
Abstract
The frontline tyrosine kinase inhibitor (TKI) imatinib has revolutionized the treatment of patients with chronic myeloid leukemia (CML). However, drug resistance is the major clinical challenge in the treatment of CML. The Hedgehog (Hh) signaling pathway and autophagy are both related to tumorigenesis, cancer therapy, and drug resistance. This study was conducted to explore whether the Hh pathway could regulate autophagy in CML cells and whether simultaneously regulating the Hh pathway and autophagy could induce cell death of drug-sensitive or -resistant BCR-ABL+ CML cells. Our results indicated that pharmacological or genetic inhibition of Hh pathway could markedly induce autophagy in BCR-ABL+ CML cells. Autophagic inhibitors or ATG5 and ATG7 silencing could significantly enhance CML cell death induced by Hh pathway suppression. Based on the above findings, our study demonstrated that simultaneously inhibiting the Hh pathway and autophagy could markedly reduce cell viability and induce apoptosis of imatinib-sensitive or -resistant BCR-ABL+ cells. Moreover, this combination had little cytotoxicity in human peripheral blood mononuclear cells (PBMCs). Furthermore, this combined strategy was related to PARP cleavage, CASP3 and CASP9 cleavage, and inhibition of the BCR-ABL oncoprotein. In conclusion, this study indicated that simultaneously inhibiting the Hh pathway and autophagy could potently kill imatinib-sensitive or -resistant BCR-ABL+ cells, providing a novel concept that simultaneously inhibiting the Hh pathway and autophagy might be a potent new strategy to overcome CML drug resistance.
Abbreviations:
- ACTB, actin
- β
- AKT/protein kinase B, v-akt murine thymoma viral oncogene homolog
- ATG, autophagy-related
- Bafi A1, bafilomycin A1
- BCC, basal cell carcinoma
- BCR-ABL, breakpoint cluster region-ABL proto-oncogene, non-receptor tyrosine kinase
- CASP, caspase
- apoptosis-related cysteine peptidase
- CML, chronic myeloid leukemia
- CQ, chloroquine
- EIF4EBP1, eukaryotic translation initiation factor 4E binding protein 1
- HCQ, hydroxychloroquine
- Hh, Hedgehog
- MAP1LC3B, microtubule-associated protein 1 light chain 3 β
- MTOR, mechanistic target of rapamycin
- PARP, poly (ADP-ribose) polymerase
- PBMC, human peripheral blood mononuclear cell
- PCR, polymerase chain reaction
- RPS6KB, ribosomal protein S6 kinase, 70kDa
- siRNA, small interfering RNA
- SQSTM1, sequestosome 1
- TKI, tyrosine kinase inhibitor
Introduction
Chronic myeloid leukemia (CML) is a clonal disease which is associated with aberrant expression of the chimeric protein BCR-ABL.Citation1 Although the frontline TKI imatinib revolutionized the treatment of patients with CML, acquired imatinib resistance mainly resulting from BCR-ABL gene mutation is an emerging problem,Citation2,3 and remains to be resolved. New TKIs dasatinib and nilotinib overcame this problem to some extent but had no effect on the drug-resistant T315I mutation in CML patients. The investigation of new regimes or combinational therapies improving the current condition of CML treatment would provide more options for patients and benefit the clinical cure of CML.
The Hedgehog (Hh) pathway, which can be categorized into 3 subgroups: Desert Hedgehog (Dhh), Indian Hedgehog (Ihh), and Sonic Hedgehog (Shh), plays crucial roles in growth, survival, and fate of vertebrate cells.Citation4,5 In mammalian cells, Hh, PTCH (patched), SMO (smoothened, frizzled class receptor), and GLI/glioma-associated oncogene (GLI family zinc finger) transcription factors (GLI1, GLI2, and GLI3) are key components of the Hh pathway.Citation6 Primary cilium provides the crucial location where Hh signaling is regulated. PTCH localizes to the primary cilium and inhibits SMO activity by preventing its trafficking and localization to the primary cilium, resulting in proteasomal cleavage of GLI and consequently leading to the inhibition of Hh signaling pathway.Citation7 The Hh ligand can bind to PTCH, displace PTCH from the primary cilium, and trigger the activation of SMO;activated SMO can subsequently lead to the activation of the Hh signaling pathway.Citation7
Increasing evidence indicates that aberrant activation of Hh pathway is closely linked to tumorigenesis.Citation6 It is well established that abnormal activation of the Hh pathway is responsible for multiple cancers including basal cell carcinoma (BCC), hematologic malignancies, breast cancer, gastric cancer, and pancreatic cancer.Citation4,6-9 Therefore, the Hh signaling pathway has been one of the most promising anti-cancer targets, and several Hh inhibitors are being evaluated in ongoing clinical trials for many types of cancers.Citation10 Vismodegib (GDC-0449, a systemic antagonist of SMO, Roche) is the first small-molecule, Hh-pathway inhibitor, which has been approved by the US. Food and Drug Administration for the treatment of BCC. There are close relationships between the CML and the Hh pathways.Citation11-15 Evidence shows that the abnormal activation of the Hh pathway is critical for the expansion of BCR-ABL+ leukemia stem cells.Citation16 The Hh pathway inhibitor cyclopamine can induce cell death of leukemia cells in vitro.Citation17 Inhibition of Hh pathway could reduce drug resistance of CD34+ leukemia cells in vitro.Citation18 Therefore, the combination of Hh pathway inhibitors and TKIs might represent a hopeful strategy for improving treatment outcomes of patients with CML. The combination of PF-04449913 (an Hh inhibitor, Pfizer) with Dasatinib for CML treatment is under phase I clinical trial (ClinicalTrials.gov: NCT00953758). A recent study reports that the combination of ponatinib and the Hh inhibitor vismodegib can induce cell death in drug-resistant CML cells.Citation19 However, this combined therapy might be based on the efficacy of TKIs themselves, since ponatinib used alone could kill drug-resistant CML cells. New strategies are needed to make any progress in the field of CML drug resistance.
Macroautophagy (autophagy hereafter) is a highly controlled bulk proteolytic process that can be induced by many conditions of stress such as nutrient or growth factor deprivation, oxidative stress, and drug treatment, and is being increasingly investigated as an anticancer target.Citation20-22 Increasing evidence indicates that autophagy plays a critical role in tumorigenesis, progression, and treatment of hematological malignancies.Citation23,24 Inhibition of autophagy enhances imatinib's cytotoxicity on Philadelphia chromosome-positive cells. However, inhibiting autophagy cannot increase the sensitivity of BCR-ABLT315I-harboring leukemia cells to TKIs.Citation24,25 Moreover, a clinical trial is ongoing to evaluate the efficacy of combination therapy of imatinib with HCQ on CML (ClinicalTrials.gov Identifier: NCT01227135, phase II).Citation26 Of note, recent reports have reported Hh pathway can regulate autophagy, while the conclusions were controversial: Activation of the Hh signaling pathway using Hh agonists can inhibit autophagy in HeLa cells, and inactivation of Hh-induced autophagy in Drosophila.Citation27 Inhibition of the Hh pathway using GANT61, an Hh pathway inhibitor which is undergoing phase II clinical trial, can induce autophagy in human hepatocellular carcinoma cells.Citation28 This evidence indicates that the Hh pathway has an inhibitory effect on autophagy. However, the Hh pathway exhibits a positive regulatory effect on autophagy in vascular smooth muscle cells and hippocampal neurons.Citation29,30 These findings suggest that there might be extensive interactions between the Hh pathway and autophagy in cancers including hematological malignancies and that these interactions might be utilized for improving cancer therapy. Therefore, we hypothesized that simultaneously targeting the Hh pathway and autophagy might represent a novel turning point in cancer therapy.
In our current study, we investigated the relationship between the Hh pathway and autophagy in CML and the potential of overcoming CML drug resistance based on combined targeting of Hh pathway and autophagy. Our results showed that inhibiting the Hh pathway and autophagy could remarkably induce cell death of drug-sensitive and -resistant BCR-ABL+ CML cells. First, we found that inhibiting the Hh pathway could induce autophagy in BCR-ABL+ CML cells. Second, compared with inhibition of the Hh pathway or autophagy alone, inhibiting the Hh pathway and autophagy simultaneously could sharply decrease the cell viability of BCR-ABL+ cells and significantly induce apoptosis. Moreover, our results suggested that inhibition of the Hh pathway and autophagy downregulated the kinase activity of the BCR-ABL oncoprotein. This study provided a novel notion that beyond TKIs, simultaneously inhibiting the Hh pathway and autophagy might be a potential new strategy to overcome CML drug resistance.
Results
Inhibition of the Hh pathway reduced cell viability of BCR-ABL+ CML cells
Initially, we determined whether the Hh inhibitor vismodegib could effectively inhibit the Hh pathway in CML cells. It is difficult to detect the change of PTCH expression in the protein level due to the lack of a specific antibody against endogenous PTCH. Therefore, we detected the relative expression of mRNA levels of PTCH. Quantitative RT-PCR results showed that 10, 20, and 40 μM of vismodegib could significantly minimize the relative expression of PTCH and GLI1 mRNA, indicating that the Hh pathway was inhibited by vismodegib (). It is well accepted that the expression level of GLI1 can reflect the activation status of the entire Hh pathway.Citation6 Our results showed that the Hh inhibitor vismodegib could appreciably decrease the protein level of GLI1 at the concentrations of 10, 20, and 40 μM, suggesting the inhibition of Hh pathway in CML cells ().
