed in a glass tube on a UV-transilluminator or scanned on a spectrophotometer.
Weight measurement and whole mount preparation
Harderian glands from both eyes were isolated and weighed (Mettler-Toledo, AJ100, Greifensee, Switzerland) and their average weight was used for subsequent calculations. For whole mount preparation organs were spread on glass slides and fixed in Carnoy fixative over night at room temperature. After fixation tissue samples were gradually changed to distilled water and stained in carmine and dehydrated in a graded ethanol series, cleared in xylene and mounted in Eukitt (Sigma, 03989). Reagents were prepared as in http://mammary.nih.gov/tools/histological/Histology/ index.html. Photographs were taken on a stereomicroscope (Leica, MZ16 A, Wetzlar, Germany).
Statistical analyses were calculated and blotted in Graphpad Prism in a Student t-test (2 tailed, unpaired samples), the error bars represent SD. N = number of independent biological samples.
Supplementary Methods
Detection of cell death by the TUNEL (TdT-mediated dUTP-biotin Nick End Labeling).
Paraffin sections were prepared as described above. After rehydration, the sections were treated with 20 μg/ml nuclease free proteinase K, in 10 mM Tris-HCl, pH 7.6, for 15 min at 37 ˚C, after which the kit components were applied according to the manufacturers’ instructions. In Situ Cell Death Detection Kit Fluorescein (Roche Diagnostics, 11 684 795 910) samples were mounted using SlowFade Light Antifade Kit (Life Technologies, S2828). The mounted sections were directly observed by fluorescence microscopy (Olympus AX70).
Oil Red O (ORO) and BODIPY Staining
Thirty ml ORO stock (500 mg, Sigma O0625) in isopropanol were combined with 20 ml destilled water, vortexed and passed through a 0.22 μm filter. Thawed, 5 μm frozen sections were incubated in ORO working solution for 3 min and rinsed in deionized water. For BODIPY staining, sections were incubated in a 1:1000 dilution of 1 μg/μl BODIPY 493/503 (Life Technologies, D3922) DMSO stock solution in 150 mM NaCl for 10 min and rinsed 3 times in PBS. Sections were counterstained with Haematoxylin or DAPI and mounted in fluorescent mounting medium (Dako, S3023).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
978221_Supplementary_Materials.zip
Download Zip (22.2 MB)Acknowledgments
We thank Erwin Tschachler for hosting and supporting this project at the Medical University of Vienna, Department of Dermatology, Masaaki Komatsu, Noboru Mizushima and Ludger Klein for providing and transferring atg7−/− and GFP-LC3 mice, Horst Robenek for giving his expertise on the ultrastructure of the cell, Supawadee Sukseree and Maria Buchberger for technically assisting the experiments, Stefanie Hartmann for graphic design advice, and Leopold Eckhart and Florian Gruber for stimulating discussions.
References
- Brookheart RT, Michel CI, Schaffer JE. As a matter of fat. Cell Metab 2009; 10:9-12; PMID:19583949; http://dx.doi.org/10.1016/j.cmet.2009.03.011
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012; 149:274-93; PMID:22500797; http://dx.doi.org/10.1016/j.cell.2012.03.017
- Ouellet J, Barral Y. Organelle segregation during mitosis: lessons from asymmetrically dividing cells. J Cell Biol 2012; 196:305-13; PMID:22312002; http://dx.doi.org/10.1083/jcb.201102078
- Lin H. Cell biology of stem cells: an enigma of asymmetry and self-renewal. J Cell Biol 2008; 180:257-60; PMID:18227277; http://dx.doi.org/10.1083/jcb.200712159
- Lippens S, Denecker G, Ovaere P, Vandenabeele P, Declercq W. Death penalty for keratinocytes: apoptosis versus cornification. Cell Death Differ 2005; 12 Suppl 2:1497-508; PMID:16247497; http://dx.doi.org/10.1038/sj.cdd.4401722
- Perlmutter DH. Autophagic disposal of the aggregation-prone protein that causes liver inflammation and carcinogenesis in alpha-1-antitrypsin deficiency. Cell Death Differ 2009; 16:39-45; PMID:18617899; http://dx.doi.org/10.1038/cdd.2008.103
