
Abstract
Both apoptosis ("self-killing") and autophagy ("self-eating") are evolutionarily conserved processes, and their crosstalk influences anticancer drug sensitivity and cell death. However, the underlying mechanism remains unclear. Here, we demonstrated that HMGB1 (high mobility group box 1), normally a nuclear protein, is a crucial regulator of TNFSF10/TRAIL (tumor necrosis factor [ligand] superfamily, member 10)-induced cancer cell death. Activation of PARP1 (poly [ADP-ribose] polymerase 1) was required for TNFSF10-induced ADP-ribosylation of HMGB1 in cancer cells. Moreover, pharmacological inhibition of PARP1 activity or knockdown of PARP1 gene expression significantly inhibited TNFSF10-induced HMGB1 cytoplasmic translocation and subsequent HMGB1-BECN1 complex formation. Furthermore, suppression of the PARP1-HMGB1 pathway diminished autophagy, increased apoptosis, and enhanced the anticancer activity of TNFSF10 in vitro and in a subcutaneous tumor model. These results indicate that PARP1 acts as a prominent upstream regulator of HMGB1-mediated autophagy and maintains a homeostatic balance between apoptosis and autophagy, which provides new insight into the mechanism of TNFSF10 resistance.
Abbreviations:
- ATG, autophagy-related
- DISC, death-inducing signaling complex
- HMGB1, high mobility group box 1
- MAP1LC3A/LC3, microtubule-associated protein 1 light chain 3 α
- PARP1, poly (ADP-ribose) polymerase 1
- PARylation, poly-ADP-ribosylation
- RIPK1/RIP, receptor (TNFRSF)-interacting serine-threonine kinase 1
- shRNA, short hairpin RNA
- TNFSF10/TRAIL, tumor necrosis factor (ligand) superfamily, member 10
- TNF, tumor necrosis factor
- TUNEL, TdT-mediated dUTP-X nick end labeling
Introduction
Chemotherapy, the use of chemical reagents to kill fast-growing tumor cells, is one of the major forms of cancer therapy. Understanding how cancer cells acquire drug resistance is a challenge in chemotherapy. Apoptosis ("self-killing"), a form of programmed cell death, includes 2 main pathways: an extrinsic pathway, which is mediated by death receptors on the plasma membrane, and an intrinsic pathway, which is mediated by mitochondria.Citation1 CASPs/caspases, a family of intracellular proteases, play central roles in the transduction and amplification of extrinsic and intrinsic apoptotic signals.Citation1 Autophagy ("self-eating"), a form of programmed cell survival, is a lysosomal degradation pathway with 3 major types: macroautophagy, microautophagy, and chaperone-mediated autophagy.Citation2 Macroautophagy (hereafter referred to as autophagy) is the best-known and most widely studied type. Autophagy-related (ATG) proteins play central roles in the regulation of various stages of the autophagic process, including phagophore assembly and autophagosome and autolysosome formation.Citation3 The balance and switch between apoptosis and autophagy significantly influence chemotherapeutic efficacy and determine whether a cell survives or dies.Citation4 In many cases, apoptosis deficiency and autophagy upregulation contribute to chemotherapy resistance.Citation5 Although the diverse functions of apoptosis and autophagy have been well elucidated, the molecular crosstalk between apoptosis and autophagy remains poorly understood.Citation6
TNFSF10/TRAIL is a member of the TNF (tumor necrosis factor) family. After it was discovered in 1995,Citation7 TNFSF10 was considered the most promising antitumor agent due to its ability to kill many tumor cells, but not most normal cells.Citation8,9 TNFSF10 induces cancer cell death through binding to its receptors to activate the CASP8-dependent apoptosis pathway.Citation8,9 By 1998, 5 human TNFSF10 receptors had been identified, including TNFRSF10A/DR4/TNFSF10-R1, TNFRSF10B/DR5/TNFSF10-R2, TNFRSF10C/DcR1/TNFSF10-R3, TNFRSF-10D/DcR2/TNFSF10-R4, and a soluble receptor called TNFRSF11B/osteoprotegerin.Citation9 Of note, TNFRSF10A and TNFRSF10B are proapoptotic TNFSF10 receptors, whereas TNFRSF10C and TNFRSF10D are decoy TNFSF10 receptors unable to induce cell death. Unfortunately, several cancer cells are resistant to TNFSF10-mediated cell death; the potential mechanisms include: decreased levels of TNFRSF10A and TNFRSF10B,Citation10 loss of CASP8,Citation11,12 post-translational modification of TNFSF10 receptors such as glycosylation,Citation13 overexpression of TNFRSF10C and TNFRSF10D,Citation14 elevates levels of negative regulators of apoptosis such as CFLAR/cFLIP and MCL1,Citation15,16 and upregulation of cell survival pathways such as NFKB/NFκB and autophagy.Citation17-19 Further insight into the molecular mechanisms determining TNFSF10 activity is important in order to use this approach for human cancer therapy.
High mobility group box 1 is a highly conserved, chromatin-associated protein and regulates nucleosome dynamics, chromosome stability, and gene transcription.Citation20 In addition, HMGB1 also functions as a damage-associated molecular pattern with cytokine and chemokine activity when released to the extracellular space.Citation20 The change in HMGB1 expression and subcellular localization has been implicated in regulating tumor development and cancer therapy.Citation21 Our recent studies demonstrated that both intracellular and extracellular HMGB1 play important roles in the regulation of autophagy in normal or cancer cells through different mechanisms. One) Nuclear HMGB1 promotes autophagy by regulation of HSPB1 (heat shock 27kDa protein 1) expression;Citation22 2) Cytoplasmic HMGB1 promotes autophagy by binding to BECN1 to initiate autophagosome formation;Citation23 3). Extracellular HMGB1 induces autophagy by AGER (advanced glycation end product-specific receptor)-mediated activation of the class III phosphatidylinositol 3-kinase.Citation24 These findings suggest that HMGB1-mediated autophagy is a potential target for chemotherapy.
The effect of HMGB1 on the anticancer activity of TNFSF10 has not yet been described. Here, we provide the first evidence that HMGB1 is specifically poly-ADP-ribosylated by PARP1 in cancer cells, which in turn contributes to TNFSF10 resistance through upregulation of autophagy and suppression of apoptosis. Notably, we demonstrated that genetic and pharmacological inhibition of the PARP1-HMGB1 pathway enhanced TNFSF10-mediated cell death in vitro and in vivo, suggesting a novel strategy to overcome TNFSF10 resistance.