Figure 1. Inhibiting the Hh pathway decreased cell viability of BCR-ABL+ CML cells. (A and B) K562 cells were treated with 10, 20, and 40 μM of vismodegib for 24 h, gene expression of PTCH (A) and GLI1 (B) were detected by quantitative RT-PCR. Data shown are mean ± SD of triplicates of one typical experiment. Similar results were obtained from 3 independent experiments * versus Control, P < 0.05, ** vs. to Control, P < 0.01. (C) K562 cells were treated with 10, 20, and 40 μM of vismodegib for 48 h, protein levels of GLI1, CCND1, MYC, p-GSK3B, GSK3B, CTNNB1, and ACTB were determined by western blot assay. Densitometric values were quantified using the ImageJ software and normalized to control. The values of control were set to 1. The data represents the mean of 3 independent experiments. (D) K562, BaF3-BCR-ABL, BaF3-BCR-ABLT315I, and BaF3-BCR-ABLY253F cells were treated with 2.5, 5, 10, 20, and 40 μM of vismodegib for 48 h, cell viability was determined by the CCK-8 assay.
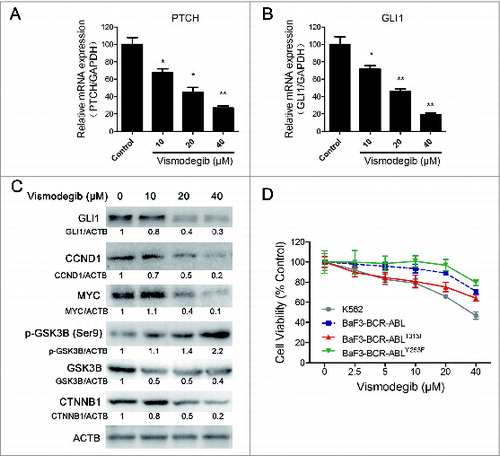
Although the comprehensive elucidation of the upstream and downstream of Hh signaling is insufficient, present evidence indicates that, in CML, the Hh pathway upregulated the canonical WNT signaling, CCND1 and MYC.Citation4,7,31 Therefore, we examined whether these protein targets were also affected by vismodegib in CML cells. Western blot results showed that the protein levels of CCND1 and MYC were decreased by vismodegib in a dose-dependent manner (). In conclusion, vismodegib effectively inhibited the Hh pathway and its downstream protein targets in CML cells.
Similarly to the Hh pathway, the WNT pathway is also one of the most important signaling pathways that plays key roles in embryonic development, and is required for the cancer stem cells (CML stem cells) and CML progression.Citation32-35 The Hh pathway can interact with the WNT pathway through phosphorylating GSK3B.Citation31 Western blot assays indicated that vismodegib augmented the phosphorylation of GSK3B and reduced the protein level of CTNNB1, the key mediator of WNT signaling, indicating the inhibition of the WNT pathway ().
We also examined the inhibitory effects of vismodegib on cell viability in drug-sensitive and -resistant CML cells. The T315I and Y253F mutations of BCR-ABL are 2 representative imatinib-resistant genotypes, while wild-type BCR-ABL is an imatinib-sensitive genotype. BaF3-BCR-ABL, BaF3-BCR-ABLT315I and BaF3-BCR-ABL YY253F cells derived from BaF3 cells (a mouse pro-B cell line) transfected with the BCR-ABL wild-type gene, the BCR-ABLT315I mutation gene and the BCR-ABLY253F mutation gene, respectively, are widely used in CML investigations.Citation36-38 Inhibiting the Hh pathway using the Hh inhibitor vismodegib could reduce the cell viability of K562, BaF3-BCR-ABL, BaF3-BCR-ABLT315I, and BaF3-BCR-ABLY253F cells in vitro at the concentration of 40 μM ().
Collectively, these data demonstrated that inhibition of the Hh pathway could reduce the cell viability of BCR-ABL+ CML cells.
Inhibiting the Hh pathway induced autophagy in BCR-ABL+ CML cells
Next, we tested whether autophagy could be induced by inhibiting the Hh pathway in BCR-ABL+ CML cells. Using transmission electron microscopy, a gold standard of autophagy assessment, we observed more autophagosomes characterized by double membranes in BCR-ABL+ CML cells exposed to vismodegib when compared with controls (), suggesting that vismodegib induced autophagy in BCR-ABL+ cells. To confirm vismodegib-induced autophagy, BCR-ABL+ CML cells were transiently transfected with an enhanced green fluorescent protein (eGFP)-MAP1LC3B plasmid, which is a well-documented model for monitoring the conversion of MAP1LC3B from a cytosolic MAP1LC3B-I form to a phagophore- and autophagosome-bound MAP1LC3B-II form.Citation39 Under normal conditions, eGFP-MAP1LC3B fluorescence is diffuse in the cytoplasm, while it converts to a punctate pattern when autophagy is activated. Treatment with 20 μM of vismodegib for 24 h appreciably increased the average number of fluorescent spots (), indicating the conversion of MAP1LC3B-I to MAP1LC3B-II.
Figure 2. Inhibiting the Hh pathway induced autophagy in CML cells. (A) K562, BaF3-BCR-ABL, BaF3-BCR-ABLT315I, and BaF3-BCR-ABLY253F cells were treated with 20 μM of vismodegib for 48 h, autophagosomes were observed using transmission electron microscopy. Arrows represent autophagosomes. (B and C) K562, BaF3-BCR-ABL, and BaF3-BCR-ABLT315I cells were transiently transfected with an eGFPMAP1LC3B plasmid for 24 h, followed by treatment of 20 μM of vismodegib for 48 h. Cells were observed under confocal microscopy and typical images are presented (B). Green fluorescence spots represent MAP1LC3B-II spots. MAP1LC3B-II spots were counted using ImageJ software (C). ** P < 0.01. n = 20.
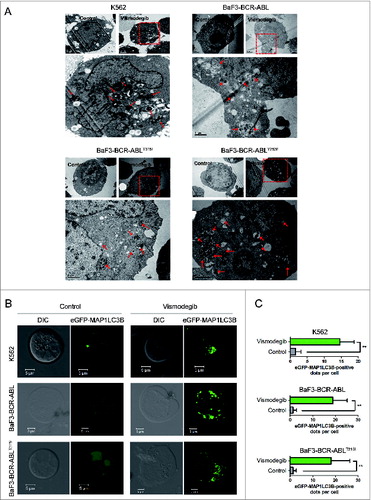
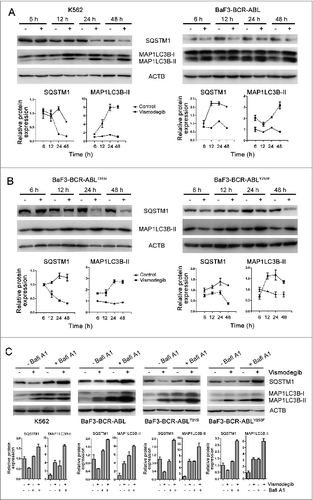
The increase of autophagosomes or MAP1LC3B-II does not represent the completion of the entire autophagy pathway. To further investigate if autophagy was induced after vismodegib treatment, we examined the autophagic flux in vismodegib-treated BCR-ABL+ CML cells. SQSTM1 is an extensively used autophagy marker. Western blot assays showed the decrease of SQSTM1 and increase of MAP1LC3B-II protein levels in vismodegib-treated CML cells at several time points (). We also observed the increase of MAP1LC3B-II in BCR-ABL+ CML cells in a dose-dependent manner (Fig. S1). Moreover, we used bafilomycin A1 (Bafi A1), a lysosomotropic agent, to prevent the fusion of autophagosomes with lysosomes. Treatment with vismodegib increased the protein levels of MAP1LC3B-II and decreased the protein levels of SQSTM1 in CML cells (). When cells were treated with vismodegib in combination with Bafi A1, the vismodegib-induced increase of MAP1LC3B-II was significantly enhanced and the vismodegib-induced decrease of SQSTM1 was recovered in CML cells (). These data indicated that vismodegib induced autophagic flux, which could trigger lysosomal degradation of MAP1LC3B-II.
Figure 3. Inhibiting the Hh pathway induced autophagic flux in CML cells. (A) K562 and BaF3-BCR-ABL cells were treated with 20 μM of vismodegib for 6 h, 12 h, 24 h, or 48 h, protein levels of SQSTM1, MAP1LC3B, and ACTB were measured by western blot assays. (B) BaF3-BCR-ABLT315I and BaF3-BCR-ABLY253F cells were treated with 20 μM of vismodegib for 6 h, 12 h, 24 h, or 48 h, protein levels of SQSTM1, MAP1LC3B, and ACTB were measured by western blot assays. (C) K562, BaF3-BCR-ABL, BaF3-BCR-ABLT315I, and BaF3-BCR-ABLY253F cells were cotreated with vismodegib (20 μM) and Bafi A1 (5 nM) or treated with these 2 agents alone for 48 h, protein levels of SQSTM1, MAP1LC3B, and ACTB were measured by western blot assays. (A–C) Densitometric values were quantified using the ImageJ software and normalized to control. The values of control were set to 1. The data are presented as means ± SD of 3 independent experiments.
