
Abstract
Fetal growth is determined by the feto-placental genome interacting with the maternal in utero environment. Failure of this interplay leads to poor placental development and fetal growth restriction (FGR), which is associated with future metabolic disease. We investigated whether whole genome methylation differences existed in umbilical cord blood and placenta, between gestational-matched, FGR, and appropriately grown (AGA) neonates. Using the Infinium HumanMethylation450 BeadChip®, we found that DNA from umbilical cord blood of FGR born at term (n = 19) had 839 differentially methylated positions (DMPs) that reached genome-wide significance compared with AGA (n = 18). Using gestational age as a continuous variable, we identified 76,249 DMPs in cord blood (adj. P < 0.05) of which 121 DMPs were common to the 839 DMPs and were still evident when comparing 12 FGR with 12 AGA [39.9 ± 1.2 vs. 40.0 ± 1.0 weeks (mean ± SD), respectively]. A total of 53 DMPs had a β methylation difference >10% and 25 genes were co-methylated more than twice within 1000 base pairs. Gene Ontology (GO) analysis of DMPs supported their involvement in gene regulation and transcription pathways related to organ development and metabolic function. A similar profile of DMPs was found across different cell types in the cord blood. At term, no DMPs between FGR and AGA placentae reached genome-wide significance, validated with an external dataset. GO analysis of 284 pre-term, placental DMPs associated with autophagy, oxidative stress and hormonal responses. Growth restricted neonates have distinct DNA methylation profiles in pre-term placenta and in cord blood at birth, which may predispose to future adult disease.
Abbreviations
| AGA | = | appropriately grown offspring |
| BMI | = | body mass index |
| CG | = | cytosine phosphate guanine loci |
| DMP | = | differentially methylated positions |
| FDR | = | false discovery rate |
| FGR | = | fetal growth restriction |
| GO | = | gene ontology |
| HOMA | = | homeostasis model assessment |
| ICR1 | = | imprinting control region 1 |
| MODY | = | maturity onset diabetes of the young |
| T2DM | = | type 2 diabetes mellitus |
| UCL | = | University College London |
| UCLH | = | University College London Hospital |
| UCSC | = | University of California Santa Cruz |
Introduction
Fetal growth restriction, defined as an estimated fetal weight <10th customized centile for gestational age and adjusted for maternal weight, height, parity, and ethnicity, is a major cause of neonatal morbidity and mortality, associated with childhood neurocognitive delay and metabolic and cardiovascular disease in later life.Citation1-3 To fulfill its growth potential, a fetus must assimilate an adequate nutritional supply from its mother across the placenta. This process is governed by a complex interplay between the feto-placental genome and the maternal environment.Citation4 In low-income nations, environmental influences on fetal growth are predominantly related to poor maternal nutrition, chronic disease or infections.Citation5 In high-income nations, fetal growth is more commonly compromised by poor placental development.Citation6
Poor placental development affects approximately 4–7% of all first time pregnancies and is often associated with the maternal gestational syndrome preeclampsia, which is characterized by maternal hypertension and proteinuria.Citation7 Inadequate placental invasion may be due to adverse immune events at the maternal-placental interface and impaired oxygen sensing by the placenta.Citation8,9 In these circumstances, high resistance to blood flow in utero-placental arteries pre-dates the onset of FGR.Citation6 Inadequate placental development might then lead to epigenetic change in the under nourished fetus. An example of this sequence of events has been observed in a rodent model when uterine artery blood flow was artificially reduced during pregnancy.Citation10 As a consequence, the growth restricted offspring developed diabetes in adulthood, associated with reduced pancreatic β cell mass.Citation10 These events were associated with DNA methylation and reduced expression of PDX1, a transcription factor critical to pancreatic cell development.Citation10
A genome wide approach has been taken to investigate methylation differences between 5 FGR and 5 AGA offspring, using hematopoietic CD34+ stem cells isolated from umbilical cord blood.Citation11 In FGR offspring, there was a low level of methylation difference (6%) in a promoter of HNF4A, a gene associated with maturity onset diabetes of the young (MODY). Altered methylation status of selected candidate genes in umbilical cord tissue has also been correlated with later life phenotype.Citation12 However, these findings have not been reproduced in prospectively collected umbilical cord blood, or placental samples from babies born with growth restriction, or across a range of birth weights investigated in later life.Citation13-15 Furthermore, gestational age at delivery has a powerful effect on fetal and placental methylation status,Citation16,17 which has not always been considered in earlier studies of FGR offspring.
Genetic factors inherited from either parent also influence placental development and fetal growth.Citation18,19 Gene variants associated with insulin resistance are associated with reduced fetal growth and provide a potential genetic link between low birth weight and future risk of type-2 diabetes.Citation20 Other genes have conflicting influences on placental development and fetal growth according to their parent of origin. Genomic imprinting describes monoallelic expression of a gene following epigenetic silencing of the other allele.Citation21 For example, hypomethylation of CpG sites on imprinting control region 1 (ICR1) of 11p15 reduces expression of IGF2 and leads to reduced fetal growth, whereas hypermethylation of ICR1 increases expression of IGF2 and is associated with fetal overgrowth.Citation22 Fetal growth restriction secondary to known imprinted gene disorders are however rare and do not explain the majority of FGR seen in the developed world.