- Nishino I, Fu J, Tanji K, Yamada T, Shimojo S, Koori T, Mora M, Riggs JE, Oh SJ, Koga Y, et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 2000; 406:906-10; PMID:10972294; http://dx.doi.org/10.1038/35022604
- Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol 1998; 143:1883-98; PMID:9864362; http://dx.doi.org/10.1083/jcb.143.7.1883
- Masters CL, Multhaup G, Simms G, Pottgiesser J, Martins RN, Beyreuther K. Neuronal origin of a cerebral amyloid: neurofibrillary tangles of Alzheimer's disease contain the same protein as the amyloid of plaque cores and blood vessels. EMBO J 1985; 4:2757-63; PMID:4065091
- Strnad P, Zatloukal K, Stumptner C, Kulaksiz H, Denk H. Mallory-Denk-bodies: lessons from keratin-containing hepatic inclusion bodies. Biochim Biophys Acta 2008; 1782:764-74; PMID:18805482; http://dx.doi.org/10.1016/j.bbadis.2008.08.008
- Vicart P, Caron A, Guicheney P, Li Z, Prevost MC, Faure A, Chateau D, Chapon F, Tome F, Dupret JM, et al. A missense mutation in the alphaB-crystallin chaperone gene causes a desmin-related myopathy. Nat Genet 1998; 20:92-5; PMID:9731540; http://dx.doi.org/10.1038/1765
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 2004; 431:805-10; PMID:15483602; http://dx.doi.org/10.1038/nature02998
- Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol Cell 2009; 34:259-69; PMID:19450525; http://dx.doi.org/10.1016/j.molcel.2009.04.026
- Mizushima N. The pleiotropic role of autophagy: from protein metabolism to bactericide. Cell Death Differ 2005; 12 Suppl 2:1535-41; PMID:16247501; http://dx.doi.org/10.1038/sj.cdd.4401728
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature 2009; 458:1131-5; PMID:19339967; http://dx.doi.org/10.1038/nature07976
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009; 43:67-93; PMID:19653858; http://dx.doi.org/10.1146/annurev-genet-102808-114910
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008; 4:151-75; PMID:18188003; http://dx.doi.org/10.4161/auto.5338
- Dolman NJ, Chambers KM, Mandavilli B, Batchelor RH, Janes MS. Tools and techniques to measure mitophagy using fluorescence microscopy. Autophagy 2013; 9:1653-62; PMID:24121704; http://dx.doi.org/10.4161/auto.24001
- Nordgren M, Wang B, Apanasets O, Fransen M. Peroxisome degradation in mammals: mechanisms of action, recent advances, and perspectives. Front Physiol 2013; 4:145; PMID:23785334; http://dx.doi.org/10.3389/fphys.2013.00145
- Bernales S, Schuck S, Walter P. ER-phagy: selective autophagy of the endoplasmic reticulum. Autophagy 2007; 3:285-7; PMID:17351330; http://dx.doi.org/10.4161/auto.3930
- Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005; 171:603-14; PMID:16286508; http://dx.doi.org/10.1083/jcb.200507002
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 2005; 169:425-34; PMID:15866887; http://dx.doi.org/10.1083/jcb.200412022
- Nedjic J, Aichinger M, Emmerich J, Mizushima N, Klein L. Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature 2008; 455:396-400; PMID:18701890; http://dx.doi.org/10.1038/nature07208
- Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K, Noon GP, Frazier OH, Taegtmeyer H. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J 2004; 18:1692-700; PMID:15522914; http://dx.doi.org/10.1096/fj.04-2263com
- Razani B, Feng C, Coleman T, Emanuel R, Wen H, Hwang S, Ting JP, Virgin HW, Kastan MB, Semenkovich CF. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab 2012; 15:534-44; PMID:22440612; http://dx.doi.org/10.1016/j.cmet.2012.02.011
- Woodhouse MA, Rhodin JA. The Ultrastructure of the Harderian Gland of the Mouse with Particular Reference to the Formation of Its Secretory Product. J Ultrastruct Res 1963; 49:76-98; PMID:14058445; http://dx.doi.org/10.1016/S0022-5320(63)80037-4
- Park SH, Kano K, Seyama Y. 1-Alkyl-2,3-diacylglycerol synthesis in primary culture cells of guinea pig harderian gland. J Biochem 1993; 114:492-7; PMID:8276758