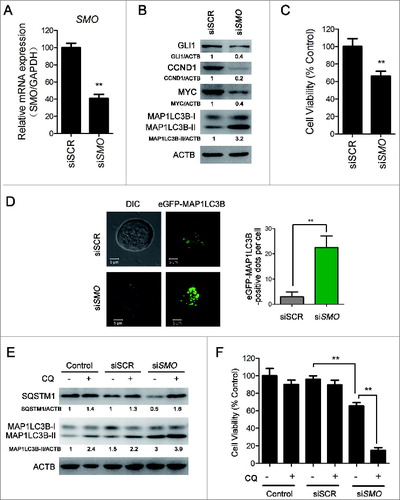
Moreover, small interfering RNA (siRNA) assays were conducted to further understand the role of inhibiting the Hh pathway in vismodegib-induced autophagy. Vismodegib inhibits the Hh pathway through binding to SMO. Therefore, we silenced SMO to inhibit the Hh pathway in CML cells. Owing to the lack of a specific antibody against endogenous SMO, to determine the efficiency of SMO silencing, the relative mRNA level of SMO was measured by quantitative RT-PCR and the protein levels of GLI1, CCND1, and MYC were determined by western blot. The results showed that the relative mRNA levels of SMO () and the protein levels of GLI1, CCND1, and MYC () were significantly attenuated in cells transfected with SMO siRNA, compared with cells transfected with the nonsilencing scrambled control (SCR) siRNA, indicating that SMO siRNA effectively silenced SMO and inhibited the Hh pathway. Consistent with vismodegib treatment, inhibiting the Hh pathway using SMO siRNA could also reduce the cell viability of CML cells (). Moreover, we explored whether Hh pathway inhibition by SMO silencing could induce autophagy in CML cells. Compared with cells transfected with SCR siRNA, the protein level of MAP1LC3B-II increased when cells were transfected with SMO siRNA (). The results showed that compared with cells transfected with SCR siRNA, cells transfected with SMO siRNA showed more eGFP-MAP1LC3B fluorescence spots () and a higher protein level of MAP1LC3B-II, and CQ could significantly further increase the protein level of MAP1LC3B-II (). These data confirmed that Hh pathway suppression could induce autophagy in CML cells. Taken together, these results demonstrated that inhibiting the Hh pathway induced autophagy in BCR-ABL+ CML cells.
Figure 4 (See previous page). SMO silencing induced autophagy in CML cells. (A to C) K562 cells were transiently transfected with SMO siRNA for 48 h. (A) The relative expression level of SMO mRNA was determined by quantitative RT-PCR. Data represents mean ± SD of 3 independent experiments. ** versus siSCR, P < 0.01. (B) The protein levels of GLI1, CCND1, MYC, MAP1LC3B, and ACTB were detected by western blot. Densitometric values were quantified using the ImageJ software and normalized to control. The values of control were set to 1. The data are presented as means of 2 independent experiments. (C) Cell viability was measured by the CCK-8 assay. Data are presented as the means ± SD of 3 independent experiments. ** vs. siSCR, P < 0.01. (D) After K562 cells were transiently transfected with an eGFPMAP1LC3B plasmid for 24 h, cells were transfected with SMO siRNA for 48 h. MAP1LC3B-II green fluorescent spots were observed under confocal microscopy and typical images are presented. ** P < 0.01. n = 20. (E and F) K562 cells were transiently transfected with SMO siRNA for 24 h and followed by the treatment of CQ (10 μM) for 48 h. (E) The protein levels of SQSTM1, MAP1LC3B, and ACTB were measured by western blot assay. Densitometric values were quantified using the ImageJ software and normalized to control. The values of control were set to 1. The data are presented as means of 2 independent experiments. (F) Cell viability was measured by CCK-8 assay. Data are presented as the means ± SD of 3 independent experiments. **P < 0.01.
Downregulation of the AKT-MTOR signaling pathway was involved in autophagy induced by inhibition of the Hh signaling pathway
The AKT-MTOR signaling pathway is a key autophagy-regulating pathway in eukaryotic cells.Citation40 MTORC1 (MTOR complex 1) is involved in both the Hh pathway and autophagy. MTORC1 is a negative regulator of autophagy and its activation status can be evaluated by measuring the phosphorylation of its downstream targets including EIF4EBP1 (eukaryotic translation initiation factor 4E binding protein 1) and RPS6KB (ribosomal S6 protein kinase, 70kDa).Citation41 Therefore, we investigated whether vismodegib affected the activation status of the MTOR signaling pathway. Vismodegib significantly decreased the protein levels of phosphorylated AKT, MTOR, RPS6KB, and EIF4EBP1 in a dose-dependent manner (). Meanwhile, treatment with vismodegib for 24 h and 48 h appreciably decreased the protein levels of phosphorylated AKT, MTOR, EIF4EBP1, and RPS6KB when compared with control groups (), indicating the inhibition of the AKT-MTOR signaling pathway in CML cells.
Figure 5. Vismodegib inhibited the AKT-MTOR signaling pathway in CML cells. (A) K562 cells were treated with 10, 20, and 40 μM of vismodegib (Vis) for 48 h, the protein levels of p-AKT (Ser473), p-AKT (Thr308), p-MTOR (Ser2448), p-RPS6KB1 (Thr389), p-EIF4EBP1 (Thr37/46), SQSTM1, MAP1LC3B, and ACTB were measured by western blot assays. (B) Densitometric values were quantified using the ImageJ software and normalized to control. The values of control were set to 1. The data are presented as means ± SD of 3 independent experiments. (C) K562 cells were treated with or without 20 μM of vismodegib for 12 h, 24 h, and 48 h, the protein levels of p-AKT (Ser473), p-AKT (Thr308), p-MTOR (Ser2448), p-RPS6KB1 (Thr389), p-EIF4EBP1 (Thr37/46), SQSTM1, MAP1LC3B, and ACTB were measured by western blot assays. (D) Densitometric values were quantified as described in (B).
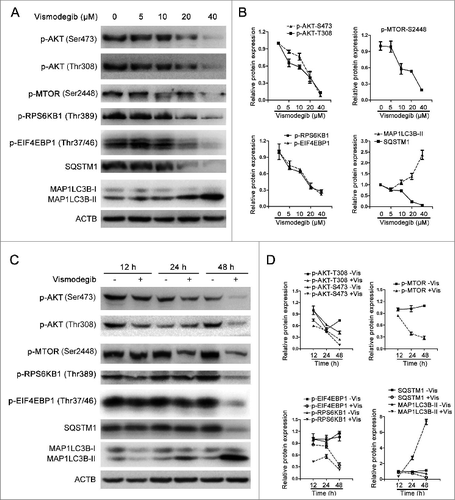
Pharmacologically blocking autophagy could significantly enhance the decrease in viability induced by the Hh inhibitor in BCR-ABL+ CML cells
We used CQ to inhibit vismodegib-induced autophagy, since CQ inhibits autophagy through blocking the fusion of autophagosome and lysosome, which leads to the block of degradation of MAP1LC3B-II and results in the increase of MAP1LC3B-II protein level.Citation39 Western blot results showed that the protein level of MAP1LC3B-II in vismodegib- and CQ-cotreated cells was higher than cells treated with vismodegib alone (), indicating that CQ inhibited vismodegib-induced autophagy in BCR-ABL+ cells. Vismodegib combined with CQ significantly decreased cell viability when compared with either agent alone (). Moreover, the combination of vismodegib and Bafi A1, another autophagy inhibitor, could also significantly decrease cell viability of BCR-ABL+ cells (Fig. S2). Furthermore, CQ could markedly enhance the inhibition of cell viability induced by SMO-silencing (). The Hh pathway is closely related to CML cell growth. Thus, we studied whether the combination of vismodegib and CQ influenced cell cycle distribution in CML cells. Results showed that the percentage of G0/G1 phase cells did not change significantly between cells treated with both vismodegib and CQ and cells treated with vismodegib alone (Fig. S3). Taken together, these results indicated that blocking autophagy could significantly enhance the inhibition of cell viability induced by the Hh inhibitor in BCR-ABL+ CML cells.
Figure 6 (See previous page). The decrease in cell viability induced by the Hh inhibitor was significantly enhanced by inhibiting autophagy. (A) K562, BaF3-BCR-ABL, BaF3-BCR-ABLT315I, and BaF3-BCR-ABLY253F cells were treated with 20 μM of vismodegib and 2.5, 5, or 10 μM of CQ for 48 h, cell viability was measured by CCK-8 assay. Data are means ± SD of triplicates. Typical results of 3 independent experiments are presented. *P < 0.05; **P < 0.01. (B and C) K562, BaF3-BCR-ABL, BaF3-BCR-ABLT315I, and BaF3-BCR-ABLY253F cells were cotreated with vismodegib (20 μM) and CQ (10 μM) or treated with vismodegib (20 μM) or CQ (10 μM) alone for 48 h, (B) then the protein levels of MAP1LC3B, and tubulin were measured by western blot assays, (C) cells were observed under inverted phase contrast microscope. Phase-contrast pictures were obtained randomly and representative pictures are presented.