In order to discover novel DNA methylation differences associated with FGR in the absence of maternal and fetal disease and in gestational age-matched subjects, we conducted a case-control study that identified otherwise healthy pregnant women with either FGR (cases) or appropriately grown offspring (AGA; controls). We used the Infinium HumanMethylation450 BeadChip® to discover differences in the whole methylome (450,000 CpG sites in 99% of genes) of umbilical cord blood and placentae from cases and controls. After controlling for gestational age, we identified the most differentially methylated positions between FGR and AGA offspring and recognized that DMPs were consistent across different cord blood cell types. Gene ontology (GO) pathway analysis associated these DMPs with gene regulation and transcription pathways related to organ development and metabolic function. These novel DNA methylation profiles provide a robust platform for further investigation into their role in the fetal origins of adult diseases.
Results
Participant phenotype
Fetal growth restricted offspring identified in utero were confirmed at birth (FGR; 1.9% ± 2.3%, customized birth weight centile) and compared with appropriately grown for gestational age offspring (AGA; 51.8% ± 30.5%, customized centile). Retrospective review of fetal ultrasound scans at 20–22 weeks gestation showed that 90% (26/29) of mothers later identified with FGR offspring had high resistance to utero placental blood flow. No abnormal recording of utero-placental blood flow was made at 20–22 weeks in pregnancies that went on to have AGA offspring.
Mothers of both cases and controls met the study inclusion criteria and were of similar age, body mass index (BMI) and parity, and did not smoke (; see Materials and Methods).
Table 1a. Baseline characteristics of the parents according to offspring birth weight. (AGA, appropriately grown for gestational age, or FGR, fetal growth restriction).
Fathers of FGR offspring were more insulin resistant, as judged by fasting HOMA IR, and had larger waist circumferences compared with fathers of AGA offspring ().
Table 1b. Pregnancy characteristics according to offspring birth weight (FGR or AGA), nature of samples analyzed (cord blood or placenta), and gestational age at birth (all samples, or term > 255 d gestation). Results expressed as mean ± SD
Differentially methylated positions in cord blood and placenta from growth restricted offspring compared with appropriately grown offspring
Initial analysis included 45 umbilical cord blood samples (FGR n = 27 and AGA n = 18) and 45 placental samples (FGR n = 23 and AGA n = 22). Differentially methylated positions (DMPs) were identified using t test-based statistics in the Limma package provided by Bioconductor 2.11.Citation23 Adjustment for genome-wide significance was performed using a False Discovery Rate (FDR) with a threshold p value for DMPs of <0 .05.Citation24 Irrespective of gestational age, there were 553 differently methylated positions in cord blood and 453 DMPs in placenta that reached genome wide significance between all FGR and AGA offspring (adj. P < 0.05). As all AGA offspring were born at term, but only 19/27 FGR offspring, it was possible that these DMPs were influenced by gestational age.
Effect of gestational age on umbilical cord blood DMPs between FGR and AGA offspring
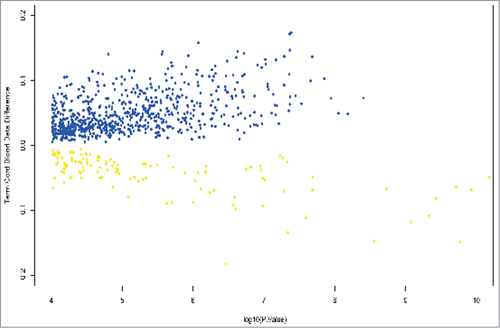
We compared genome-wide methylation differences between FGR cases (n = 19) with AGA controls (n = 18) that only delivered >255 d (36.4 weeks). This analysis yielded 839 DMPs in the cord blood between FGR and AGA offspring, of which the majority (86%) were hypermethylated (). From the original 553 cord blood DMPs, 371 DMPs (67%) were also common to the 839 term DMPs and may therefore be secondary to fetal growth restriction, rather than the effect of gestational age.
Figure 1. Volcano plot showing differentially methylated positions in umbilical cord blood between growth restricted (FGR) and appropriately grown offspring (AGA) at term gestation. β values are plotted against log P values. Hypermethylated positions are shown in blue and hypomethylated positions in yellow. A β methylation value of 10% is represented as 0.1.
To further elucidate the effect of gestational age on DMPs between FGR and AGA cord blood, we ran a linear model on all cord blood samples (FGR and AGA) using gestational age as a continuous variable. Across the gestational age range (202 d to 299 days) there were 76,249 DMPs in cord blood (adjusted P < 0.05). The majority of DMPs were characterized by hypomethylation (68%), an assessment that was heavily influenced by samples from pre-term FGR offspring.
We then compared the 839 DMPs evident between FGR and AGA offspring at term with the 76,249 DMPs found in the gestational age cord blood model and identified 121 DMPs that were common to both. We compared the direction of methylation of these 121 DMPs from both groups and found that in all cases their direction of methylation was identical. Furthermore, the same 121 DMPs were still evident when comparing 12 FGR and 12 AGA offspring that were more exactly matched for gestational age (279.1 ± 8.3 vs. 280.3 ± 7.2 days, respectively).
We also compared our results to another study of a separate cohort of AGA offspring that investigated DMPs within whole cord blood between 32–43 weeks gestation.Citation16 Using the HumanMethylation27 BeadChip, 25 DMPs were identified in whole umbilical cord blood, related to gestational age. None of these 25 gestational age-related DMPs were present in our 121 FGR cord blood DMPs at term. This observation does not diminish the possibility that the 121 DMPs relate to fetal growth rather than gestational age.