Results
Inhibition of HMGB1 expression and cytoplasmic translocation enhanced TNFSF10-mediated cell death
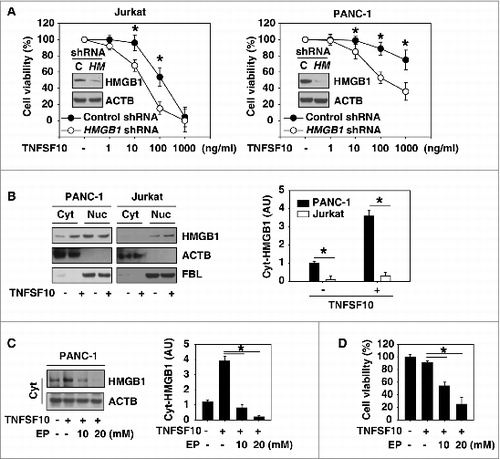
To explore the potential role of HMGB1 in the regulation of TNFSF10s anticancer activity, specific short hairpin RNA (shRNA) against HMGB1 was transfected into the TNFSF10-sensitive human leukemic cell line JurkatCitation11 and the TNFSF10-resistant human pancreatic cancer cell line PANC-1.Citation25 Knockdown of HMGB1 by shRNA enhanced TNFSF10-mediated cell death in Jurkat cells and reversed TNFSF10 resistance in PANC-1 cells (), suggesting an important role for HMGB1 in the regulation of the TNFSF10 response. HMGB1 cytoplasmic translocation () was increased after TNFSF10 treatment in PANC-1 cells compared with Jurkat cells. To explore the role of cytoplasmic HMGB1 in the regulation of TNFSF10s anticancer activity, we treated cells with ethyl pyruvate, a potential inhibitor of HMGB1 cytoplasmic translocation and release.Citation26 Indeed, ethyl pyruvate limited TNFSF10-induced HMGB1 cytoplasmic translocation () and enhanced TNFSF10-mediated cell death () in PANC-1 cells. Collectively, these findings suggest that increased cytosolic HMGB1 levels contribute to TNFSF10 resistance.
Figure 1. Inhibition of HMGB1 expression and cytoplasmic translocation enhance TNFSF10-mediated cell death. (A) The indicated HMGB1 wild-type (control shRNA) and knockdown (HMGB1 shRNA) cells were treated with TNFSF10 (1 to 1000 ng/ml) for 24 h, and then cell viability was assayed with the CCK-8 kit (n = 3 , *, P < 0.05). Western blot analysis of the expression of HMGB1 is shown in the inserts. (B) PANC-1 and Jurkat cells were treated with TNFSF10 (100 ng/ml) for 24 h and then cytosolic (Cyt) and nuclear (Nuc) HMGB1 were assayed by western blot. Relative band intensities of cytosolic HMGB1 were quantified and normalized to actin expression (P < 0.05). AU, arbitrary unit. (C and D) PANC-1 cells were treated with TNFSF10 (100 ng/ml) in the absence or presence of indicated ethyl pyruvate (EP) for 24 h. Cytosolic (Cyt) HMGB1 (C) was assayed by western blot and cell viability (D) was assayed by CCK-8 kit (n = 3 , *, P < 0.05). AU, arbitrary unit.
PARP1 is required for TNFSF10-induced Poly-ADP-ribosylation and subsequent cytoplasmic translocation of HMGB1
Next, we investigated the mechanism of HMGB1 cytoplasmic translocation following TNFSF10 treatment. Previous studies have shown that post-translational modification (e.g., acetylation and phosphorylation) is critical for translocation of HMGB1 from the nucleus to the cytoplasm in immune cells such as macrophages and monocytes.Citation27,28 However, we did not observe significant HMGB1 acetylation and phosphorylation after treatment with TNFSF10 in cancer cells (data not shown). In contrast, TNFSF10 significantly induced HMGB1 poly-ADP-ribosylation (PARylation) in cancer cells, especially in TNFSF10-resistant PANC-1 cells (), suggesting a potential role for PARylation in the regulation of HMGB1 cytoplasmic translocation as well as TNFSF10 resistance. PARP1 is the master regulator of PARylation.Citation29 We found that inhibition of PARP1 expression/activity through shRNA knockdown () or the pharmacological inhibitor PJ-34 () significantly reduced TNFSF10-induced PARylation and the subsequent cytoplasmic translocation of HMGB1 in PANC-1 cells. These findings suggest that PARP1 is required for TNFSF10-induced HMGB1 PARylation and cytoplasmic translocation.
Figure 2. PARP1 is required for TNFSF10-induced poly-ADP-ribosylation and subsequent cytoplasmic translocation of HMGB1. (A) PANC-1 and Jurkat cells were treated with TNFSF10 (100 ng/ml) for 8 to 24 h. Samples were pulled down with anti-HMGB1 and immunoblotted with anti-PARylation and anti-HMGB1 antibodies. Relative band intensities of HMGB1 PARylation were quantified (*, P < 0.05 versus untreated group). (B) PANC-1 cells were transfected with control shRNA and PARP1 shRNA for 48 h and then treated with TNFSF10 (100 ng/ml) for 24 h. The levels of HMGB1 PARylation and cytoplasmic HMGB1 were assayed (*, P < 0.05). AU, arbitrary unit. (C) PANC-1 cells were treated with TNFSF10 (100 ng/ml) in the absence or presence of PJ-34 (20 μM) for 24 h. The levels of HMGB1 PARylation and cytoplasmic HMGB1 were assayed (*, P < 0.05). AU=arbitrary unit. (D–G) Knockdown of FADD and CASP8 by shRNA (D) increased TNFSF10 (100 ng/ml)-induced PARP1 activity (E), HMGB1 PARylation (F), and HMGB1 cytosolic translocation (G) in PANC-1 cells. In contrast, necrostatin-1 (10 μM), a specific inhibitor of RIPK1, inhibits this process. AU, arbitrary unit.
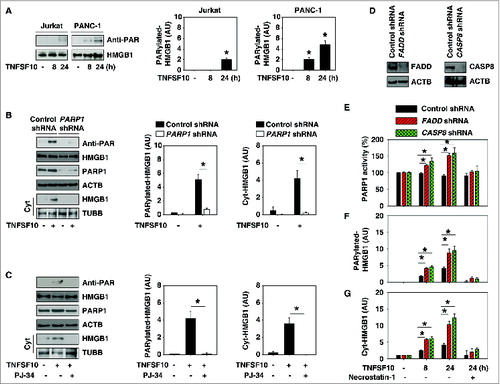
The death-inducing signaling complex (DISC) is essential for induction of cell death receptor-mediated apoptosis. To explore whether DISC is required for TNFSF10-induced PARylation and translocation of HMGB1, we knocked down the components of DISC complex such as FADD and CASP8 by specific shRNA (). Suppression of expression of FADD and CASP8 increased TNFSF10-induced PARP1 activity (), HMGB1 PARylation (), and HMGB1 cytosolic translocation () at 8 and 24 h. RIPK1/RIP1 (receptor [TNFRSF]-interacting serine-threonine kinase 1) is recruited to the DISC that drives CASP8-dependent apoptosis in response to TRAIL.Citation30 In contrast, RIPK1 is required for PARP1 activation and necroptosis when CASP8 activation is inhibited or FADD is deficient.Citation31 We found that necrostatin-1, a specific inhibitor of RIPK1, inhibits TNFSF10-induced PARP1 activity (), HMGB1 PARylation (), and HMGB1 cytosolic translocation () when downregulation of FADD and CASP8 occurs. These findings suggest that DISC-associated CASP8 activation negatively regulates PARP1-mediated PARylation and that HMGB1 is translocated in TNFSF10-resistant cells partly in a RIPK1-dependent manner. Indeed, activation of both PARP and RIPK1 is required for epipolythiodioxopiperazine-mediated autophagy induction in human colon cancer cells, which is closely associated with CASP-dependent cell death.Citation32 PARP1 can collectively regulate both autophagic and apoptotic processes depending on cell type and stimulus.