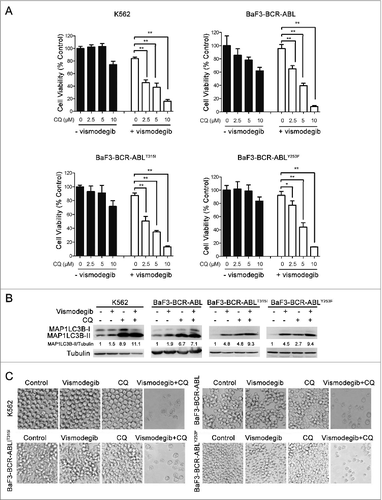
ATG5 and ATG7 silencing significantly enhanced the inhibition of cell viability induced by the Hh inhibitor in BCR-ABL+ CML cells
To further corroborate the crucial role of autophagy in the induction of cell death induced by Hh inhibition, we used siRNA targeting 2 established core autophagy molecules, namely ATG5 and ATG7, in K562 (imatinib-sensitive) and BaF3-BCR-ABLT315I (imatinib-resistant) cells, respectively. As shown in , compared with transfection with the SCR siRNA, a robust downregulation of ATG5 and ATG7 protein levels was observed 24 h after ATG5 or ATG7 siRNA transfection. Meanwhile, Hh inhibiting-mediated autophagosome formation was significantly abrogated following ATG5 and ATG7 silencing as evaluated by measuring MAP1LC3B conversion (). Next, we examined the cell viability under similar experiment conditions. Although, in the absence of vismodegib, there were significant inhibitions of cell viability after ATG5 and ATG7 silencing when compared with SCR-transfected cells, robust cell viability inhibition was obtained in the presence of vismodegib (), demonstrating the critical role of autophagy in the induction of CML cell death induced by Hh inhibition.
Figure 7. ATG5 and ATG7 silencing significantly enhanced cell death induced by Hh inhibition in CML cells. (A) K562 cells were transiently transfected with ATG5 or ATG7 siRNAs for 48 h, the protein levels of ATG5, ATG7, and ACTB were detected by western blot assays. (B) After K562 cells were transiently transfected with ATG5 or ATG7 siRNAs for 24 h, cells were treated with 20 μM of vismodegib for 12 h, 24 h, and 48 h, the protein levels of SQSTM1, MAP1LC3B, and ACTB were measured by western blot assay. (C) K562 cells were treated as described in (B), cell viability was determined by CCK-8 assay. (D) BaF3-BCR-ABLT315I cells were treated as described in (A), the indicated protein levels were detected by western blot assay. (E) BaF3-BCR-ABLT315I cells were treated as described in (B), the indicated protein levels were detected by western blot (E) and cell viability was measured by CCK-8 assay (F). Relative expression levels in (A, B, D and E), shown below the blot are representative of 2 independent experiments. Densitometric values were quantified using the ImageJ software and normalized to control. The values of control were set to 1. Data in (C and F) are means ± SD of 3 independent experiments performed in triplicate. **P < 0.01.
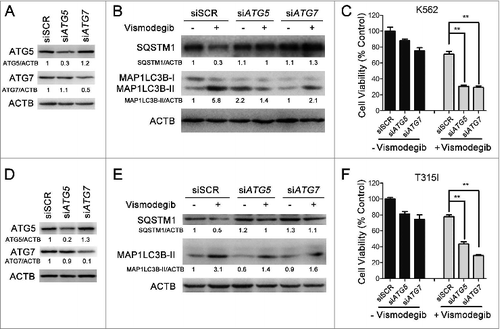
The simultaneous inhibition of the Hh pathway and autophagy could significantly inhibit cell viability of drug-resistant BCR-ABL+ CML cells
We next focused our study on evaluating the effect of simultaneously inhibiting the Hh pathway and autophagy on drug-resistant cells. Currently, drug resistance resulting from gene mutations of BCR-ABL is a drawback of CML treatment. Among a variety of mutations, the T315I mutation of BCR-ABL is resistant to most TKIs. Therefore, BaF3-BCR-ABLT315I cells were used to evaluate the efficacy of overcoming CML drug resistance through the simultaneous inhibition of the Hh pathway and autophagy. The results showed that the combination of vismodegib with CQ could significantly inhibit cell viability of drug-sensitive (K562) and -resistant BCR-ABL+ (BaF3-BCR-ABLT315I) cells, whereas imatinib alone had no effect on drug-resistant BCR-ABL+ cells (). Consistent with the results of the previous study, our data indicated that the combination of imatinib with CQ could significantly inhibit cell viability of drug-sensitive BCR-ABL+ cells when compared with the treatment of imatinib or CQ alone, except for drug -resistant BCR-ABL+ cells. Moreover, the combination of vismodegib with imatinib could also significantly inhibit cell viability of drug-sensitive BCR-ABL+ cells, except for drug-resistant BCR-ABL+ cells. Importantly, the combination of vismodegib and CQ could significantly inhibit cell viability of drug-sensitive and -resistant BCR-ABL+ cells (). Furthermore, although vismodegib could inhibit cell viability of PBMCs, there was no significant difference between the group of cells treated with vismodegib alone and the group of cells cotreated with vismodegib and CQ (). Since vismodegib has proven to be a safe drug in clinical use, this result suggested that this combined therapy might be a relative safe treatment strategy. Collectively, these data indicated that simultaneously inhibiting the Hh pathway and autophagy could effectively inhibit cell viability of drug-resistant BCR-ABL+ CML cells.
Figure 8. Simultaneous inhibition of the Hh pathway and autophagy can significantly inhibit cell viability of drug-resistant CML cells. (A) K562, BaF3-BCR-ABL, BaF3-BCR-ABLT315I, and BaF3-BCR-ABLY253F cells were treated with imatinib or cotreated with vismodegib and CQ for 48 h, cell viability was measured by CCK-8 assay. Error bars represent SD from triplicates. A typical result from 3 independent experiments is presented. (B) Phase-contrast images of cells treated with 1 μM of inmatinib or the combination of vismodegib (20 μM) and CQ (10 μM). (C) K562 and BaF3-BCR-ABLT315I cells were treated with imatinib (1 μM) or vismodegib (20 μM) or CQ (10 μM) or the indicated combinations of these agents for 48 h, cell viability was measured using CCK-8. Error bars represent SD from triplicates. A typical result from 3 independent experiments is presented. **P < 0.01. (D) Fresh PBMCs were treated with DMSO, vismodegib, CQ or the combination of vismodegib and CQ for 24 h, Cell viability was measured using CCK-8. Error bars represent SD from triplicates. A typical result from 3 independent experiments is presented. *P < 0.05 versus to the group of cells treated with DMSO.
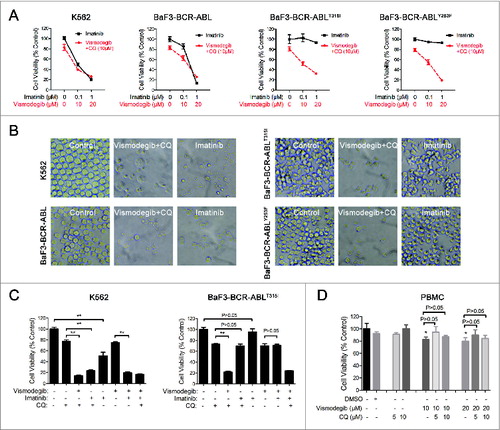
Simultaneous inhibition of the Hh pathway and autophagy sharply induced apoptosis of BCR-ABL+ CML cells
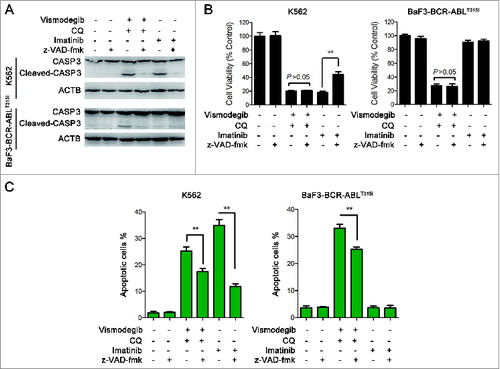
The Hedgehog pathway inhibitor can induce apoptosis in K562 cells.Citation17 We investigated if simultaneously inhibiting the Hh pathway and autophagy could induce apoptosis of BCR-ABL+ CML cells. The effect of the combination of vismodegib with CQ on BCR-ABL+ CML cells was determined by western blot and flow cytometry. Western blot assays showed that the combination of vismodegib with CQ noticeably induced the cleavage of CASP9, CASP3, and its substrate PARP1 (poly [ADP-ribose] polymerase 1) (), suggesting that this combination activated the apoptosis pathway. Flow cytometry analysis showed that the proportions of apoptotic cells (ANXA5/annexin V+ PI−) and necrotic cells (ANXA5+ PtdIns+) increased significantly when BCR-ABL+ cells were cotreated with vismodegib and CQ for 24 h and 48 h (). Moreover, the collapse of mitochondrial membrane potential, an indicator of early apoptosis, was detected after K562 cells were treated with vismodegib and CQ (Fig. S4). These results suggested that combination of vismodegib with CQ markedly induced apoptosis in BCR-ABL+ cells. To confirm this observation, z-VAD-fmk, a pan-CASP inhibitor, was used to inhibit the cleavage of CASP. Imatinib used as positive and negative controls of CASP activation in K562 (imatinib sensitive) and BaF3-BCR-ABLT315I (imatinib resistant) cells, respectively. Western blot results indicated that the cleavage of CASP3 was abolished by 20 μM of z-VAD-fmk (). However, 20 μM of z-VAD-fmk could not rescue CML cells from the reduction of cell viability induced by combination of vismodegib and CQ (), while z-VAD-fmk could partially but significantly weaken the apoptosis induced by combination of vismodegib and CQ (). These data indicated that the CML cell death induced by combination of vismodegib and CQ was partially depended on CASP-dependent apoptosis.