Effect of cell type on umbilical cord blood DMPs between FGR and AGA offspring
We investigated whether the 839 DMPs between FGR and AGA might be due to differences in cellular composition of umbilical cord blood. We downloaded 450K data from the publicly available database Marmal-aid,Citation25 which incorporates data from multiple studies that has been normalized and quality controlled. Data was available on circulating cell subtypes identified as CD4, CD14, CD8, CD19, CD56, peripheral blood leucocytes, whole blood or peripheral blood mononuclear cells from 109 different samples. We plotted the 839 different probes for the 109 different samples and grouped the samples into their cellular subtypes (). Overall, there was very little difference in methylation between the 839 DMPs according to cell subtype. This is portrayed as similar color profiles for each of the cell types (blue, yellow, and green) that correspond to the level of β methylation (far column) of the 839 probes ().
Figure 2. Heat map of the 839 DMPs between FGR and AGA in circulating cord blood cell subtypes identified as CD4, CD14, CD8, CD19, CD56, peripheral blood leucocytes, whole blood or peripheral blood mononuclear cells from 109 different samples. 450K data from a publicly available database Marmal-aid (http://marmal-aid.org.)Citation25 (see methods). There is very little difference in methylation between the 839 DMPs according to cell subtype. This is portrayed as similar color profiles for each of the cell types (blue, yellow and green) that correspond to the level of β methylation (far column) of the 839 probes.
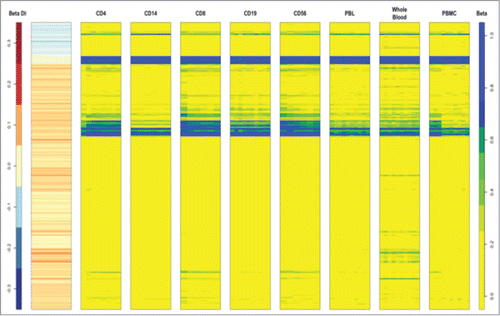
In order to identify whether there was a pattern of methylation within a single cell subtype, we plotted the 839 DMPs between FGR and AGA in cord blood (second column, ). There was no such correlation with a single cell subtype, which supports the conclusion that DMPs between FGR and AGA in cord blood are unlikely to be due to different cell proportions.
Leucocyte number is increased in neonates compared with adults, but the absolute number of CD4 and CD19 lymphocytes or dendritic cells is not different between pre-term or term neonates.Citation26 The absolute CD14 monocyte count is lower in very pre-term neonates compared with late pre-term and term neonates, but the relative frequency of CD14 monocyte cells is not different, at around 3.5% of all leucocytes.Citation26
Identity of most differentially methylated positions between FGR and AGA offspring in umbilical cord blood
We stratified umbilical cord blood DMPs between FGR and AGA offspring born at term, according to their degree of methylation difference. Of the 839 DMPs, 304 had a β methylation difference of >5% and 53 DMPs had a β methylation value difference of >10% (). The majority of DMPs with a β methylation >10% were hypermethylated (41/53, 77%; ) and 8/53 were hypomethylated >-10% (). Of the 839 DMPs that were identified on Ref-seq annotated genes, 658 DMPs were within 1000 bp of each other and 25 of these DMPs were co-methylated in the same direction. Out of the 53 most methylated DMPs, 8 were co-methylated in the same direction. These included C5orf39 that had 4 DMPs, FOXP1 and NRN1 that had 3 DMPs, while RIOK3 had 2 DMPs within 5 base pairs with β methylation values of +11 and +17% ().
Table 2a. The most methylated DMPs (β value >+10%) in umbilical cord blood from FGR compared with AGA offspring. Columns show CG identifying site, chromosome number, chromosome coordinate, gene identifier, chromosome number, methylation β value, and adjusted P value for difference in mean β value between FGR and AGA offspring and M value
Table 2b. Table shows the most hypomethylated DMPs (β value >−10%) in umbilical cord blood from FGR compared with AGA offspring. Columns show CG identifying site, chromosome number, chromosome coordinate, gene identifier, chromosome number, methylation β value, and adjusted P value for difference in mean β value between FGR and AGA offspring and M value
We also include the M value, calculated as the log2 ratio of the intensities of methylated versus unmethylated probes. Citation27 The top M values did not exactly correlate with the β values, but we have categorized the most differentially methylated CpG sites according to β value difference, which reflect biological activity, while M value difference more accurately assess statistical difference. ().
Potential role of SNPs influencing DMPs
Underlying genetic polymorphisms can influence differences in methylation of CpG sites.Citation28 We assessed the potential influence of SNPs on DNA methylation, by cross-referencing the 53 DMPs within term cord blood that had a β value difference of >10% with more than 150,000 SNPs in the Illumina database of SNPs known to influence methylation (http://support.illumina.com/downloads/infinium_hd_methylation_snp_list.ilmn). We found 13 hypermethylated and hypomethylated positions in close proximity to SNPs. These included a hypomethylated CpG site (β value -0.15) within 7 and 24 base pairs of 2 C5orf39 SNPs and one hypermethylated site (β value +0.14) within 47 base pairs of a TAF5 gene variant.