PARP1 promotes TNFSF10-induced autophagy by sustaining formation of the HMGB1-BECN1 complex
Microtubule-associated protein 1 light chain 3 α (MAP1LC3A/LC3) is a widely used marker to assay autophagy because the amount of MAP1LC3A-II correlates well with the number of autophagosomes.Citation33,34 Similar to findings from a previous study,Citation19 TNFSF10 treatment induced MAP1LC3A-II expression () and MAP1LC3A puncta formation (). As an adaptor between the autophagic machinery and its substrates, SQSTM1/p62 is degraded following TNFSF10 treatment (). In contrast, knockdown of PARP1 by RNAi significantly inhibits TNFSF10-induced MAP1LC3A-II expression (), MAP1LC3A puncta formation (), and SQSTM1 degradation (). These findings indicate that PARP1 function is a positive regulator of TNFSF10-induced autophagy. Cytoplasmic HMGB1 is a BECN1-binding protein, which sustains autophagosome formation.Citation23 Given that PARP1 is required for TNFSF10-induced HMGB1 cytoplasmic translocation (), we therefore explored whether PARP1 is required for HMGB1-BECN1 complex formation during TNFSF10 treatment. As expected, knockdown of PARP1 significantly inhibited the TNFSF10-induced formation of the HMGB1-BECN1 complex (). Similar to results from the knockdown of PARP1, knockdown of both HMGB1 and BECN1 decreased TNFSF10-induced MAP1LC3A puncta formation ().
Figure 3. PARP1 promotes TNFSF10-induced autophagy by sustaining formation of the HMGB1-BECN1 complex. (A) PANC-1 cells were transfected with control shRNA and PARP1 shRNA for 48 h and then treated with TNFSF10 (100 ng/ml) for 24 h. The levels of MAP1LC3A-I/II and SQSTM1 were assayed by western blot. Relative band intensities of MAP1LC3A-II and SQSTM1 were quantified (P < 0.05). AU, arbitrary unit. (B and C) The indicated PANC-1 cells were treated with TNFSF10 (100 ng/ml) for 24 h. The number of MAP1LC3A puncta (yellow arrows) (B) was assayed by confocal microscopic analysis (* P < 0.05 vs. control shRNA group; bar = 10 μm). The interaction between HMGB1 and BECN1 (C) was assayed by coimmunoprecipitation analysis. (D) PANC-1 cells were transfected with wild-type and HMGB1C106S cDNA for 48 h and then treated with TNFSF10 (100 ng/ml) for 24 h. The levels of indicated proteins were assayed by western blot. (E) PANC-1 cells were transfected with the indicated control shRNA, PARP1 shRNA, and HMGB1C106S cDNA for 48 h and then treated with TNFSF10 (100 ng/ml) for 24 h. The levels of the indicated proteins were assayed by western blot. Relative band intensities of MAP1LC3A-II and cyt-HMGB1 were quantified. In parallel, cell viability was assayed (n = 3 , * P < 0.05). (F) PANC-1 cells were transfected with wild-type and HMGB1[3KA] cDNA for 48 h and then treated with TNFSF10 (100 ng/ml) for 24 h. The levels of indicated proteins were assayed by western blot.
![Figure 3. PARP1 promotes TNFSF10-induced autophagy by sustaining formation of the HMGB1-BECN1 complex. (A) PANC-1 cells were transfected with control shRNA and PARP1 shRNA for 48 h and then treated with TNFSF10 (100 ng/ml) for 24 h. The levels of MAP1LC3A-I/II and SQSTM1 were assayed by western blot. Relative band intensities of MAP1LC3A-II and SQSTM1 were quantified (P < 0.05). AU, arbitrary unit. (B and C) The indicated PANC-1 cells were treated with TNFSF10 (100 ng/ml) for 24 h. The number of MAP1LC3A puncta (yellow arrows) (B) was assayed by confocal microscopic analysis (* P < 0.05 vs. control shRNA group; bar = 10 μm). The interaction between HMGB1 and BECN1 (C) was assayed by coimmunoprecipitation analysis. (D) PANC-1 cells were transfected with wild-type and HMGB1C106S cDNA for 48 h and then treated with TNFSF10 (100 ng/ml) for 24 h. The levels of indicated proteins were assayed by western blot. (E) PANC-1 cells were transfected with the indicated control shRNA, PARP1 shRNA, and HMGB1C106S cDNA for 48 h and then treated with TNFSF10 (100 ng/ml) for 24 h. The levels of the indicated proteins were assayed by western blot. Relative band intensities of MAP1LC3A-II and cyt-HMGB1 were quantified. In parallel, cell viability was assayed (n = 3 , * P < 0.05). (F) PANC-1 cells were transfected with wild-type and HMGB1[3KA] cDNA for 48 h and then treated with TNFSF10 (100 ng/ml) for 24 h. The levels of indicated proteins were assayed by western blot.](/cms/asset/39d49ad3-18ef-46fb-8dac-ca1d9e60e34b/kaup_a_994400_f0003_c.jpg)
Our previous study demonstrates that the mutation of HMGB1 at Cys106 increases HMGB1 translocation and subsequent HMGB1-BECN1 interaction during autophagy.Citation23 We therefore investigated whether PARylation of HMGB1 is increased when HMGB1 Cys106 is mutated. Indeed, PARylation and cytosolic translocation of HMGB1 were significantly increased when wild-type PANC-1 cells were transfected with HMGB1C106S mutant cDNA in the absence or presence of TNFSF10 (). Moreover, transfection of HMGB1C106S mutant cDNA into PANC-1 cells with PARP1 knockdown, increased cytosolic HMGB1 level, autophagy (MAP1LC3A-II), and TNFSF10 resistance (), suggesting that HMGB1C106S is PARylated by other PARPs and that cytoplasmic HMGB1 is sufficient to trigger autophagy and TNFSF10 resistance in PARP1-deficient cells. To determine whether PARylation per se is required for HMGB1 translocation, we mutated lysine at 27, 28, and 29 into alanine (HMGB1[3KA] henceforth). Compared with C106S (), these lysine mutations inhibited TNFSF10-induced PARylation and cytosolic translocation of HMGB1 and autophagy (MAP1LC3A-II) ().
Collectively, these findings suggest that PARP1-mediated PARylation is required for TNFSF10-induced autophagy partly through promoting cytosolic HMGB1-BECN1 complex formation.