Figure 9. Simultaneous inhibition of the Hh pathway and autophagy significantly induced apoptosis in CML cells. (A) K562, BaF3-BCR-ABL, and BaF3-BCR-ABLT315I cells were cotreated with vismodegib (20 μM) and CQ (10 μM) for 12 h, 24 h, and 48 h. The expression levels of PARP, CASP9, CASP3 and ACTB were analyzed by western blot. (B) K562, BaF3-BCR-ABL, and BaF3-BCR-ABLT315I cells were treated as described in (A), apoptosis was detected by ANXA5 and PI staining and flow cytometry. The percentages of ANXA5+ PtdIns− cells (apoptosis) and ANXA5+ PI+ cells (necrosis) are presented in bar charts. Error bars represent SD from triplicate. A typical result from 3 independent experiments is presented. *P < 0.05; **P < 0.01; ***P < 0.001.
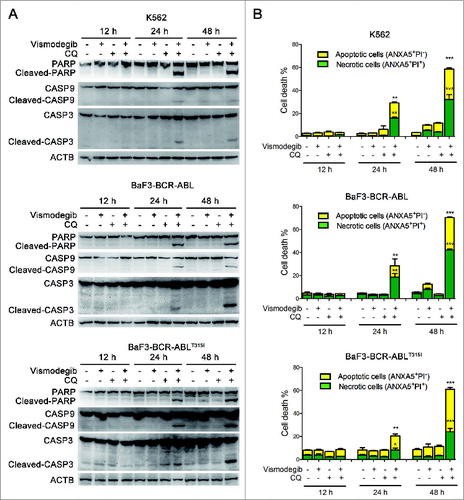
Figure 10. Apoptosis induced by simultaneous inhibition of the Hh pathway and autophagy can be partially rescued by z-VAD-fmk. (A) K562 and BaF3-BCR-ABLT315I cells were pretreated with z-VAD-fmk for 1 h, then cells were cotreated with vismodegib (20 μM) and CQ (10 μM) or imatinib (1 μM) for 48 h. Cells were subjected to western blot to detect the protein levels of CASP3 and ACTB. (B) K562 and BaF3-BCR-ABLT315I cells were treated as described in (A), cell viability was measured by CCK-8 assay. Error bars shown are SD from triplicates. A typical result from 3 independent experiments is presented. **P < 0.01. (C) K562 and BaF3-BCR-ABLT315I cells were treated as described in (A), apoptosis (ANXA5+ PtdIns− cells) was measured by flow cytometry. Error bars shown are SD from triplicates. A typical result from 3 independent experiments is presented. **P < 0.01.
Furthermore, HL-60 cells, a BCR-ABL negative leukemia cell line, were used as independent control cells. Another group has reported that the Hh inhibitor cyclopamine can induce cell death in HL-60 cells.Citation42 We observed that vismodegib upregulated the expression level of MAP1LC3B-II in HL-60 cells (Fig. S5A), indicating that inhibiting the Hh pathway could induce autophagy in HL-60 cells. The results showed that the combination of vismodegib with CQ could also significantly inhibit cell viability of HL-60 cells (Fig. S5B). Although CQ could significantly enhance the inhibition of cell viability by vismodegib in HL-60 cells compared with the vismodegib treatment alone, this enhancement was relatively weak when compared with the increase in the inhibition of cell viability by CQ in vismodegib-treated BCR-ABL+ cells. Moreover, inhibiting autophagy using CQ could not enhance vismodegib-induced apoptosis in HL-60 cells (Fig. S5C). Taken together, these results demonstrated that simultaneously inhibiting the Hh pathway and autophagy could sharply induce apoptosis in BCR-ABL+ CML cells.
Simultaneous inhibition of the Hh pathway and autophagy also inhibited the BCR-ABL pathway in BCR-ABL+ CML cells
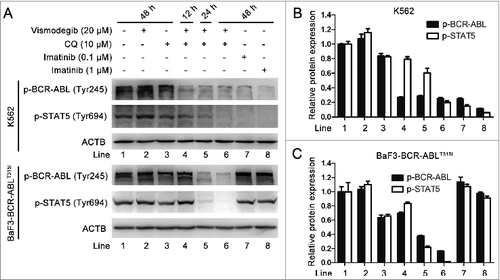
The expression of the BCR-ABL oncoprotein is responsible for the development of CML.Citation1 New generations of TKIs including nilotinib, dasatinib, bosutinib, exhibit effective inhibitory effects on the BCR-ABL oncoprotein; however, these TKIs have no effect on BCR-ABL with the T315I mutation.Citation43 Given that the combination of vismodegib with CQ could potently induce imatinib-resistant CML cell death, we determined whether this combined therapy could influence the kinase activity of the BCR-ABL oncoprotein, particularly the T315I mutation of BCR-ABL. The phospholation of BCR-ABL and its downstream protein STAT5 is widely used to assess the kinase activity of the BCR-ABL oncoprotein, while inhibition of the kinase activity of BCR-ABL is an important indicator representing the pharmacologically inhibitory activity of TKIs on CML.Citation36,37,44 Western blot showed that combination of vismodegib with CQ decreased the phosphorylation of BCR-ABL and its downstream protein STAT5 in both K562 and BaF3-BCR-ABLT315I cells, while imatinib did not exhibit similar effects in BaF3-BCR-ABLT315I cells (), suggesting that a combination of vismodegib with CQ could inhibit the kinase activity of BCR-ABL. Collectively, these results indicated that simultaneously inhibiting the Hh pathway and autophagy could inhibit the BCR-ABL pathway in BCR-ABL+ CML cells.
Figure 11. Simultaneous inhibition of the Hh pathway and autophagy also inhibited the kinase activity of BCR-ABL oncoprotein in BCR-ABL+ CML cells. K562 and BaF3-BCR-ABLT315I cells were cotreated with vismodegib (20 μM) and CQ (10 μM) for 48 h. (A) The protein levels of p-BCR-ABL and p-STAT5 were analyzed by western blot. (B and C) The relative protein expression levels are shown in bar chart. Densitometric values were quantified using the ImageJ software and normalized to control. The values of control were set to 1. Data are means ± SD of 3 independent experiments.
Discussion
CML, which is becoming more intractable due to drug resistance, is one of the most prevalent hematological malignancies all over the world. The aberrant expression of the chimeric protein BCR-ABL is well known to mediate the development of CML.Citation1 Over the past decades, accumulating knowledge of the mechanism of BCR-ABL has led to the development of TKIs for this disease, which has changed the history of CML therapy.Citation2 Even so, the majority of patients with CML exhibits drug resistance are likely to relapse within several years.Citation3 Today, drug resistance is becoming the biggest challenge of CML treatment. It is urgent to investigate new treatment strategies or combination therapies for CML.
Our current study suggested that simultaneously inhibiting the Hh pathway and autophagy might be a promising combined therapy candidate for overcoming the drug resistance of CML. First, our results revealed that inhibiting the Hh pathway could induce autophagy in CML cells. Autophagy is a basic physiological process in eukaryotes that is closely related to a variety of human disorders including neurodegenerative diseases, autoimmune diseases, cardiovascular diseases, and cancer.Citation45-47 In particular, the relationship between autophagy and cancer treatment has become a research hot spot in the field of cancer research.Citation48 Several clinical trials are ongoing to evaluate the potential of enhancing the antitumor effect of clinical drugs by targeting autophagy.Citation49-54 The Hh pathway plays key roles in embryonic pattern formation and adult tissue renewal and maintenance.Citation4 Evidence shows that the Hh pathway regulates autophagy in Drosophila and in several tumor types.Citation27-30 This study reports the relationship between the Hh pathway and autophagy in CML for the first time.
Second, we investigated the role of autophagy induced by inhibition of the Hh pathway, on its effect in the reduction of cell viability in CML cells. Although other groups have reported that Hh inhibitors such as cyclopamine can reduce the cell viability of CML cells,Citation17 treating with 20 μM of vismodegib for 48 h only inhibited cell viability of CML cells to about 80% of control in the current study. When CML cells were cotreated with vismodegib (20 μM) and CQ (10 μM) for 48 h, the cell viability was about 15% of control, while cells of control groups (treated with DMSO, vismodegib or CQ alone) exhibited relatively normal cell growth. Apparently, this combined strategy exhibited a potent synergistic cell killing effect on CML cells.
Moreover, we focused on the effects of simultaneously inhibiting the Hh pathway and autophagy on drug resistant BCR-ABL+ cells. BaF3-BCR-ABLT315I and BaF3-BCR-ABLY253F cells were insensitive to imatinib treatment, while the combination of vismodegib and CQ could nearly eliminate all these cells. Meanwhile, our results showed that the combination of vismodegib and CQ had little cytotoxicity on PBMCs. Furthermore, Hh inhibition could not influence the self-renewal and function of adult haematopoietic stem cells.Citation55,56 CQ, used in clinical practice for more than half a century for the treatment of malaria and autoimmune diseases, has been approved by FDA as an autophagy inhibitor for clinical applications. This body of evidence indicates that the combination of vismodegib and CQ might be a relatively safe option to solve the problem of CML drug resistance. Even if inhibiting autophagy could significantly enhance imatinib-induced K562 cells and primary CML stem cells death, this combination strategy could not kill drug-resistant CML cells.Citation25 The combination of Hh pathway inhibitors with autophagy inhibitors might be an effective combined therapy to overcome CML drug resistance.