Furthermore, within the 839 DMPs found in umbilical cord blood between FGR and AGA at term, we did not observe the methylation pattern typically associated with SNPs (0%, 50%, or 100%). It is also of note that cord blood and placenta are of fetal origin and share the same genotype, but we found no overlap in DMPs between these tissues. Taken together, these observations suggest that the methylation differences between FGR and AGA offspring that we found are unlikely to be driven by SNPs.
Functional pathways of DMPs discovered in cord blood between FGR and AGA offspring using the Marmal-aid database
The Marmal-aid DNA methylation database, which contains >10,000 samples in one repository,Citation25 was used to gain insight into the functionality of the 839 differentially methylated CpG sites that we discovered in cord blood from FGR offspring born at term. The Marmal-aid output suggested that the 839 DMPs are found in normally very stable unmethylated or hypomethylated gene loci consistent across all blood cell types. Also, these same positions vary little in methylation status between tissue type and through aging and are located in highly conserved, invariate regions of the genome.
To establish whether the 839 term DMPs linked to Ref Seq-annotated genes had significant functional roles, we used GO pathway analysis. Using an R based package and thresholds described in the materials and methods section, 23 GO pathways were identified (). Many of these pathways contained DMPs with >10% β value differences between FGR and AGA offspring, supporting a potential functional role for these DMPs. The majority of pathways identified through GO analysis related to regulation of gene transcription and expression and control of metabolic processes. Pathways with the greatest methylation difference were involved in the regulation and transcription of RNA polymerase II.
Table 3. Twenty-three GO pathways identified using 839 DMPs within FGR term umbilical cord blood samples. Fold change refers to the number of DMP on genes actually present in the pathway compared with the number expected and that met the P-value < 0.0001 and cut off of 2-fold change (rounded up). The last 2 columns report the most differentially methylated genes (>10 %) found in the respective GO pathway.
Placental DMPs between FGR and AGA offspring
In the placenta from term deliveries, there were no DMPs between FGR and AGA offspring that reached genome-wide significance. To elucidate whether the 453 placental DMPs initially identified between FGR and AGA offspring were primarily driven by differences in gestational age, we ran a linear model on all placental samples using gestational age as a continuous variable across the gestational age range (202 to 299 days). We identified 20,893 placental DMPs associated with gestational age, heavily influenced by placentae from early gestational FGR pregnancies. The majority of placental DMPs were hypomethylated (93%), but with advancing gestation the placenta became more methylated (adjusted P < 0.05), consistent with the findings of others.Citation17
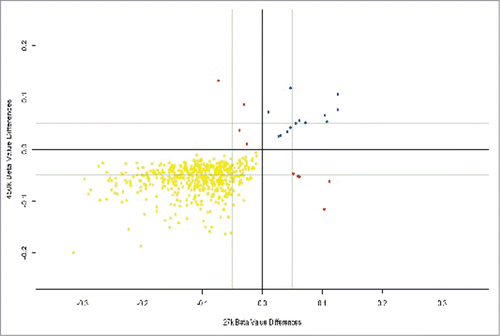
We validated placental results using methylation data from a study of placental tissue from healthy pregnancies, across all 3 trimesters and run on an Illumina HumanMethylation27 BeadChip, a precursor of the 450K array.Citation17 Using the same cutoff for statistical significance as our study (FDR-adjusted P < 0.05) we identified placental DMPs between the second trimester and term groups in the published data setCitation17 and searched for similar placental DMPs between the second trimester and term in our own cohort. Out of 23,677 positions available for direct comparison between the 27K and 450K platforms, 825 DMPs were called from these positions using the 450K analysis and of these, 541 (65%) were also identified using the 27K analysis.Citation17 Both data sets contained 519 similarly hypomethylated and 13 similarly hypermethylated positions. Only 9 probes showed methylation in opposite directions (). The remaining 284/825 DMPs unique to our FGR cases have the potential to have a role in fetal growth independent of gestational age. GO pathway analysis of these 284 placental DMPs showed 16 pathways that met the criteria for reporting. These included pathways involved in the regulation of autophagy, response to oxidative stress and hormonal stimuli ().
Table 4. GO pathway analysis of 284 placental DMPs associated with our FGR cohort. These DMPs were identified after validation analysis with an external data set of placentas from healthy pregnancies across 3 trimesters.Citation17 After excluding DMPs that were present on both platforms and assumed to be associated with gestational age, the remaining 284 DMPs remained unique to our FGR cohort. Sixteen functional pathways met the criteria for reporting. Pathways known to be associated with fetal growth restriction and that were evident in the placenta of our FGR cohort included autophagy, response to oxidative stress and the regulation of monocyte chemotaxis. The actual and expected number of DMPs on genes in the pathway is recorded and the fold change between these counts that met the P-value <0.0001 and cut off of >2-fold change
Figure 3. Placental DMPs between 2nd and 3rd trimesters in our data set using the Infinium HumanMethylation450 Beadchip® and also present in an external data set using the Infinium HumanMethylation27.Citation17 There were 519 hypomethylated DMPs common to both platforms (yellow), 13 hypermethylated DMPs common to both platforms (blue) and 9 DMPs that were mismatched with regards direction of methylation (red).