PARP1-mediated autophagy plays a cytoprotective role in TNFSF10-mediated apoptosis
TNFSF10-mediated apoptosis requires CASP8 activation.Citation35 We first tested CASP8 activity using ELISA-based colorimetric methods. Knockdown of PARP1, HMGB1, and BECN1 significantly induced TNFSF10-induced CASP8 activation (), suggesting a role of PARP1-mediated autophagy in the regulation of CASP8 activity. To investigate the role of PARP activity in TNFSF10-induced CASP8 activation, the PARP inhibitor PJ-34 was used. Treatment with PJ-34 significantly increased TNFSF10-induced CASP8 activation and apoptosis in PANC-1 cells, but not in normal mouse primary lung fibroblasts and pancreatic acinar cells (). Consistently, western blot analysis revealed that knockdown of PAPR1, HMGB1, and BECN1 or treatment with PJ-34 increased the proteolytic cleavage of the active CASP8 as well as decreased MAP1LC3A-II in PANC-1 cells (). In contrast, flow cytometric analysis revealed that the CASP8 inhibitor Z-IETD-FMK reversed TNFSF10-induced apoptosis in PANC-1 cells with knockdown of PARP1, HMGB1, and BECN1 (). Moreover, knockdown of HMGB1, PARP1, and BECN1 by shRNA increased TNFSF10-induced apoptosis in 2 other TNFSF10-resistant cell lines (HeLa and MCF7) (), suggesting a wider role of the PARP1-HMGB1-BECN1 pathway in the regulation of TNFSF10 resistance. In contrast, knockdown of Hmgb1 by lentivirus-mediated shRNA in normal mouse primary lung fibroblasts did not increase cytotoxicity of TNFSF10 and PJ-34 (). These findings suggest that suppression of HMGB1 expression appears to be a relatively safe way to increase anticancer activity of TNFSF10 in vitro.
Figure 4. PARP1-mediated autophagy plays a cytoprotective role in TNFSF10-mediated apoptosis. (A–C) The indicated PANC-1 cells, mouse primary lung fibroblasts, and pancreatic acinar cells were treated with TNFSF10 (100 ng/ml) with or without PARP1 inhibitor PJ-34 for 24 h. CASP8 activity (A and B) was assayed by CASP8 Assay Kit (n = 3 , * P < 0.05 versus control shRNA group). In parallel, apoptosis was assayed by flow cytometry (n = 3 , * P < 0.05) and the cleaved CASP8 (C) was assayed by western blot. (D) The indicated PANC-1 cells were treated with TNFSF10 (100 ng/ml) with or without CASP8 inhibitor Z-IETD-FMK (20 μM) for 8 and 24 h. The apoptosis was assayed by flow cytometry (n = 3 , * P < 0.05). (E) HeLa and MCF7 cells were transfected with the indicated shRNA for 48 h and then treated with TNFSF10 (100 ng/ml) for 24 h. The apoptosis was assayed by flow cytometry (n = 3 , * P < 0.05 vs. control shRNA group). (F) Mouse primary lung fibroblasts were transfected with lentivirus-mediated control shRNA and Hmgb1 shRNA for 48 h and then treated with TNFSF10 (100 ng/ml) and PJ-34 (40 μM) for 24 h. Cell viability was assayed with the CCK-8 kit. (G) PANC-1 cells with HMGB1-knockdown were transfected with HMGB1C23S, HMGB1C45S, and HMGB1C106S cDNA for 48 h and then treated with TNFSF10 (100 ng/ml) for 24 h. The interaction between HMGB1 (GFP) and BECN1 was assayed by immunoprecipitation (IP). (H) Indicated PANC-1 cells were transfected with HMGB1C23S and HMGB1C106S cDNA for 48 h and then treated with TNFSF10 (100 ng/ml) for 24 h. The levels of the indicated proteins were assayed by western blot.
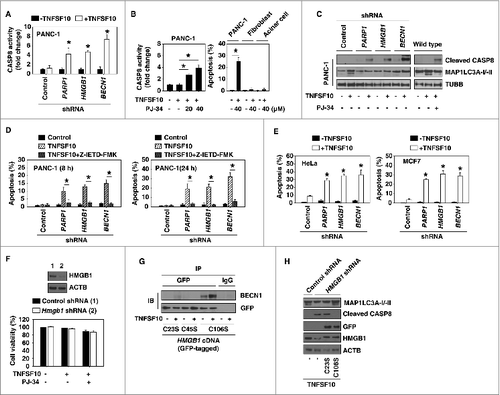
Our previous studies have demonstrated that Cys23 and Cys45 of HMGB1 are required for interaction between HMGB1 and BECN1 during autophagy.Citation23 Compared with the C106S mutation, the C23S and C45S mutations failed to restore TNFSF10-induced interaction between HMGB1 and BECN1 (), autophagy (MAP1LC3A-II) (), and resistance to apoptosis (cleaved CASP8) () in pancreatic cancer cells with HMGB1 knockdown. Collectively, these findings suggest that PARP1-HMGB1-BECN1 mediated autophagy plays a cytoprotective role in TNFSF10-mediated apoptosis by inhibition of CASP8 activity.
Suppression of the PARP1-HMGB1 pathway increases sensitivity to TNFSF10 in vivo
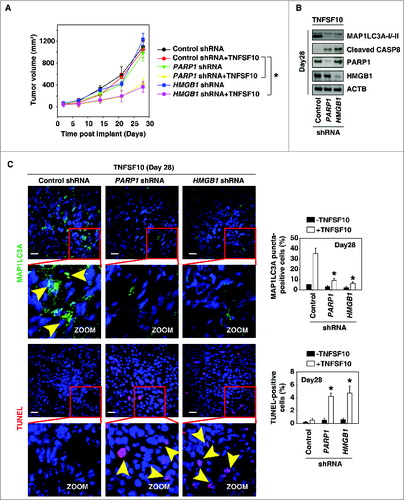
To test if suppression of PARP1 and HMGB1 expression also increased sensitivity to chemotherapy in vivo, PANC-1 cells with knockdown of PARP1 and HMGB1 were implanted into the subcutaneous space of the right flank of mice. Beginning at d 7, these mice were treated with TNFSF10. Compared with the control shRNA group, TNFSF10 shRNA treatment effectively reduced the tumor size of the cells with the PARP1 and HMGB1 knockdown (). Western blot analysis of the expression of MAP1LC3A and cleaved CASP8 in isolated tumors () indicated that knockdown of PARP1 and HMGB1 inhibits autophagy and increases apoptosis. Moreover, TUNEL staining of tumor sections showed significant increases in TNFSF10-induced apoptosis in the tumors with PARP1 and HMGB1 knockdown (). In contrast, the MAP1LC3A puncta were decreased in the tumors with PARP1 and HMGB1 knockdown (). Together, these findings demonstrated that PARP1 and HMGB1 are important in modulating the anticancer activity of TNFSF10 in vivo.
Figure 5. Suppression of the PARP1-HMGB1 pathway increases sensitivity to TNFSF10 in vivo. (A) Pancreatic tumor cells with PARP1 and HMGB1 knockdown are more sensitive to TNFSF10 in vivo. SCID mice were injected subcutaneously with indicated human pancreas cancer PANC-1 cells (2 × 106 cells/mouse) and treated with TNFSF10 (10 mg/kg/ i.v. twice daily) at d 7 for 2 wk. Tumor volume was calculated weekly. Data represent mean ± SE (n = 5 to 8 mice/group, * P < 0.05). (B) Western blot analysis of the indicated protein expression in isolated tumor at d 28 in TNFSF10 treatment group. (C) Autophagy was assayed by MAP1LC3A staining and apoptosis was assayed by TUNEL staining in isolated tumor atd 28. Yellow arrows show MAP1LC3A punctate and TUNEL positive cells, respectively. Quantifications of MAP1LC3A- and TUNEL-positive tumor cells were performed in the tumor sections (n=5 mice/group, * P < 0.05 versus control shRNA group; bar: 30 μm).