Apoptosis is a type of typical programmed cell death, which the majority of anticancer agents can induce. We next explored whether the combination of vismodegib and CQ could induce apoptosis in CML cells. Caspase family proteins are key regulators of canonical apoptosis. Our results demonstrated that the combination of vismodegib and CQ could potently induce apoptosis but was partially dependent on caspase activation in CML cells.
Evidence shows that an activated MTOR pathway can promote GLI1 transcriptional activity and oncogenic function which lead to Hh pathway activation.Citation57 The MTOR pathway also plays a key role in autophagy. In general, inhibiting the MTOR pathway induces autophagy.Citation40 Our results indicated that inhibiting the Hh pathway induced autophagy and concomitantly downregulated the AKT-MTOR pathway. Moreover, the evidence indicated that both the Hh pathway and autophagy were closely related in modulating the BCR-ABL oncoprotein.Citation58 Our results indicated that the combination of vismodegib and CQ could inhibit the kinase activity of the BCR-ABL oncoprotein. It seems that there are complicated mechanisms in the combination of vismodegib and CQ-induced cell death of BCR-ABL+ CML cells. Killing drug-sensitive or -resistant BCR-ABL+ CML cells through simultaneously inhibiting the Hh pathway and autophagy was an interesting finding. Potential molecular mechanisms underlying this phenomenon need deeper investigation in the future.
It is consequential to mention that extensive academic and industrial efforts have contributed to the ongoing clinical trials of Hh inhibitors, such as vismodegib (GDC-0449, Roche), saridegib (IPI-926, Infinity), sonidegib (LDE225, Novartis), BMS-833923 (Bristol-Myers Squibb), PF04449913 (Pfizer), and LY2940680 (Eli Lilly), for the treatment of a variety of cancers.Citation7 Moreover, CQ and HCQ have been approved by the FDA for autophagy inhibition in clinical use. Thus, developing a large number of possible therapeutic combinations will be feasible to simultaneously target the Hh pathway and autophagy for CML treatment.
Taken together, the current study indicated that inhibition of the Hh pathway could induce autophagy in CML cells. Our results also demonstrated that combination of inhibitors of these 2 pathways could potently induce cell death in drug-sensitive or -resistant CML cells, while a single agent alone had mild effects on these cancer cells. This study suggested that the combined therapy, targeting both the Hh signaling pathway and autophagy, might be an effective strategy to overcome CML drug resistance.
Materials and Methods
Cell lines and culture
The BCR-ABL-expressing K562 cells and the BCR-ABL-nonexpressing HL-60 cells were obtained from the Cell Bank of Chinese Academy of Sciences, Shanghai Branch (Shanghai, China). BaF3 cells stably expressing wild-type (p210) or mutant forms of BCR-ABL (T315I and Y253F) were generously provided by Prof. Xiaoguang Chen (Department of Pharmacology, Institute of Materia Medica, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, China). All cells were passaged for fewer than 6 mo (fewer than passage number 100) after receipt from cell bank or resuscitation. Human peripheral blood mononuclear cells (PBMCs) were purchased from Shanghai Blood Center (Shanghai, China). Fresh PBMCs were separated from peripheral blood of healthy donors and subjected to experiments within 24 h. All cells were cultured in RPMI-1640 medium (Invitrogen, 21875–091) supplemented with 10% heat-inactivated fetal bovine serum (Invitrogen, 10099–141), 100 U/ml penicillin and 100 μg/ml streptomycin (Invitrogen, 15140–122) at 37°C in a humidified incubator with 5% CO2.
Reagents
The Hh inhibitor vismodegib (GDC-0449) (Shanghai biochempartner, 20120611) was dissolved in DMSO (40 mM) for storage. Other reagents were purchased as follows: CQ (Sigma, C6628), Bafi A1 (Sigma, B1793), RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific, K1622), TRlzol Reagent (Invitrogen, 15596026), and FastStart Universal SYBR Green Master (Rox) (Roche Diagnostics, 4913850001). Monoclonal antibodies were purchased as follows: MAP1LC3B (Cell Signaling Technology, 3868), ATG5 (Epitomics, 3167–1), ATG7 (Cell Signaling Technology, 8558), SQSTM1 (Epitomics, 3340–1), GLI1 (Epitomics, 6588–1), CTNNB1 (abcam, ab32572), GSK3B (Cell Signaling Technology, 12456), p-GSK3B (Ser9) (Cell Signaling Technology, 5558), tubulin (Cell Signaling Technology, 2148), ACTB (Cell Signaling Technology, 4970), CASP3 (Cell Signaling Technology, 9665), CASP9 (Cell Signaling Technology, 9502), MYC (Cell Signaling Technology, 5605), CCND1 (Cell Signaling Technology, 2978), PARP1 (Cell Signaling Technology, 9542), p-AKT (Thr308) (Cell Signaling Technology, 2965), p-AKT (Ser473) (Cell Signaling Technology, 4058), p-RPS6KB1 (Thr389) (Cell Signaling Technology, 9234), p-EIF4EBP1 (Thr37/46) (Cell Signaling Technology, 2855), p-MTOR (Ser2448) (Cell Signaling Technology, 2971), and BCR-ABL activity assay kit (contains p-BCR-ABL [Tyr245] and p-STAT5 [Tyr694]) (Cell Signaling Technology, 5300). Secondary antibodies were peroxidase-conjugated AffiniPure goat anti-mouse (Jackson ImmunoResearch Laboratories, 103757) and anti-rabbit IgG (Jackson ImmunoResearch Laboratories, 104122). All other reagents were purchased from Sigma (St Louis, MO, USA).
Cell viability assay
Cell viability was measured using Cell Counting Kit-8 (CCK-8) (Dojin Laboratories, CK04), as previously described.Citation59 Briefly, about 5,000 cells per well were seeded in 96-well plates and then exposed to vismodegib or autophagy inhibitors at indicated concentrations. These cells were incubated with CCK-8 solution for 0.5 to 1 h at 37°C. The optical density (O.D.) was measured at an absorbance wavelength of 450 nm. Images of phase contrast microscope were obtained using Nikon ECLIPSE TS100 microscopy (Nikon Corporation, Japan).
Flow cytometry analysis
Apoptosis was measured using the Annexin V (ANXA5)-FITC and PI Apoptosis Detection Kit (BD Bioscience, 556547) according to the manufacturer's instructions. Analysis was performed using a FACSCalibur flow cytometer (Becton–Dickinson, Fullerton, CA). Mitochondrial membrane potential was monitored after cells were staining with JC-1 mitochondrial membrane potential assay kit (Beyotime, C2006) as previously described.Citation60 For apoptosis and mitochondrial membrane potential analysis, 30,000 cells were analyzed per sample. Cells were stained with PtdIns (Beyotime, C1052) and cell cycle was analyzed using flow cytometry as previously described.Citation60 For cell cycle distribution analysis, 10,000 cells were analyzed per sample.
Western blot analysis
Western blot analysis was performed as previously described.Citation61 Breifly, cells were harvested and subjected to protein extraction using Cell Lysis Buffer. Equal amounts of the extracted protein were run on SDS-PAGE gels and followed by transferring membranes, blocking membrane, and incubating with primary and secondary antibodies. The signals were developed using an enhanced chemiluminescent detection reagent.
Quantitative real-time PCR
Total RNA extraction, reverse transcription, and quantitative RT-PCR were performed as previously described.Citation60 The following primers were used: GLI1, forward: AGCGTGAGCCTGAATCTGTG; reverse: CAGCATGTACTGGGCTTTGAA. PTCH, forward: CTCTGGAGCAGATTTCCAA GG; reverse: TGCCGCAGTTCTTTTGAATG. SMO, forward: TGCCACCAGAAGA ACAAGC, reverse: GGAGATCTCTGCCTCAACCA. These primers were synthesized by Shanghai Generay Biotech Co., Ltd. (Shanghai, China). Relative mRNA expression was estimated by normalization with GAPDH expression.
Small interfering RNA transfection
Cells were transfected with 50 nM siRNA using LipofectamineTM 2000 Transfection Reagent (Invitrogen, 11668) according to the manufacturer's instructions. Human SMO (Q000006608–1-A), ATG5 (siG10726164423), and ATG7 (siB111124164552) siRNA, were purchased from Guangzhou RiboBio Co., Ltd. Mouse Atg5 (siB081013135244), Atg7 (siB10108143630) siRNA, and nonsilencing scrambled control (SCR) siRNA (siNO5815122147–1–10) were purchased from Guangzhou RiboBio Co., Ltd.
Confocal immunofluorescence
Cells were transiently transfected with 50 nM of eGFP-MAP1LC3B plasmid (Addgene, 11546) using LipofectamineTM 2000 Transfection Reagent (Invitrogen, 11668). After 24 h transfection, cells were treated with 20 μM of vismodegib for 48 h. Green fluorescence was observed under confocal microscopy (Carl Zeiss LSM710, Carl Zeiss, Germany). Green fluorescent spots were counted using ImageJ software.
Transmission electron microscopy
BCR-ABL+ cells were incubated with 20 μM of vismodegib for 24 h, then harvested and processed as described.Citation62 Samples were analyzed with a JEM 1230 transmission electron microscope (JEOL Co., Ltd., Japan). Micrographs were taken at 7,000 or 20,000 magnification.
Statistical analysis
GraphPad Prism 5 (GraphPad Software Inc.., San Diego, CA) was used to perform statistical analysis. The results were expressed as means ± standard deviations (SD). Comparisons were performed using the Student t test (2-tailed). P-value < 0.05 was considered statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on thepublisher's website.