Discussion
We found that cord blood from newborn offspring affected by fetal growth restriction (FGR) had significant DNA methylation differences compared with appropriately grown offspring (AGA). Methylation differences persisted after accounting for differences in gestational age and were consistent across leucocyte populations in umbilical cord blood. DMPs were associated with genes involved in pathways crucial to key cellular processes, particularly regulation of gene expression and transcription. Several of the 53 DMPs with the greatest methylation differences were methylated at more than one CpG site, emphasizing the potential for methylation differences to influence gene expression.
Three of the most differentially methylated positions between FGR and AGA cord blood were co-methylated at several CpG sites on genes that control ubiquitous transcription factors. FOXP1, which was hypermethylated at 3 CpG sites, is expressed in many tissues and is important for organogenesis, metabolism, and immunity.Citation29 RIOK3 was hypermethylated on 2 CpG sites just 5 base pairs apart (β values +11% and +17%). RIOK3 is an atypical protein kinase that interacts with caspase-10 and negatively regulates NF-Kappa B.Citation30 Inhibition of NF-Kappa B reduces angiogenesis, but permits apoptosis with the potential to compromise fetal growth and development.Citation31 TAF5 encodes a key subunit that is critical to the formation of basal transcription factor, Transcription Initiation Factor TFIID subunit 5 (TFIID).Citation32 TFIID is used by RNA polymerase II for transcript initiation and is critical for reprogramming of pluripotent embryonic stem cells.Citation33 Enhanced expression of TFIID during fetal life may therefore generate plasticity in gene expression that could influence embryonic stem cells to pass into different cellular states. RNA polymerase II pathways also featured heavily in our GO analysis of term DMPs in cord blood from FGR offspring. Epigenetic interference with the expression of these transcription factors has the potential for far reaching effects during a time of developmental plasticity.
A minority of DMPs on genes from FGR cord blood were hypomethylated compared with AGA offspring. Gene variants of APOL1, which we found to be hypomethylated (β value, −18%) are associated with accelerated kidney disease and lower HDL concentrations, especially in African Americans.Citation34,35 Methylation differences on APOL1 were also found in cord blood mononuclear cells in twins and correlate with birth weight.Citation36 Interestingly, a near neighbor of APOL1 on chromosome 22 is PNPLA3, which we found to be hypermethylated (β value +0.12). Gene variants of PNPLA3 produce excess adiponutrin, which promotes lipid synthesis and increases the risk of non-alcoholic fatty liver disease.Citation37
We also found hypomethylation on C5orf39 (β value -15% and co-methylated in 4 positions) is a gene unique to humans that encodes Annexin II receptor, a novel activator of apoptosis.Citation38 Low birth weight offspring show increased lymphocyte apoptosis, which correlates with insulin resistance.Citation39
Genes that encode histone proteins were found to be differentially methylated in both directions, HIST1H4J (β value −22%) and HIST1H3I (β value +11%). There were 7 DMPS regulating histone modification, which may suggest a role for epigenetic events other than DNA methylation influencing gene expression in FGR offspring. Histone modification, which most often involves post-translational changes to the tail of a histone protein, influences transcription of the associated gene and has a role in human development.Citation40 Cellular pluripotency during fetal life is partly determined by histone modification and therefore factors that influence histone gene expression may play an important role in this critical element of fetal development.Citation41
Umbilical cord blood from over 1000 infants in the Norwegian Mother and Child Cohort assessed using the Infinium HumanMethylation450 BeadChip showed differential methylation at 19 CpGs associated with either increased or decreased birth weight across a range of normal birth weights.Citation42 Two genes, ARID5B and XRCC3, were particularly associated with either decreased or increased birth weight, respectively. These genes did not appear among the strongly differentially methylated genes in our cohort of pathologically growth restricted offspring. It is possible that the influence of DNA methylation is different within a spectrum of healthy birth weight offspring as compared with pathological growth restriction.
We studied well-phenotyped FGR offspring identified in utero according to customized fetal growth scales in otherwise healthy mothers and in the absence of other fetal disease. In all but 1 of the 29 cases for whom we have data, there was elevated utero-placental perfusion pressures with poor placental perfusion, before the identification of FGR. In an animal model that mimics human fetal growth restriction by ligating both uterine arteries, epigenetic dysregulation was seen at conserved intergenic sequences in 7-week old offspring.Citation43 These epigenetic events were near genes that regulate vascularization, pancreatic β cell proliferation, and apoptosis, which may underpin the vulnerability of FGR offspring to type-2 diabetes.Citation33 Similarities between this animal model and the placental driven FGR in our cases, supports the concept that the methylation differences seen in FGR offspring are secondary to reduced utero-placental perfusion.
Animal models have also shown that maternal and paternal diet can alter offspring phenotype through epigenetic mechanisms.Citation44,45 We studied FGR offspring from well-nourished mothers who had a similar phenotype to mothers of AGA offspring, but in whom there was poor placental development. Interestingly, we found sub-clinical insulin resistance in the fathers of FGR pregnancies, as we previously reported.Citation46 We did not assess the paternal epigenome, but animal studies support the possibility that paternal diet-induced epigenetic change can be inherited by offspring and lead to reduced fetal growth.Citation45 It is also possible that gene variants that predispose the father to insulin resistance are inherited by offspring. Under these circumstances, insulin cannot act as an effective growth factor in utero and predisposes the fetus to diabetes in later life.Citation47
To date, the study of rare imprinted gene disorders has shown an influential role for altered methylation of genes essential to fetal growth.Citation19,48 Our study has taken a broader, unbiased whole-genome approach to identify methylation changes on genes of FGR offspring secondary to poor placental development. In our cases, we found low-level methylation differences in 2 imprinted genes Plagl and Kcnq1, but despite good coverage in the HM450 data set, we found no differences in methylation of Igf2/H19. Our study suggests that imprinted genes have little influence on FGR secondary to poor placental development, but rather genes encoding key transcription factors are differentially methylated. Future studies might investigate the parent of origin of these novel regions of differential methylation.