Discussion
In the present study, we demonstrated that PARP1-HMGB1 pathway-mediated autophagy inhibits TNFSF10-mediated cell death. In addition, we demonstrated that PARP1 is an essential regulator of TNFSF10-induced HMGB1 PARylation and subsequent cytoplasmic translocation. Cytoplasmic HMGB1 binding to BECN1 induced autophagy to diminish TNFSF10-mediated CASP8 activity. Our findings identify the translocation of HMGB1 as a critical step in the mechanism of TNFSF10 resistance and provide molecular links between autophagy and apoptosis.
Over 40 y ago, HMGB1 was discovered as a non-histone, chromatin-binding nuclear protein in calf thymus.Citation36 It is now increasingly evident that HMGB1 can be present in the cytoplasm and extracellular space. The complex functions of HMGB1 in tumor development and therapy have been reported to depend on its subcellular location.Citation37 We and others have demonstrated that upregulated HMGB1 expression or HMGB1 release contributes to drug resistance in several cancer cells such as leukemia, colon cancer cells, osteosarcoma, retinoblastoma cells, and liver cancers.Citation38-41 In this study, we demonstrated that the level of HMGB1 cytoplasmic translocation correlates well with TNFSF10 resistance in leukemia and pancreatic cancer cells. TNFSF10 induced even more HMGB1 cytoplasmic translocation in TNFSF10-resistant PANC-1 cells, but not in TNFSF10-sensitive Jurkat cells. In contrast, TNFSF10 did not significantly induce HMGB1 release in TNFSF10-resistant cancer cells (data not shown), although HMGB1 is recognized as a member of the damage-associated molecular pattern molecules in many cell death types.Citation42 Moreover, inhibition of HMGB1 expression by shRNA or inhibition of HMGB1 cytoplasmic translocation by ethyl pyruvate significantly enhanced TNFSF10-mediated cell death in TNFSF10-resistant cancer cells. These findings suggest that HMGB1-mediated TNFSF10 resistance occurs partly through its cytoplasmic translocation.
Next, we investigated the mechanism of HMGB1 translocation from the nucleus to the cytoplasm in response to TNFSF10. We demonstrated that PARP1-mediated HMGB1 PARylation is required for TNFSF10-induced HMGB1 cytoplasmic translocation. DNA damage was shown to induce HMGB1 ADP-ribosylation in tumor cells.Citation43 Several recent studies indicate that TNFSF10 treatment causes DNA damage not only in dying cancer cells, but also in surviving cells,Citation44,45 suggesting that DNA damage maybe a direct signal to drive HMGB1 translocation from the nucleus to the cytosol. PARP activation is an immediate cellular response to DNA damage and can increase DNA repair. PARP1-mediated PARylations play a dual role in the regulation both of cell survival and cell death, which each depend on the substrate for PARylations.Citation29 Our findings suggest that PARP1-mediated PARylation of HMGB1 is required for HMGB1 cytoplasmic translocation during TNFSF10-mediated cell survival. Gene deletion of PARP1 or pharmacological inhibition of PARP1 decreased HMGB1 cytoplasmic translocation and increased the anticancer effect of TNFSF10. In addition, PARP1-mediated PARylation of HMGB1 is also required for passive HMGB1 release during necrosis and injury. HMGB1 release during inflammation and necrosis promotes growth of tumors such as pancreatic cancer and malignant mesothelioma.Citation46 Thus, targeting PARP1-mediated HMGB1 translocation and release could be a novel strategy in cancer therapy.
The induction of autophagy is a common response to many forms chemotherapy, including TNFSF10. The role of autophagy in chemotherapy is complex and depends on tumor typeCitation47 and drug type,Citation47 as well as baseline autophagy level.Citation17 Autophagy has been considered a programmed cell survival process, although its deregulation has been implicated in nonapoptotic/necrotic cell death. Several studies have demonstrated that inhibition of autophagy enhanced TNFSF10-induced apoptotic cancer cell death by mechanisms such as degradation of antiapoptotic factors (e.g.,, BIRC2/CIAP1 and BIRC3/CIAP2, respectively), XIAP, and CFLAR)Citation48 and degradation of activated CASP8.Citation18,19 However, the elucidation of the molecular mechanism of TNFSF10-induced autophagy is still elusive.Citation49-51 Recent studies have indicated that PARP1 activation is required for DNA damage and oxidative injury-mediated autophagy in normal or cancer cells.Citation32,52,53 In the present study, we show that HMGB1 function is downstream of PARP1 activation to mediate TNFSF10-induced autophagy. The activity of PARP1 is not significantly impaired in TNFSF10-resistant cells compared to TNFSF10-sensitive cells. Increased PARP1 activity may be involved in the mechanism of impaired intracellular mitochondrial functionCitation25,54 and decreased extracellular pH.Citation31 The autophagy dynamic process is tightly regulated at different stages by ATG and other proteins.Citation3 We have demonstrated that autophagic stimuli promote translocation of HMGB1 to the cytosol and that cytosolic HMGB1 directly interacts with BECN1 to induce autophagy.Citation23 Our data show that suppression of PARP1 expression or activity inhibits TNFSF10-induced HMGB1-BECN1 complex formation, as well as autophagy.
Collectively, our results demonstrate that inhibition of the PARP1-HMGB1 pathway diminished autophagy and increased apoptosis following TNFSF10 treatment in vitro and in vivo. These findings provide mechanistic insight into TNFSF10 resistance and may contribute to the identification of novel targets to improve TNFSF10 activity.Citation8
Materials and Methods
Reagents
The antibodies to MAP1LC3A (NB100–2220) and HMGB1 (H00003146-M08) were bought from NOVUS. The antibodies to FBL/fibrillarin (Ab4566) and FADD (Ab24533) were bought from Abcam. The antibodies to ACTB/actin (3700), BECN1 (3738), TUBB/tubulin (2128), PARP1 (5625 or 9532), cleaved CASP8 (9496), and GFP (2555) were bought from Cell Signaling Technology. The antibodies to poly-ADP ribosylation/PAR (sc-8205 or sc-5598), SQSTM1 (Sc-28359), and CASP8 (Sc-5263) were bought from Santa Cruz Biotechnology. TNFSF10 (T9701), PJ-34 (P4365), Z-IETD-FMK (C1230), and necrostatin-1 (N9037) were bought from Sigma. The nuclear and cytoplasmic protein extraction kit (78833) was bought from Pierce.
Cell culture
Jurkat (TIB-152), PANC-1 (CRL-1469), HeLa (CCL-2), and MCF7 (HTB-22) cells were obtained from ATCC. Primary lung fibroblasts and pancreatic acinar cells were isolated from C57BL/6 mice as previously described.Citation55,56 Jurkat cells were grown in RPMI-1640 with 10% fetal bovine serum. PANC-1, HeLa, and MCF7 cells were grown in Dulbecco's Modified Eagle's Medium with 10% fetal bovine serum, 2 mM L-glutamine, and 100 U/ml of penicillin and streptomycin.
Cell viability analysis
Cell viability was evaluated with Cell Counting Kit-8 (CCK-8; Dojindo Molecular Technologies, CK04) according to the manufacturer's instructions.