- Payne AP. The harderian gland: a tercentennial review. J Anat 1994; 185 (Pt 1):1-49; PMID:7559104
- Johnston HS, McGadey J, Payne AP, Thompson GG, Moore MR. The Harderian gland, its secretory duct and porphyrin content in the woodmouse (Apodemus sylvaticus). J Anat 1987; 153:17-30; PMID:3429316
- Chen W, Kelly MA, Opitz-Araya X, Thomas RE, Low MJ, Cone RD. Exocrine gland dysfunction in MC5-R-deficient mice: evidence for coordinated regulation of exocrine gland function by melanocortin peptides. Cell 1997; 91:789-98; PMID:9413988; http://dx.doi.org/10.1016/S0092-8674(00)80467-5
- Baldan A, Tarr P, Vales CS, Frank J, Shimotake TK, Hawgood S, Edwards PA. Deletion of the transmembrane transporter ABCG1 results in progressive pulmonary lipidosis. J Biol Chem 2006; 281:29401-10; PMID:16887795; http://dx.doi.org/10.1074/jbc.M606597200
- Carriere R. Ultrastructural visualization of intracellular porphyrin in the rat Harderian gland. Anat Rec 1985; 213:496-504; PMID:4083530; http://dx.doi.org/10.1002/ar.1092130404
- Reiter RJ, Klein DC. Observations on the pineal gland, the Harderian glands, the retina, and the reproductive organs of adult female rats exposed to continuous light. J Endocrinol 1971; 51:117-25; PMID:4108164; http://dx.doi.org/10.1677/joe.0.0510117
- Tomas-Zapico C, Coto-Montes A, Martinez-Fraga J, Rodriguez-Colunga MJ, Tolivia D. Effects of continuous light exposure on antioxidant enzymes, porphyric enzymes and cellular damage in the Harderian gland of the Syrian hamster. J Pineal Res 2003; 34:60-8; PMID:12485373; http://dx.doi.org/10.1034/j.1600-079X.2003.02951.x
- Tomas-Zapico C, Caballero B, Sierra V, Vega-Naredo I, Alvarez-Garcia O, Tolivia D, Rodriguez-Colunga MJ, Coto-Montes A. Survival mechanisms in a physiological oxidative stress model. FASEB J 2005; 19:2066-8; PMID:16186173
- Rossiter H, Konig U, Barresi C, Buchberger M, Ghannadan M, Zhang CF, Mlitz V, Gmeiner R, Sukseree S, Fodinger D, et al. Epidermal keratinocytes form a functional skin barrier in the absence of Atg7 dependent autophagy. J Dermatol Sci 2013; 71:67-75; PMID:23669018
- Kuma A, Matsui M, Mizushima N. LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: caution in the interpretation of LC3 localization. Autophagy 2007; 3:323-8; PMID:17387262; http://dx.doi.org/10.4161/auto.4012
- Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, Yin XM. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol 2007; 171:513-24; PMID:17620365; http://dx.doi.org/10.2353/ajpath.2007.070188
- Sukseree S, Mildner M, Rossiter H, Pammer J, Zhang CF, Watanapokasin R, Tschachler E, Eckhart L. Autophagy in the thymic epithelium is dispensable for the development of self-tolerance in a novel mouse model. PLoS One 2013; 7:e38933; http://dx.doi.org/10.1371/journal.pone.0038933
- Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007; 131:1149-63; PMID:18083104; http://dx.doi.org/10.1016/j.cell.2007.10.035
- Thiele C, Spandl J. Cell biology of lipid droplets. Curr Opin Cell Biol 2008; 20:378-85; PMID:18606534; http://dx.doi.org/10.1016/j.ceb.2008.05.009
- Nuotio K, Isoviita PM, Saksi J, Ijas P, Pitkaniemi J, Sonninen R, Soinne L, Saimanen E, Salonen O, Kovanen PT, et al. Adipophilin expression is increased in symptomatic carotid atherosclerosis: correlation with red blood cells and cholesterol crystals. Stroke; a J Cerebral Circul 2007; 38:1791-8; PMID:17446422; http://dx.doi.org/10.1161/STROKEAHA.106.478867
- de Jong A, Cheng TY, Huang S, Gras S, Birkinshaw RW, Kasmar AG, Van Rhijn I, Pena-Cruz V, Ruan DT, Altman JD, et al. CD1a-autoreactive T cells recognize natural skin oils that function as headless antigens. Nature immunology 2014; 15:177-85; PMID:24362891; http://dx.doi.org/10.1038/ni.2790
- Monick MM, Powers LS, Walters K, Lovan N, Zhang M, Gerke A, Hansdottir S, Hunninghake GW. Identification of an autophagy defect in smokers’ alveolar macrophages. J Immunol 2010; 185:5425-35; PMID:20921532; http://dx.doi.org/10.4049/jimmunol.1001603