Fetal growth restriction often results in iatrogenic premature childbirth, secondary to failure of the fetus to thrive in utero. This leads to the dual problems of low birth weight and prematurity. In our study we dissected apart the influence of gestational age from impaired fetal growth by matching FGR cases that delivered at term with the AGA offspring and by excluding DMPs associated with increasing gestational age. We also compared our placental methylation results with a published data set.Citation17 Although we showed methylation status of the placenta and cord blood altered with advancing gestational age, we identified DMPs that remained unique to FGR offspring. Further investigation of the effects of these DMPs on placental gene function is necessary to understand how they might influence fetal growth and predispose to disease in later life.
In placentas of pre-term FGR offspring, GO pathway analysis identified differential methylation on genes implicated in pathways important to oxidative stress, hormonal stimulation, and autophagy. These pathways are crucial to normal placental development, while abnormal autophagy and oxidative stress are recognized features of FGR placentas.Citation49-51
At term, we did not observe methylation differences between the placentas from FGR and AGA that were evident in earlier pregnancy. It is possible that given time, pathological differences in the placental methylome that influence fetal growth give way to gestational influences in anticipation of childbirth. Differences in placental methylation in pregnancies destined to result in fetal growth restriction appear to be more pronounced in early pregnancy.
The Infinium HumanMethylation27 BeadChip® platform, was previously used on 206 term placentas and identified 22 methylation loci that could act as markers for poor fetal growth.Citation52 Notably, placentas from growth restricted (FGR) and constitutionally small offspring (SGA) were combined in this analysis.Citation52 Although we did not replicate methylation loci using the Infinium HumanMethylation450 BeadChip® with this cohort, methylation differences on pathways associated with basic cellular functions including transcription and DNA repairCitation52 were similar to those we found in pre-term placentas. However, 6 of the 22 DMPs in the FGR and SGA cohort combinedCitation52 were present in the second and third trimester of the external placental dataset,Citation17 supporting a possible influence of these loci on gestational age.
Placentas from pregnancies affected by pre-term preeclampsia, a condition that shares very similar placental characteristics to FGR pregnancies,Citation53 have been found to have distinct methylation profiles using the 450 BeadChip.Citation54 However, no further differences in placental methylation were found according to whether the preeclamptic pregnancies were also affected by FGR or AGA offspring.Citation54 The differences we found in methylation profile in umbilical cord blood between FGR and AGA suggests that the fetal response to the same placental pathology is distinctive for those who cannot grow to their full potential.
Placentas from monochorionic twins discordant for fetal growth, have shown increased DNA methylation of the leptin promoter region and upregulation of leptin mRNA in the smaller twin.Citation55 Similarly, expression of LEPROT1 (Leptin receptor overlapping transcript-like 1) from cord blood mononuclear cells of dichorionic twins showed a negative correlation with birth weight.Citation56 These observations may explain the future vulnerability of low birth weight offspring to metabolic syndrome. However, we did not find any DMPs associated with leptin that reached genome wide significance between FGR and AGA placentae.
Although there was no difference in the mode of delivery of FGR and AGA offspring born at term, the mode of delivery has been shown to influence offspring methylation status.Citation57 Offspring born by caesarean section have higher DNA methylation in umbilical cord blood leucocytes compared with those born by vaginal delivery.Citation57 Interestingly, methylation differences associated with mode of delivery did not persist when re-examined in neonatal leucocytes 3–5 d postpartum.Citation57 It is possible that transient environmental exposure such as mode of delivery causes short-lived epigenetic changes, while persistent environmental influences during a time of high developmental plasticity in utero, cause more enduring effects.
A potential limitation of our study was that we assessed DNA methylation in umbilical cord whole blood at birth, while fetal circulating cell types change according to birth weight and gestation at delivery.Citation58 However, at term nucleated red blood cells comprise a small percentage of total cell number (3 per 100 wbcs), which limits their influence on the methylation of cells from cord blood.Citation59 Furthermore, according to the Marmal-aid data set, we showed a similar pattern of DMPs in areas normally unmethylated and across cord blood cell types. Differences in cell type that might exist at term between FGR and AGA are unlikely to play a significant role in observed DMPs.Citation26 Cell composition is however likely to explain some of the 76,249 DMPs that we identified in cord blood of pre-term FGR offspring.