Western blotting analysis
Cell lysates for western blotting were prepared as previously described.Citation24,38 All lysates from each sample were resolved on a denaturing 10% SDS-PAGE gel and transferred onto polyvinylidene difluoride membrane. After blocking with 3% to 5% nonfat dry milk in phosphate-buffered saline (PBS; Sigma, P5493) for 2 h and washing with 0.1% Tween-20 (Sigma, P1379) in PBS 3 times at room temperature, the membrane was incubated overnight at 4°C with primary antibodies, and then with peroxidase-conjugated secondary antibodies for 1 h at room temperature. Following incubation, signals were detected using enhanced chemiluminescence (Pierce, 32132) and then exposed to X-ray film. The relative quantification of bands on western blot was performed with Gel-pro Analyzer software (Media Cybernetics).
RNAi and gene transfection
The human PARP1 shRNA (SHCLND-NM_001618), human HMGB1 shRNA (SHCLND-NM_002128), mouse Hmgb1-shRNA (SHCLNG-NM_010439), human BECN1 shRNA (SHCLND-NM_003766), human FADD shRNA (SHCLND-NM_003824), human CASP8 shRNA (SHCLND-NM_033356), and control shRNA (SHC001) were obtained from Sigma. HMGB1-GFP (wild type) or mutant (C23S, C45S, or C106S) were a kind gift from Dr. George Hoppe (Cleveland Clinic, USA). The HMGB1-GFP mutant (K27/K28/K29→A27/A28/A29, "HMGB1[3KA]") was a kind gift from Dr. Alessandra Agresti (San Raffaele Scientific Institute, Italy). Transfections were performed by Lipofectamine™ 2000 (Invitrogen, 11668–019) or lentivirus as previously described.Citation24,38
Immunoprecipitation analysis
Cells were lysed in ice-cold RIPA lysis buffer (Pierce, 89900) for 5 min with periodic mixing. Cell lysates were centrifuged and the protein concentrations of supernatants were determined. The samples were precleared with protein A/G agarose for 3 h at 4°C, and then incubated with specific antibodies (3 to 5 μg/ml) or irrelevant IgG (control group) in the presence of protein A/G agarose beads overnight at 4°C with gentle shaking. Following incubation, samples were washed 4 times with PBS and subjected to western blot analysis.
MAP1LC3A image analysis
Formalin-fixed cells were firstly permeabilized with 0.1% Triton X-100 (Sigma, T8787) in PBS for 5 min, and then saturated with 2% bovine serum albumin in PBS for 1 h at room temperature. Subsequently, cells were processed for immunofluorescence with anti-MAP1LC3A antibody (4°C, overnight) followed by Alexa Fluor 488-conjugated Ig (37°C, 1 h). Nuclei were counterstained with Hoechst 33258 for 30 seconds. After the image was acquired by an Olympus confocal microscope (FV1000, Tokyo, Japan), the average number of MAP1LC3A puncta per cell (from at least 100 cells) was analyzed by Image-Pro Plus 5.1 software (Media Cybernetics) as previously described.Citation22, 23
Apoptosis assays
Apoptosis in cells was assessed using an ANXA5/annexin V-PI Apoptosis Detection Kit (BD PharMingen, 556547) by flow cytometric analysis (BD Accuri® C6, San Jose, CA, USA) according to the manufacturer's instructions. CASP8/caspase 8 activity was assessed using a CASP8 Assay Kit (Abcam, ab39700) according to the manufacturer's instructions. Apoptosis in tissue sample was assessed using a TUNEL (TdT-mediated dUTP-X nick end labeling) Assay Kit (Roche, 11684795910) according to the manufacturer's instructions.
PARP1 activity assay
PARP1 activity was measured with the PARP1 Enzyme Activity Assay Kit (EMD Millipore Corporation, 17–10149) according to the manufacturer's instructions. This assays employed biotinylated NAD+ as a substrate for PARP1 to incorporate biotinyl-ADP-ribose into the PAR chain on an immobilized histone substrate. The quantity of incorporated biotinyl-ADP-ribose was determined with streptavidin-horseradish peroxidase.
Mouse tumor model
An amount of 2 × 106 PANC-1 cells with PARP1 shRNA knockdown, HMGB1 shRNA knockdown, or control shRNA was subcutaneously injected into the dorsal flanks right of the midline in SCID mice. At d 7, mice were injected with TNFSF10 (10 mg/kg/i.v., twice daily) for 2 wk. Tumors were measured once a week. The volumes were calculated using the following formula: volume (mm3) = length × width2 × π/6. Experiments were performed according to the Xiangya Hospital Institutional Ethics Committee principles and guidelines for animal experimentation.
Statistical analysis
Unless otherwise indicated, data are expressed as means ± SD of 3 independent experiments. When the ANOVA was significant, post hoc testing of differences between groups was performed using an LSD test. Statistical significance was considered when P < 0.05.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Christine Heiner (Department of Surgery, University of Pittsburgh) for her critical reading of the manuscript.
Funding
This work was supported by grants from The National Natural Sciences Foundation of China (81200378 to L.L; 31171328 to L.C; 81270616 to Y.Y; 81100359 to M.Y), a grant from the National Institutes of Health (R01CA160417 to D.T.), a 2013 Pancreatic Cancer Action Network-AACR Career Development Award (Grant Number 13–20–25-TANG) and The National Natural Science Foundation-Guangdong Joint Fund (U1132005 to X.S.), and Science and Information Technology of Guangzhou Key Project (2011Y1–00038 to X.S.). This project used University of Pittsburgh Cancer Institute shared resources that are supported in part by award P30CA047904.