- Vega-Naredo I, Caballero B, Sierra V, Huidobro-Fernandez C, de Gonzalo-Calvo D, Garcia-Macia M, Tolivia D, Rodriguez-Colunga MJ, Coto-Montes A. Sexual dimorphism of autophagy in Syrian hamster Harderian gland culminates in a holocrine secretion in female glands. Autophagy 2009; 5:1004-17; PMID:19736526; http://dx.doi.org/10.4161/auto.5.7.9610
- Rodriguez A, Duran A, Selloum M, Champy MF, Diez-Guerra FJ, Flores JM, Serrano M, Auwerx J, Diaz-Meco MT, Moscat J. Mature-onset obesity and insulin resistance in mice deficient in the signaling adapter p62. Cell Metab 2006; 3:211-22; PMID:16517408; http://dx.doi.org/10.1016/j.cmet.2006.01.011
- Wetzel R. Mutations and off-pathway aggregation of proteins. Trends Biotechnol 1994; 12:193-8; PMID:7764903; http://dx.doi.org/10.1016/0167-7799(94)90082-5
- Djeridane Y. The harderian gland and its excretory duct in the Wistar rat. A histological and ultrastructural study. J Anat 1994; 184 (Pt 3):553-66; PMID:7928644
- Lajoie P, Guay G, Dennis JW, Nabi IR. The lipid composition of autophagic vacuoles regulates expression of multilamellar bodies. J Cell Sci 2005; 118:1991-2003; PMID:15840653; http://dx.doi.org/10.1242/jcs.02324
- Platt FM, Boland B, van der Spoel AC. The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction. J Cell Biol 2012; 199:723-34; PMID:23185029; http://dx.doi.org/10.1083/jcb.201208152
- Greenberg AS, Coleman RA, Kraemer FB, McManaman JL, Obin MS, Puri V, Yan QW, Miyoshi H, Mashek DG. The role of lipid droplets in metabolic disease in rodents and humans. J Clin Invest 2011; 121:2102-10; PMID:21633178; http://dx.doi.org/10.1172/JCI46069
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441:885-9; PMID:16625204; http://dx.doi.org/10.1038/nature04724
- Kovacs GG, Botond G, Budka H. Protein coding of neurodegenerative dementias: the neuropathological basis of biomarker diagnostics. Acta Neuropathol 2010; 119:389-408; PMID:20198481; http://dx.doi.org/10.1007/s00401-010-0658-1
- Masters SL, Latz E, O’Neill LA. The inflammasome in atherosclerosis and type 2 diabetes. Sci Trans Med 2011; 3:81 ps17; http://dx.doi.org/10.1126/scitranslmed.3001902
- Dahlqvist J, Torma H, Badhai J, Dahl N. siRNA silencing of proteasome maturation protein (POMP) activates the unfolded protein response and constitutes a model for KLICK genodermatosis. PLoS One 2012; 7:e29471; PMID:22235297; http://dx.doi.org/10.1371/journal.pone.0029471
- Raymond AA, Gonzalez de Peredo A, Stella A, Ishida-Yamamoto A, Bouyssie D, Serre G, Monsarrat B, Simon M. Lamellar bodies of human epidermis: proteomics characterization by high throughput mass spectrometry and possible involvement of CLIP-170 in their trafficking/secretion. Mol Cell Proteomics : MCP 2008; 7:2151-75; http://dx.doi.org/10.1074/mcp.M700334-MCP200
- Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003; 115:727-38; PMID:14675537; http://dx.doi.org/10.1016/S0092-8674(03)00939-5
- Boulares AH, Yakovlev AG, Ivanova V, Stoica BA, Wang G, Iyer S, Smulson M. Role of poly(ADP-ribose) polymerase (PARP) cleavage in apoptosis. Caspase 3-resistant PARP mutant increases rates of apoptosis in transfected cells. J Biol Chem 1999; 274:22932-40; PMID:10438458; http://dx.doi.org/10.1074/jbc.274.33.22932
- Leist M, Gantner F, Bohlinger I, Germann PG, Tiegs G, Wendel A. Murine hepatocyte apoptosis induced in vitro and in vivo by TNF-alpha requires transcriptional arrest. J Immunol 1994; 153:1778-88; PMID:8046244
- Listenberger LL, Han X, Lewis SE, Cases S, Farese RV, Jr., Ory DS, Schaffer JE. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci U S A 2003; 100:3077-82; PMID:12629214; http://dx.doi.org/10.1073/pnas.0630588100
- Papaccio G, Sellitti S, Salvatore G, Latronico VG, Baccari GC. The harderian gland in autoimmune diabetes of the nonobese diabetic mouse. Microsc Res Tech 1996; 34:156-65; PMID:8722711; http://dx.doi.org/10.1002/(SICI)1097-0029(19960601)34 :2%3c156::AID-JEMT10%3e3.0.CO;2-P.
- Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, Metzger D, Reggiani C, Schiaffino S, Sandri M. Autophagy is required to maintain muscle mass. Cell Metab 2009; 10:507-15; PMID:19945408; http://dx.doi.org/10.1016/j.cmet.2009.10.008
- Singh R, Xiang Y, Wang Y, Baikati K, Cuervo AM, Luu YK, Tang Y, Pessin JE, Schwartz GJ, Czaja MJ. Autophagy regulates adipose mass and differentiation in mice. J Clin Invest 2009; 119:3329-39; PMID:19855132
- Choi SE, Lee SM, Lee YJ, Li LJ, Lee SJ, Lee JH, Kim Y, Jun HS, Lee KW, Kang Y. Protective role of autophagy in palmitate-induced INS-1 beta-cell death. Endocrinology 2009; 150:126-34; PMID:18772242; http://dx.doi.org/10.1210/en.2008-0483
- Khan MJ, Rizwan Alam M, Waldeck-Weiermair M, Karsten F, Groschner L, Riederer M, Hallstrom S, Rockenfeller P, Konya V, Heinemann A, et al. Inhibition of autophagy rescues palmitic acid-induced necroptosis of endothelial cells. J Biol Chem 2012; 287:21110-20; PMID:22556413; http://dx.doi.org/10.1074/jbc.M111.319129
- Bush KT, Goldberg AL, Nigam SK. Proteasome inhibition leads to a heat-shock response, induction of endoplasmic reticulum chaperones, and thermotolerance. J Biol Chem 1997; 272:9086-92; PMID:9083035; http://dx.doi.org/10.1074/jbc.272.14.9086
- Pan X, Hussain MM. Gut triglyceride production. Biochim Biophys Acta 2012; 1821:727-35; PMID:21989069; http://dx.doi.org/10.1016/j.bbalip.2011.09.013
- Fischer H, Koenig U, Eckhart L, Tschachler E. Human caspase 12 has acquired deleterious mutations. Biochem Biophys Res Commun 2002; 293:722-6; PMID:12054529; http://dx.doi.org/10.1016/S0006-291X(02)00289-9
- Yancey PG, Jerome WG. Lysosomal cholesterol derived from mildly oxidized low density lipoprotein is resistant to efflux. J Lipid Res 2001; 42:317-27; PMID:11254742
- Ohara S, Ukita Y, Ninomiya H, Ohno K. Axonal dystrophy of dorsal root ganglion sensory neurons in a mouse model of Niemann-Pick disease type C. Exp Neurol 2004; 187:289-98; PMID:15144855; http://dx.doi.org/10.1016/j.expneurol.2004.03.002
- Bernales S, McDonald KL, Walter P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol 2006; 4:e423; PMID:17132049
- Narita M, Young AR, Arakawa S, Samarajiwa SA, Nakashima T, Yoshida S, Hong S, Berry LS, Reichelt S, Ferreira M, et al. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science 2011; 332:966-70; PMID:21512002; http://dx.doi.org/10.1126/science.1205407
- Duran JM, Anjard C, Stefan C, Loomis WF, Malhotra V. Unconventional secretion of Acb1 is mediated by autophagosomes. J Cell Biol 2010; 188:527-36; PMID:20156967; http://dx.doi.org/10.1083/jcb.200911154
- Manjithaya R, Anjard C, Loomis WF, Subramani S. Unconventional secretion of Pichia pastoris Acb1 is dependent on GRASP protein, peroxisomal functions, and autophagosome formation. J Cell Biol 2010; 188:537-46; PMID:20156962; http://dx.doi.org/10.1083/jcb.200911149
- Hung YH, Chen LM, Yang JY, Yang WY. Spatiotemporally controlled induction of autophagy-mediated lysosome turnover. Nat Commun 2013; 4:2111; PMID:23817530; http://dx.doi.org/10.1038/ncomms3111