KAUP_A_994368_supplemental_files.zip
Download Zip (3.1 MB)Acknowledgments
We would like to appreciate the generosity of Prof. Xiaoguang Chen (Department of Pharmacology, Institute of Materia Medica, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, China) for friendly providing BaF3 cells expressed wild type (p210) and mutant forms of BCR-ABL (T315I and Y253F).
Funding
This study was supported by grants from National Key Basic Research Program of China (2015CB931800, 2013CB932502) and Shanghai Science and Technology Funds (14431900200, 13431900303).
References
- Hehlmann R, Hochhaus A, Baccarani M. Chronic myeloid leukaemia. Lancet 2007; 370:342-50; PMID:17662883; http://dx.doi.org/10.1016/S0140-6736(07)61165-9
- Jabbour E, Mathisen MS, O'Brien S. Ten years of progress in chronic myelogenous leukemia. J Natl Compr Canc Netw 2012; 10:1049-53; PMID:22956803
- Kimura S, Ando T, Kojima K. Ever-advancing chronic myeloid leukemia treatment. Int J Clin Oncol 2014; 19:3-9; PMID:24258348; http://dx.doi.org/10.1007/s10147-013-0641-7
- Briscoe J, Therond PP. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol 2013; 14:416-29; PMID:24258348; http://dx.doi.org/10.1038/nrm3598
- Atwood SX, Chang AL, Oro AE. Hedgehog pathway inhibition and the race against tumor evolution. J Cell Biol 2012; 199:193-7; PMID:23071148; http://dx.doi.org/10.1083/jcb.201207140
- Scales SJ, de Sauvage FJ. Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol Sci 2009; 30:303-12; PMID:19443052; http://dx.doi.org/10.1016/j.tips.2009.03.007
- Amakye D, Jagani Z, Dorsch M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nature Medicine 2013; 19:1410-22; PMID:24202394; http://dx.doi.org/10.1038/nm.3389
- Merchant AA, Matsui W. Targeting Hedgehog–a cancer stem cell pathway. Clin Cancer Res 2010; 16:3130-40; PMID:20530699; http://dx.doi.org/10.1158/1078-0432.CCR-09-2846
- Dierks C, Grbic J, Zirlik K, Beigi R, Englund NP, Guo GR, Veelken H, Engelhardt M, Mertelsmann R, Kelleher JF, et al. Essential role of stromally induced hedgehog signaling in B-cell malignancies. Nat Med 2007; 13:944-51; PMID:17632527; http://dx.doi.org/10.1038/nm1614
- Low JA, de Sauvage FJ. Clinical experience with Hedgehog pathway inhibitors. J Clin Oncol 2010; 28:5321-6; PMID:21041712; http://dx.doi.org/10.1200/JCO.2010.27.9943
- Mar BG, Amakye D, Aifantis I, Buonamici S. The controversial role of the Hedgehog pathway in normal and malignant hematopoiesis. Leukemia 2011; 25:1665-73; PMID:21660044; http://dx.doi.org/10.1038/leu.2011.143
- Irvine DA, Copland M. Targeting hedgehog in hematologic malignancy. Blood 2012; 119:2196-204; PMID:22223823; http://dx.doi.org/10.1182/blood-2011-10-383752
- Jagani Z, Dorsch M, Warmuth M. Hedgehog pathway activation in chronic myeloid leukemia. Cell Cycle 2010; 9:3449-56; PMID:20928937; http://dx.doi.org/10.4161/cc.9.17.12945
- Hamad A, Sahli Z, El Sabban M, Mouteirik M, Nasr R. Emerging therapeutic strategies for targeting chronic myeloid leukemia stem cells. Stem Cells Int 2013; 2013:724360; PMID:23935640; http://dx.doi.org/10.1155/2013/724360
- Queiroz KC, Ruela-de-Sousa RR, Fuhler GM, Aberson HL, Ferreira CV, Peppelenbosch MP, Spek CA. Hedgehog signaling maintains chemoresistance in myeloid leukemic cells. Oncogene 2010; 29:6314-22; PMID:20802532; http://dx.doi.org/10.1038/onc.2010.375
- Dierks C, Beigi R, Guo G-R, Zirlik K, Stegert MR, Manley P, Trussell C, Schmitt-Graeff A, Landwerlin K, Veelken H, et al. Expansion of Bcr-Abl-Positive Leukemic Stem Cells Is Dependent on Hedgehog Pathway Activation. Cancer Cell 2008; 14:238-49; PMID:18772113; http://dx.doi.org/10.1016/j.ccr.2008.08.003
- Warzecha J, Bonke L, Koehl U, Munkelt D, Gottig S, Percic D, Arabmotlagh M, Kurth A. The hedgehog inhibitor cyclopamine induces apoptosis in leukemic cells in vitro. Leuk Lymphoma 2008; 49:2383-6; PMID:19052992; http://dx.doi.org/10.1080/10428190802510315
- Kobune M, Takimoto R, Murase K, Iyama S, Sato T, Kikuchi S, Kawano Y, Miyanishi K, Sato Y, Niitsu Y, et al. Drug resistance is dramatically restored by hedgehog inhibitors in CD34+ leukemic cells. Cancer Sci 2009; 100:948-55; PMID:19245435; http://dx.doi.org/10.1111/j.1349-7006.2009.01111.x
- Katagiri S, Tauchi T, Okabe S, Minami Y, Kimura S, Maekawa T, Naoe T, Ohyashiki K. Combination of ponatinib with Hedgehog antagonist vismodegib for therapy-resistant BCR-ABL1-positive leukemia. Clin Cancer Res 2013; 19:1422-32; PMID:23319824; http://dx.doi.org/10.1158/1078-0432.CCR-12-1777
- Swampillai AL, Salomoni P, Short SC. The role of autophagy in clinical practice. Clin Oncol (R Coll Radiol) 2012; 24:387-95; PMID:22032864; http://dx.doi.org/10.1016/j.clon.2011.09.010
- Shanware NP, Bray K, Abraham RT. The PI3K, metabolic, and autophagy networks: interactive partners in cellular health and disease. Annu Rev Pharmacol Toxicol 2013; 53:89-106; PMID:23294306; http://dx.doi.org/10.1146/annurev-pharmtox-010611-134717
- Gewirtz DA. Cytoprotective and nonprotective autophagy in cancer therapy. Autophagy 2013; 9:1263-5; PMID:23800720; http://dx.doi.org/10.4161/auto.25233
- Helgason GV, Karvela M, Holyoake TL. Kill one bird with two stones: potential efficacy of BCR-ABL and autophagy inhibition in CML. Blood 2011; 118:2035-43; PMID:21693757; http://dx.doi.org/10.1182/blood-2011-01-330621
- Mishima Y, Terui Y, Taniyama A, Kuniyoshi R, Takizawa T, Kimura S, Ozawa K, Hatake K. Autophagy and autophagic cell death are next targets for elimination of the resistance to tyrosine kinase inhibitors. Cancer Sci 2008; 99:2200-8; PMID:18823378; http://dx.doi.org/10.1111/j.1349-7006.2008.00932.x
- Bellodi C, Lidonnici MR, Hamilton A, Helgason GV, Soliera AR, Ronchetti M, Galavotti S, Young KW, Selmi T, Yacobi R, et al. Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J Clin Invest 2009; 119:1109-23; PMID:19363292; http://dx.doi.org/10.1172/JCI35660
- Nencioni A, Cea M, Montecucco F, Longo VD, Patrone F, Carella AM, Holyoake TL, Helgason GV. Autophagy in blood cancers: biological role and therapeutic implications. Haematologica 2013; 98:1335-43; PMID:24006406; http://dx.doi.org/10.3324/haematol.2012.079061
- Jimenez-Sanchez M, Menzies FM, Chang YY, Simecek N, Neufeld TP, Rubinsztein DC. The Hedgehog signalling pathway regulates autophagy. Nat Commun 2012; 3:1200; PMID:23149744; http://dx.doi.org/10.1038/ncomms2212
- Wang Y, Han C, Lu L, Magliato S, Wu T. Hedgehog signaling pathway regulates autophagy in human hepatocellular carcinoma cells. Hepatology 2013; 58:995-1010; PMID:23504944; http://dx.doi.org/10.1002/hep.26394
- Li H, Li J, Li Y, Singh P, Cao L, Xu LJ, Li D, Wang Y, Xie Z, Gui Y. Sonic hedgehog promotes autophagy of vascular smooth muscle cells. Am J Physiol Heart Circ Physiol 2012; 303:H1319-31; PMID:23023870; http://dx.doi.org/10.1152/ajpheart.00160.2012
- Petralia RS, Schwartz CM, Wang YX, Kawamoto EM, Mattson MP, Yao PJ. Sonic hedgehog promotes autophagy in hippocampal neurons. Biol Open 2013; 2:499-504; PMID:23789099; http://dx.doi.org/10.1242/bio.20134275
- Sarkar FH, Li Y, Wang Z, Kong D. The role of nutraceuticals in the regulation of Wnt and Hedgehog signaling in cancer. Cancer and Metastasis Reviews 2010; 29:383-94; PMID:20711635; http://dx.doi.org/10.1007/s10555-010-9233-4
- Clevers H. Wnt/β-Catenin Signaling in Development and Disease. Cell 2006; 127:469-80; PMID:17081971; http://dx.doi.org/10.1016/j.cell.2006.10.018