References
- Hengartner MO. The biochemistry of apoptosis. Nature 2000; 407:770-6; PMID:11048727; http://dx.doi.org/10.1038/35037710
- Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science 2000; 290:1717-21; PMID:11099404; http://dx.doi.org/10.1126/science.290.5497.1717
- Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol 2010; 12:814-22; PMID:20811353; http://dx.doi.org/10.1038/ncb0910-814
- Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 2007; 8:741-52; PMID:17717517; http://dx.doi.org/10.1038/nrm2239
- Amaravadi RK, Lippincott-Schwartz J, Yin XM, Weiss WA, Takebe N, Timmer W, DiPaola RS, Lotze MT, White E. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res 2011; 17:654-66; PMID:21325294; http://dx.doi.org/10.1158/1078-0432.CCR-10-2634
- Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 2011; 18:571-80; PMID:21311563; http://dx.doi.org/10.1038/cdd.2010.191
- Wiley SR, Schooley K, Smolak PJ, Din WS, Huang CP, Nicholl JK, Sutherland GR, Smith TD, Rauch C, Smith CA, et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 1995; 3:673-82; PMID:8777713; http://dx.doi.org/10.1016/1074-7613(95)90057-8
- Johnstone RW, Frew AJ, Smyth MJ. The TRAIL apoptotic pathway in cancer onset, progression and therapy. Nat Rev Cancer 2008; 8:782-98; PMID:18813321; http://dx.doi.org/10.1038/nrc2465
- Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science 1998; 281:1305-8; PMID:9721089; http://dx.doi.org/10.1126/science.281.5381.1305
- Jin Z, McDonald ER, 3rd, Dicker DT, El-Deiry WS. Deficient tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) death receptor transport to the cell surface in human colon cancer cells selected for resistance to TRAIL-induced apoptosis. J Biol Chem 2004; 279:35829-39; PMID:15155747; http://dx.doi.org/10.1074/jbc.M405538200
- Eggert A, Grotzer MA, Zuzak TJ, Wiewrodt BR, Ho R, Ikegaki N, Brodeur GM. Resistance to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in neuroblastoma cells correlates with a loss of caspase-8 expression. Cancer Res 2001; 61:1314-9; PMID:11245427
- Hopkins-Donaldson S, Bodmer JL, Bourloud KB, Brognara CB, Tschopp J, Gross N. Loss of caspase-8 expression in highly malignant human neuroblastoma cells correlates with resistance to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis. Cancer Res 2000; 60:4315-9; PMID:10969767
- Wagner KW, Punnoose EA, Januario T, Lawrence DA, Pitti RM, Lancaster K, Lee D, von Goetz M, Yee SF, Totpal K, et al. Death-receptor O-glycosylation controls tumor-cell sensitivity to the proapoptotic ligand Apo2L/TRAIL. Nat Med 2007; 13:1070-7; PMID:17767167; http://dx.doi.org/10.1038/nm1627
- Pan G, Ni J, Yu G, Wei YF, Dixit VM. TRUNDD, a new member of the TRAIL receptor family that antagonizes TRAIL signalling. FEBS Lett 1998; 424:41-5; PMID:9537512; http://dx.doi.org/10.1016/S0014-5793(98)00135-5
- Krueger A, Baumann S, Krammer PH, Kirchhoff S. FLICE-inhibitory proteins: regulators of death receptor-mediated apoptosis. Mol Cell Biol 2001; 21:8247-54; PMID:11713262; http://dx.doi.org/10.1128/MCB.21.24.8247-8254.2001
- Ricci MS, Kim SH, Ogi K, Plastaras JP, Ling J, Wang W, Jin Z, Liu YY, Dicker DT, Chiao PJ, et al. Reduction of TRAIL-induced Mcl-1 and cIAP2 by c-Myc or sorafenib sensitizes resistant human cancer cells to TRAIL-induced death. Cancer Cell 2007; 12:66-80; PMID:17613437; http://dx.doi.org/10.1016/j.ccr.2007.05.006
- Gump JM, Staskiewicz L, Morgan MJ, Bamberg A, Riches DW, Thorburn A. Autophagy variation within a cell population determines cell fate through selective degradation of Fap-1. Nat Cell Biol 2014; 16:47-54; PMID:24316673; http://dx.doi.org/10.1038/ncb2886
- Hou W, Han J, Lu C, Goldstein LA, Rabinowich H. Autophagic degradation of active caspase-8: A crosstalk mechanism between autophagy and apoptosis. Autophagy 2010; 6:891-900; PMID:20724831; http://dx.doi.org/10.4161/auto.6.7.13038
- Han J, Hou W, Goldstein LA, Lu C, Stolz DB, Yin XM, Rabinowich H. Involvement of protective autophagy in TRAIL resistance of apoptosis-defective tumor cells. J Biol Chem 2008; 283:19665-77; PMID:18375389; http://dx.doi.org/10.1074/jbc.M710169200
- Kang R, Chen R, Zhang Q, Hou W, Wu S, Cao L, Huang J, Yu Y, Fan XG, Yan Z, et al. HMGB1 in health and disease. Mol Aspects Med 2014; PMID:25010388
- Tang D, Kang R, Zeh HJ, 3rd, Lotze MT. High-mobility group box 1 and cancer. Biochim Biophys Acta 2010; 1799:131-40; PMID:20123075; http://dx.doi.org/10.1016/j.bbagrm.2009.11.014
- Tang D, Kang R, Livesey KM, Kroemer G, Billiar TR, Van Houten B, Zeh HJ, 3rd, Lotze MT. High-mobility group box 1 is essential for mitochondrial quality control. Cell Metab 2011; 13:701-11; PMID:21641551; http://dx.doi.org/10.1016/j.cmet.2011.04.008
- Tang D, Kang R, Livesey KM, Cheh CW, Farkas A, Loughran P, Hoppe G, Bianchi ME, Tracey KJ, Zeh HJ, 3rd, Lotze MT. Endogenous HMGB1 regulates autophagy. J Cell Biol 2010; 190:881-92; PMID:20819940; http://dx.doi.org/10.1083/jcb.200911078
- Tang D, Kang R, Cheh CW, Livesey KM, Liang X, Schapiro NE, Benschop R, Sparvero LJ, Amoscato AA, Tracey KJ, et al. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene 2010; 29:5299-310; PMID:20622903; http://dx.doi.org/10.1038/onc.2010.261
- Yuan K, Sun Y, Zhou T, McDonald J, Chen Y. PARP-1 regulates resistance of pancreatic cancer to TRAIL therapy. Clin Cancer Res 2013; 19:4750-9; PMID:23833311; http://dx.doi.org/10.1158/1078-0432.CCR-13-0516
- Ulloa L, Ochani M, Yang H, Tanovic M, Halperin D, Yang R, Czura CJ, Fink MP, Tracey KJ. Ethyl pyruvate prevents lethality in mice with established lethal sepsis and systemic inflammation. Proc Natl Acad Sci U S A 2002; 99:12351-6; PMID:12209006; http://dx.doi.org/10.1073/pnas.192222999
- Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, Rubartelli A, Agresti A, Bianchi ME. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. Embo J 2003; 22:5551-60; PMID:14532127; http://dx.doi.org/10.1093/emboj/cdg516
- Youn JH, Shin JS. Nucleocytoplasmic Shuttling of HMGB1 Is Regulated by Phosphorylation That Redirects It toward Secretion. J Immunol 2006; 177:7889-97; PMID:17114460; http://dx.doi.org/10.4049/jimmunol.177.11.7889
- Luo X, Kraus WL. On PAR with PARP: cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev 2012; 26:417-32; PMID:22391446; http://dx.doi.org/10.1101/gad.183509.111