- Dahlqvist J, Klar J, Tiwari N, Schuster J, Torma H, Badhai J, Pujol R, van Steensel MA, Brinkhuizen T, Gijezen L, et al. A single-nucleotide deletion in the POMP 5’ UTR causes a transcriptional switch and altered epidermal proteasome distribution in KLICK genodermatosis. Am J Hum Genet 2010; 86:596-603; PMID:20226437; http://dx.doi.org/10.1016/j.ajhg.2010.02.018
- Feingold KR, Elias PM. Role of lipids in the formation and maintenance of the cutaneous permeability barrier. Biochim Biophys Acta 2014; 1841:280-94; PMID:24262790; http://dx.doi.org/10.1016/j.bbalip.2013.11.007
- Coto-Montes A, Garcia-Macia M, Caballero B, Sierra V, Rodriguez-Colunga MJ, Reiter RJ, Vega-Naredo I. Analysis of constant tissue remodeling in Syrian hamster Harderian gland: intra-tubular and inter-tubular syncytial masses. J Anat 2013; 222:558-69; PMID:23496762; http://dx.doi.org/10.1111/joa.12040
- Vega-Naredo I, Caballero B, Sierra V, Garcia-Macia M, de Gonzalo-Calvo D, Oliveira PJ, Rodriguez-Colunga MJ, Coto-Montes A. Melatonin modulates autophagy through a redox-mediated action in female Syrian hamster Harderian gland controlling cell types and gland activity. J Pineal Res 2012; 52:80-92; PMID:21771054; http://dx.doi.org/10.1111/j.1600-079X.2011.00922.x
- Fung C, Lock R, Gao S, Salas E, Debnath J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol Biol Cell 2008; 19:797-806; PMID:18094039; http://dx.doi.org/10.1091/mbc.E07-10-1092
- Berry DL, Baehrecke EH. Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell 2007; 131:1137-48; PMID:18083103; http://dx.doi.org/10.1016/j.cell.2007.10.048
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 2004; 15:1101-11; PMID:14699058; http://dx.doi.org/10.1091/mbc.E03-09-0704
- Hager B, Bickenbach JR, Fleckman P. Long-term culture of murine epidermal keratinocytes. J Invest Dermatol 1999; 112:971-6; PMID:10383747; http://dx.doi.org/10.1046/j.1523-1747.1999.00605.x
- Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol 1959; 37:911-7; PMID:13671378; http://dx.doi.org/10.1139/o59-099
- Pappinen S, Hermansson M, Kuntsche J, Somerharju P, Wertz P, Urtti A, Suhonen M. Comparison of rat epidermal keratinocyte organotypic culture (ROC) with intact human skin: lipid composition and thermal phase behavior of the stratum corneum. Biochim Biophys Acta 2008; 1778:824-34; PMID:18211819; http://dx.doi.org/10.1016/j.bbamem.2007.12.019
- Cullen P, Fobker M, Tegelkamp K, Meyer K, Kannenberg F, Cignarella A, Benninghoven A, Assmann G. An improved method for quantification of cholesterol and cholesteryl esters in human monocyte-derived macrophages by high performance liquid chromatography with identification of unassigned cholesteryl ester species by means of secondary ion mass spectrometry. J Lipid Res 1997; 38:401-9; PMID:9162758
- Allain CC, Poon LS, Chan CS, Richmond W, Fu PC. Enzymatic determination of total serum cholesterol. Clin Chem 1974; 20:470-5; PMID:4818200