- Sengupta A, Banerjee D, Chandra S, Banerji SK, Ghosh R, Roy R, Banerjee S. Deregulation and cross talk among Sonic hedgehog, Wnt, Hox and Notch signaling in chronic myeloid leukemia progression. Leukemia 2007; 21:949-55; PMID:17361218; http://dx.doi.org/10.1038/sj.leu.2404657
- Zhao C, Blum J, Chen A, Kwon HY, Jung SH, Cook JM, Lagoo A, Reya T. Loss of β-Catenin Impairs the Renewal of Normal and CML Stem Cells In Vivo. Cancer Cell 2007; 12:528-41; PMID:18068630; http://dx.doi.org/10.1016/j.ccr.2007.11.003
- Heidel Florian H, Bullinger L, Feng Z, Wang Z, Neff Tobias A, Stein L, Kalaitzidis D, Lane SW, Armstrong SA. Genetic and Pharmacologic Inhibition of β-Catenin Targets Imatinib-Resistant Leukemia Stem Cells in CML. Cell Stem Cell 2012; 10:412-24; PMID:22482506; http://dx.doi.org/10.1016/j.stem.2012.02.017
- O'Hare T. Inhibition of wild-type and mutant Bcr-Abl by AP23464, a potent ATP-based oncogenic protein kinase inhibitor: implications for CML. Blood 2004; 104:2532-9; PMID:15256422; http://dx.doi.org/10.1182/blood-2004-05-1851
- O'Hare T, Walters DK, Stoffregen EP, Jia T, Manley PW, Mestan J, Cowan-Jacob SW, Lee FY, Heinrich MC, Deininger MW, et al. In vitro activity of Bcr-Abl inhibitors AMN107 and BMS-354825 against clinically relevant imatinib-resistant Abl kinase domain mutants. Cancer Res 2005; 65:4500-5; PMID:15930265; http://dx.doi.org/10.1158/0008-5472.CAN-05-0259
- Yuan X, Zhang Y, Zhang H, Jin J, Li X, Liu H, Feng Z, Chen X. Activity of the potent dual Abl/Src tyrosine kinase inhibitor FB2 against Bcr-Abl positive cell lines in vitro and in vivo. Leuk Res 2011; 35:237-42; PMID:20739063; http://dx.doi.org/10.1016/j.leukres.2010.07.041
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/10.4161/auto.19496
- Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett 2010; 584:1287-95; PMID:20083114; http://dx.doi.org/10.1016/j.febslet.2010.01.017
- Ambjørn M, Ejlerskov P, Liu Y, Lees M, Jäättelä M, Issazadeh-Navikas S. IFNB1/interferon-β-induced autophagy in MCF-7 breast cancer cells counteracts its proapoptotic function. Autophagy 2013; 9:287-302; PMID:23221969; http://dx.doi.org/10.4161/auto.22831
- Kawahara T, Kawaguchi-Ihara N, Okuhashi Y, Itoh M, Nara N, Tohda S. Cyclopamine and quercetin suppress the growth of leukemia and lymphoma cells. Anticancer Res 2009; 29:4629-32; PMID:20032413
- Kim JE, Yoon S, Choi BR, Kim KP, Cho YH, Jung W, Kim DW, Oh S, Kim DE. Cleavage of BCR-ABL transcripts at the T315I point mutation by DNAzyme promotes apoptotic cell death in imatinib-resistant BCR-ABL leukemic cells. Leukemia 2013; 27:1650-8; PMID:23434731; http://dx.doi.org/10.1038/leu.2013.60
- Shi X, Jin Y, Cheng C, Zhang H, Zou W, Zheng Q, Lu Z, Chen Q, Lai Y, Pan J. Triptolide inhibits Bcr-Abl transcription and induces apoptosis in STI571-resistant chronic myelogenous leukemia cells harboring T315I mutation. Clin Cancer Res 2009; 15:1686-97; PMID:19240172; http://dx.doi.org/10.1158/1078-0432.CCR-08-2141
- Weiner LM, Lotze MT. Tumor-cell death, autophagy, and immunity. N Engl J Med 2012; 366:1156-8; PMID:22435376; http://dx.doi.org/10.1056/NEJMcibr1114526
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011; 147:728-41; PMID:22078875; http://dx.doi.org/10.1016/j.cell.2011.10.026
- Choi AMK, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med 2013; 368:651-62; PMID:23406030; http://dx.doi.org/10.1056/NEJMra1205406
- Janku F, McConkey DJ, Hong DS, Kurzrock R. Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol 2011; 8:528-39; PMID:21587219; http://dx.doi.org/10.1038/nrclinonc.2011.71
- Rosenfeld MR, Ye X, Supko JG, Desideri S, Grossman SA, Brem S, Mikkelson T, Wang D, Chang YC, Hu J, et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2014; 10:1359-68; PMID:24991840; http://dx.doi.org/10.4161/auto.28984
- Rangwala R, Leone R, Chang YC, Fecher L, Schuchter L, Kramer A, Tan KS, Heitjan DF, Rodgers G, Gallagher M, et al. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy 2014; 10:1369-79; PMID:24991839; http://dx.doi.org/10.4161/auto.29118
- Vogl DT, Stadtmauer EA, Tan KS, Heitjan DF, Davis LE, Pontiggia L, Rangwala R, Piao S, Chang YC, Scott EC, et al. Combined autophagy and proteasome inhibition: a phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy 2014; 10:1380-90; PMID:24991834; http://dx.doi.org/10.4161/auto.29264
- Rangwala R, Chang YC, Hu J, Algazy KM, Evans TL, Fecher LA, Schuchter LM, Torigian DA, Panosian JT, Troxel AB, et al. Combined MTOR and autophagy inhibition: phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy 2014; 10:1391-402; PMID:24991838; http://dx.doi.org/10.4161/auto.29119
- Mahalingam D, Mita M, Sarantopoulos J, Wood L, Amaravadi R, Davis LE, Mita AC, Curiel TJ, Espitia CM, Nawrocki ST, et al. Combined autophagy and HDAC inhibition: a phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy 2014; 10:1403-14; PMID:24991835; http://dx.doi.org/10.4161/auto.29231
- Barnard RA, Wittenburg LA, Amaravadi RK, Gustafson DL, Thorburn A, Thamm DH. Phase I clinical trial and pharmacodynamic evaluation of combination hydroxychloroquine and doxorubicin treatment in pet dogs treated for spontaneously occurring lymphoma. Autophagy 2014; 10:1415-25; PMID:24991836; http://dx.doi.org/10.4161/auto.29165
- Gao J, Graves S, Koch U, Liu S, Jankovic V, Buonamici S, El Andaloussi A, Nimer SD, Kee BL, Taichman R, et al. Hedgehog signaling is dispensable for adult hematopoietic stem cell function. Cell Stem Cell 2009; 4:548-58; PMID:19497283; http://dx.doi.org/10.1016/j.stem.2009.03.015
- Hofmann I, Stover EH, Cullen DE, Mao J, Morgan KJ, Lee BH, Kharas MG, Miller PG, Cornejo MG, Okabe R, et al. Hedgehog signaling is dispensable for adult murine hematopoietic stem cell function and hematopoiesis. Cell Stem Cell 2009; 4:559-67; PMID:19497284; http://dx.doi.org/10.1016/j.stem.2009.03.016
- Wang Y, Ding Q, Yen CJ, Xia W, Izzo JG, Lang JY, Li CW, Hsu JL, Miller SA, Wang X, et al. The crosstalk of mTOR/S6K1 and Hedgehog pathways. Cancer Cell 2012; 21:374-87; PMID:22439934; http://dx.doi.org/10.1016/j.ccr.2011.12.028
- Goussetis DJ, Gounaris E, Wu EJ, Vakana E, Sharma B, Bogyo M, Altman JK, Platanias LC. Autophagic degradation of the BCR-ABL oncoprotein and generation of antileukemic responses by arsenic trioxide. Blood 2012; 120:3555-62; PMID:22898604; http://dx.doi.org/10.1182/blood-2012-01-402578
- Zhai B, Hu F, Jiang X, Xu J, Zhao D, Liu B, Pan S, Dong X, Tan G, Wei Z. Inhibition of Akt reverses the acquired resistance to sorafenib by switching protective autophagy to autophagic cell death in hepatocellular carcinoma. Mol Cancer Ther 2014; 13:1589-98; PMID:24705351; http://dx.doi.org/10.1158/1535-7163.MCT-13-1043
- Zeng X, Li Y, Fan J, Zhao H, Xian Z, Sun Y, Wang Z, Wang S, Zhang G, Ju D. Recombinant human arginase induced caspase-dependent apoptosis and autophagy in non-Hodgkin's lymphoma cells. Cell Death Dis 2013; 4:e840; PMID:24113174; http://dx.doi.org/10.1038/cddis.2013.359
- Li Y, Zhu H, Zeng X, Fan J, Qian X, Wang S, Wang Z, Sun Y, Wang X, Wang W, et al. Suppression of autophagy enhanced growth inhibition and apoptosis of interferon-beta in human glioma cells. Mol Neurobiol 2013; 47:1000-10; PMID:23329343; http://dx.doi.org/10.1007/s12035-013-8403-0
- Fan J, Zeng X, Li Y, Wang S, Wang Z, Sun Y, Gao H, Zhang G, Feng M, Ju D, et al. Autophagy plays a critical role in ChLym-1-induced cytotoxicity of non-hodgkin's lymphoma cells. PLoS One 2013; 8:e72478; PMID:24015249; http://dx.doi.org/10.1371/journal.pone.0072478