- Abhari BA, Cristofanon S, Kappler R, von Schweinitz D, Humphreys R, Fulda S. RIP1 is required for IAP inhibitor-mediated sensitization for TRAIL-induced apoptosis via a RIP1/FADD/caspase-8 cell death complex. Oncogene 2013; 32:3263-73; PMID:22890322; http://dx.doi.org/10.1038/onc.2012.337
- Jouan-Lanhouet S, Arshad MI, Piquet-Pellorce C, Martin-Chouly C, Le Moigne-Muller G, Van Herreweghe F, Takahashi N, Sergent O, Lagadic-Gossmann D, Vandenabeele P, et al. TRAIL induces necroptosis involving RIPK1/RIPK3-dependent PARP-1 activation. Cell Death Differ 2012; 19:2003-14; PMID:22814620; http://dx.doi.org/10.1038/cdd.2012.90
- Zhang N, Chen Y, Jiang R, Li E, Chen X, Xi Z, Guo Y, Liu X, Zhou Y, Che Y, et al. PARP and RIP 1 are required for autophagy induced by 11'-deoxyverticillin A, which precedes caspase-dependent apoptosis. Autophagy 2011; 7:598-612; PMID:21460625; http://dx.doi.org/10.4161/auto.7.6.15103
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo J 2000; 19:5720-8; PMID:11060023; http://dx.doi.org/10.1093/emboj/19.21.5720
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/10.4161/auto.19496
- Seol DW, Li J, Seol MH, Park SY, Talanian RV, Billiar TR. Signaling events triggered by tumor necrosis factor-related apoptosis-inducing ligand (TRAIL): caspase-8 is required for TRAIL-induced apoptosis. Cancer Res 2001; 61:1138-43; PMID:11221844
- Goodwin GH, Sanders C, Johns EW. A new group of chromatin-associated proteins with a high content of acidic and basic amino acids. Eur J Biochem 1973; 38:14-9; PMID:4774120; http://dx.doi.org/10.1111/j.1432-1033.1973.tb03026.x
- Kang R, Zhang Q, Zeh HJ, 3rd, Lotze MT, Tang D. HMGB1 in cancer: good, bad, or both? Clin Cancer Res 2013; 19:4046-57; PMID:23723299; http://dx.doi.org/10.1158/1078-0432.CCR-13-0495
- Liu L, Yang M, Kang R, Wang Z, Zhao Y, Yu Y, Xie M, Yin X, Livesey KM, Lotze MT, et al. HMGB1-induced autophagy promotes chemotherapy resistance in leukemia cells. Leukemia 2011; 25:23-31; PMID:20927132; http://dx.doi.org/10.1038/leu.2010.225
- Livesey KM, Kang R, Vernon P, Buchser W, Loughran P, Watkins SC, Zhang L, Manfredi JJ, Zeh HJ, 3rd, Li L, et al. p53/HMGB1 complexes regulate autophagy and apoptosis. Cancer Res 2012; 72:1996-2005; PMID:22345153; http://dx.doi.org/10.1158/0008-5472.CAN-11-2291
- Huang J, Ni J, Liu K, Yu Y, Xie M, Kang R, Vernon P, Cao L, Tang D. HMGB1 promotes drug resistance in osteosarcoma. Cancer Res 2012; 72:230-8; PMID:22102692; http://dx.doi.org/10.1158/0008-5472.CAN-11-2001
- Liu K, Huang J, Xie M, Yu Y, Zhu S, Kang R, Cao L, Tang D, Duan X. MIR34A regulates autophagy and apoptosis by targeting HMGB1 in the retinoblastoma cell. Autophagy 2014 Mar; 10(3):442-52.
- Hou W, Zhang Q, Yan Z, Chen R, Zeh Iii HJ, Kang R, Lotze MT, Tang D. Strange attractors: DAMPs and autophagy link tumor cell death and immunity. Cell Death Dis 2013; 4:e966.
- Tanuma S, Yagi T, Johnson GS. Endogenous ADP ribosylation of high mobility group proteins 1 and 2 and histone H1 following DNA damage in intact cells. Arch Biochem Biophys 1985; 237:38-42; PMID:3970545; http://dx.doi.org/10.1016/0003-9861(85)90251-6
- Lovric MM, Hawkins CJ. TRAIL treatment provokes mutations in surviving cells. Oncogene 2010; 29:5048-60; PMID:20639907; http://dx.doi.org/10.1038/onc.2010.242
- Solier S, Sordet O, Kohn KW, Pommier Y. Death receptor-induced activation of the Chk2- and histone H2AX-associated DNA damage response pathways. Mol Cell Biol 2009; 29:68-82; PMID:18955500; http://dx.doi.org/10.1128/MCB.00581-08
- Kang R, Tang D, Schapiro NE, Loux T, Livesey KM, Billiar TR, Wang H, Van Houten B, Lotze MT, Zeh HJ. The HMGB1/RAGE inflammatory pathway promotes pancreatic tumor growth by regulating mitochondrial bioenergetics. Oncogene 2014; 33:567-77; PMID:23318458; http://dx.doi.org/10.1038/onc.2012.631
- White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer 2012; 12:401-10; PMID:22534666; http://dx.doi.org/10.1038/nrc3262
- He W, Wang Q, Xu J, Xu X, Padilla MT, Ren G, Gou X, Lin Y. Attenuation of TNFSF10/TRAIL-induced apoptosis by an autophagic survival pathway involving TRAF2- and RIPK1/RIP1-mediated MAPK8/JNK activation. Autophagy 2012; 8:1811-21; PMID:23051914; http://dx.doi.org/10.4161/auto.22145
- Herrero-Martin G, Hoyer-Hansen M, Garcia-Garcia C, Fumarola C, Farkas T, Lopez-Rivas A, Jaattela M. TAK1 activates AMPK-dependent cytoprotective autophagy in TRAIL-treated epithelial cells. Embo J 2009; 28:677-85; PMID:19197243; http://dx.doi.org/10.1038/emboj.2009.8
- Mills KR, Reginato M, Debnath J, Queenan B, Brugge JS. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is required for induction of autophagy during lumen formation in vitro. Proc Natl Acad Sci U S A 2004; 101:3438-43; PMID:14993595; http://dx.doi.org/10.1073/pnas.0400443101
- Monma H, Harashima N, Inao T, Okano S, Tajima Y, Harada M. The HSP70 and Autophagy Inhibitor Pifithrin-mu Enhances the Antitumor Effects of TRAIL on Human Pancreatic Cancer. Mol Cancer Ther 2013; 12:341-51; PMID:23371857; http://dx.doi.org/10.1158/1535-7163.MCT-12-0954
- Munoz-Gamez JA, Rodriguez-Vargas JM, Quiles-Perez R, Aguilar-Quesada R, Martin-Oliva D, de Murcia G, Menissier de Murcia J, Almendros A, Ruiz de Almodovar M, Oliver FJ. PARP-1 is involved in autophagy induced by DNA damage. Autophagy 2009; 5:61-74; PMID:19001878; http://dx.doi.org/10.4161/auto.5.1.7272
- Yoon JH, Ahn SG, Lee BH, Jung SH, Oh SH. Role of autophagy in chemoresistance: regulation of the ATM-mediated DNA-damage signaling pathway through activation of DNA-PKcs and PARP-1. Biochem Pharmacol 2012; 83:747-57; PMID:22226932; http://dx.doi.org/10.1016/j.bcp.2011.12.029
- Mathieu J, Flexor M, Lanotte M, Besancon F. A PARP-1/JNK1 cascade participates in the synergistic apoptotic effect of TNFalpha and all-trans retinoic acid in APL cells. Oncogene 2008; 27:3361-70; PMID:18084321; http://dx.doi.org/10.1038/sj.onc.1210997
- Kang R, Zhang Q, Hou W, Yan Z, Chen R, Bonaroti J, Bansal P, Billiar TR, Tsung A, Wang Q, et al. Intracellular Hmgb1 inhibits inflammatory nucleosome release and limits acute pancreatitis in mice. Gastroenterology 2014; 146:1097-107; PMID:24361123; http://dx.doi.org/10.1053/j.gastro.2013.12.015
- Seluanov A, Vaidya A, Gorbunova V. Establishing primary adult fibroblast cultures from rodents. J Vis Exp 2010 Oct 5; (44). pii: 2033. doi: 10.3791/2033.