Despite many human studies investigating genetic and environmental mechanisms that might link low birth weight with risk of adult disease, there is very little reproducible evidence to support a unifying pathophysiological process. This may partly reflect the multi-factorial elements that lead to FGR as well as subsequent environmental influences that promote vulnerability to polygenic disease, such as type-2 diabetes mellitus (T2DM). Although gene variants can have pleiotropic effects on different organs, organ specific diseases need to be investigated using the relevant tissue. In this regard, the study of the placenta is relevant to fetal growth restriction, but the study of less accessible tissue, such as the pancreas, is needed to investigate the metabolic syndrome or T2DM. Altered methylation status of selected candidate genes from umbilical cord tissue has however been correlated with later life phenotype.Citation12 Conversely, saliva from monozygotic monochorionic twins aged 34 years, showed very similar genome-wide DNA methylation profiles between twins that were discordant for birth weight.Citation15
The novel cord blood DMPs in well-phenotyped FGR offspring that we observed warrant further targeted study that might provide a pathophysiological link between low birth weight and adult disease. DMPs in FGR cord blood may be a consequence of placental dysfunction or inherited from insulin resistant fathers. Reassessment of FGR neonates in adulthood would determine if DMPs identified at birth have endured into later life to influence vulnerability to adult disease.
Patients and Methods
Selection of cases and controls
A case-control collection of samples from pregnancies affected by fetal growth restriction and appropriately grown offspring was undertaken at University College London Hospital (UCLH) between September 2009 and May 2011. The Joint UCLH/UCL (α) Ethical Committee (09/H0715/28) granted ethical approval for the study. All participants gave informed consent. Caucasian women were included as cases when an ultrasound scan identified fetal growth restriction (FGR), defined as an estimated fetal weight <10th customized centile for gestational age and adjusted for maternal weight, height, parity, and ethnicity (available from Perinatal Institute, UK - www.gestation.net). Pregnancies were excluded if fetal growth restriction was associated with fetal structural or chromosomal abnormalities, infections, chronic maternal disease, or multiple pregnancies. Pregnant women were included in the control group if they were found to have an appropriately grown for gestational age (AGA) fetus, with an estimated fetal weight between 10th and 95th customized centile, born at more than 255 d and only if predicted normal fetal growth matched neonatal measures after delivery. After a diagnosis of FGR, we examined case notes for information on uterine artery blood flow at 20–22 week gestation.
At childbirth, umbilical cord blood was taken from the umbilical vein, and stored at −80oC. Placental samples were taken from the fetal surface within 2 cm of the cord insertion and stored at −80°C. Gestational age, offspring gender, weight, length, and delivery details were recorded.
Within 4 weeks of childbirth, paternal phenotype was also assessed. Specifically, we measured paternal blood pressure, waist circumference, fasting glucose and insulin levels.
DNA extraction
DNA was extracted from paired placenta and cord blood samples using standard kits and protocols (DNeasy Blood & Tissue Kit, Qiagen, UK). After cleanup (DNA Clean & Concentrator Kit, Zymo Research, Cambridge Biosciences, UK), samples with a 260/280 ratio >1.75 and a 260/230 ratio >1.8 were discarded from further analysis.
Bisulphite conversion
High-quality genomic DNA (500ng) was bisulphite converted using the EZ-96 DNA Methylation Deep-well Kit (Zymo Research, Cambridge Biosciences, UK). Bisulphite conversion efficiency was assessed using qPCR with primers to converted and non-converted DNA (Primers and cycling parameters in appendix) and samples that showed <97% bisulphite conversion were excluded. A total of 56 cord blood and 54 placental samples were suitable for methylation analysis, including 49 participants with both cord blood and placental samples and 12 further tissue samples (7 cord blood and 5 placenta).
Illumina human methylation450 beadChip
Bisulphite-converted samples were hybridized to the Illumina HumanMethylation450 BeadChip (Illumina, Inc., CA, USA)Citation60 using standard protocols. Cases and controls and tissue type were arranged randomly on each chip and processed in a blinded fashion. Arrays were scanned using the Illumina iScan SQ 2-color laser scanner.
Quality Control
We evaluated the quality of the data based on the signals of control probes built in to the assay. This resulted in 90 samples that were suitable for downstream analysis. Multiple Dimensional Scaling analysis (MDS) demonstrated no significant positional or chip batch effect between samples.
Quantile normalization was performed separately for individual color channels, probe types, and tissue types (cord blood and placenta) due to their variable distribution of methylation values (β values). Probe β values (methylated probe intensity/[methylated probe intensity + unmethylated probe intensity + 100]) were converted to M-values by Logit transformation to reduce heteroscedasticity.Citation27
Of 486,428 probes present on the array, 11,648 X and Y chromosome probes were removed; 1,927 were removed for failing to meet the detection P-value cut-off (P = 0.05) and a further 43,097 were removed due to probe cross-hybridization. This resulted in a total of 429,756 probes, representing individual positions of methylation available for downstream analysis.
Statistical Analysis
The influence of gestational age on DMPs between FGR and AGA was assessed using a continuous linear regression model in the Limma package, incorporating all samples and gestational age. Results were validated against an external data set of placental tissue from AGA pregnancies that were investigated using the Illumina 27K methylation array.Citation17
To further characterize the DMPs, we utilized Marmal-aid (http://marmal-aid.org),Citation25 a recently developed meta-analysis tool. In Marmal-aid, all publicly available Illumina 450K data (>10,000 different arrays as of June, 2013) is incorporated into a single repository and re-processed to enable customizable meta-analyses.
GO pathway analysis was also performed in order to deduce gene function of identified DMPs. We only reported those pathways that had a 2-fold (rounded up) enrichment of genes more than expected and that reached a significant P-value of <0.0001.
Author Contributions
SH, SF, VR, GH, and DW designed the study. SH recruited patients and collected samples. SH and MS extracted DNA, bisulphite converted and performed QC on DNA. SH, CM, RL, and VR carried out the bioinformatics analysis. All authors contributed to interpretation of the data. SH and DW wrote the first and last drafts of the manuscript and all authors made critical revisions. GH and DW take responsibility for the integrity of the data and accuracy of the data analysis.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the Wellbeing of Women and a British Medical Association Joan Dawkins award. The National Institute for Health Research, University College London Hospitals, Biomedical Research Center provided part of DW's salary. VR and RL are supported by BLUEPRINT (EU FP7 - Grant number 282510) and VR is also supported by the BBSRC (BB/H012494/1).
References
- Roberts G, Anderson PJ, Doyle LW and the Victorian Infant Collaborative Study Group. Neurosensory disabilities at school age in geographic cohorts of extremely low birth weight children born between the 1970s and the 1990s. J Pediatr, 2009; 154:829-34; PMID:19230899; http://dx.doi.org/10.1016/j.jpeds.2008.12.036
- Gluckman PD, Hanson MA, Cooper C, Thornburg KL Effect of in utero and early life conditions on adult health and disease. N Engl J Med, 2008; 359:61-73; PMID:18596274; http://dx.doi.org/10.1056/NEJMra0708473
- Wang G, Divall S, Radovick S, Paige D, Ning Y, Chen Z, Yuelong J, Hong X, Walker SO, Caruso D etal Preterm birth and random plasma insulin levels at birth and in early childhood. JAMA 2014; 311:587-96; PMID:24519298; http://dx.doi.org/10.1001/jama.2014.1
- Gaccioli F, Lager S, Powell TL, Jansson T Placental transport in response to altered maternal nutrition. J Dev Orig Health Dis 2013; 4:101-15; PMID:25054676; http://dx.doi.org/10.1017/S2040174412000529
- Black RE, Victora CG, Walker SP, Bhutta ZA, Christian P, de Onis M, Ezzati M, Grantham-McGregor S, Katz J, Martorell R, et al. Maternal and Child Nutrition Study Group. Maternal and child undernutrition and overweight in low-income and middle-income countries. Lancet 2013; 382:427-51; PMID:23746772; http://dx.doi.org/10.1016/S0140-6736(13)60937-X
- Sankaran S, Kyle PM Aetiology and pathogenesis of IUGR. Best Pract Res Clin Obstet Gynaecol 2009; 23:765-77; PMID:19666240; http://dx.doi.org/10.1016/j.bpobgyn.2009.05.003
- Williams D, Craft N. Pre-eclampsia. BMJ 2012; 345:e4437; PMID:22815427; http://dx.doi.org/10.1136/bmj.e4437
- Erlebacher A. Immunology of the maternal-fetal interface. Annu Rev Immunol 2013; 31:387-411; PMID:23298207; http://dx.doi.org/10.1146/annurev-immunol-032712-100003
- Jauniaux E, Poston L, Burton GJ. Placental-related diseases of pregnancy: Involvement of oxidative stress and implications in human evolution. Hum Reprod Update 2006; 12:747-75; PMID:16682385; http://dx.doi.org/10.1093/humupd/dml016
- Park JH, Stoffers DA, Nicholls RD, Simmons RA. Development of type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing of Pdx1. J Clin Invest 2008; 118:2316-24; PMID:18464933; http://dx.doi.org/10.1172/JCI32011
- Einstein F, Thompson RF, Bhagat TD, Fazzari MJ, Verma A, Barzilai N, Greally JM Cytosine methylation dysregulation in neonates following intrauterine growth restriction. Plos One 2010; 5:e8887; PMID:20126273; http://dx.doi.org/10.1371/journal.pone.0008887
- Godfrey KM, Sheppard A, Gluckman PD, Lillycorp KA, Burdge GC, McLean C, Rodford J, Slater-Jefferies JL, Garratt E, Crozier SR, et al. Epigenetic gene promoter methylation at birth is associated with child's later adiposity. Diabetes 2011; 60:1528-34; PMID:21471513; http://dx.doi.org/10.2337/db10-0979
- Tobi EW, Heijmans BT, Kremer D, Putter H, Delemarre-van de Waal HA, Finken MJJ, Wit JM, Slagboom PE DNA methylation of IGF2, GNASAS, INSIGF and LEP and being born small for gestational age. Epigenetics 2011; 6:171-76; PMID:20930547; http://dx.doi.org/10.4161/epi.6.2.13516
- Adkins RM, Tylavsky FA, Krushkal J. Newborn umbilical cord blood DNA methylation and gene expression levels exhibit limited association with birth weight. Chem Biodivers 2012; 9:888 - 99; PMID:22589090; http://dx.doi.org/10.1002/cbdv.201100395
- Souren NYP, Lutsik P, Gasparoni G, Tierling S, Gries J, Riemenschneider M, Fryns J-P, Derom C, Zeegers MP, Walter J. Adult monozygotic twins discordant for intra-uterine growth have indistinguishable genome-wide DNA methylation profiles. Genome Biol 2013; 14:R44; PMID:23706164; http://dx.doi.org/10.1186/gb-2013-14-5-r44
- Schroeder JW, Conneely KN, Cubells JC, Kilaru V, Newport J, Knight BT, Stowe ZN, Brennan PA, Krushkal J, Tylavsky S, et al. Neonatal DNA methylation patterns associate with gestational age. Epigenetics 2011; 6:1498-1504; PMID:22139580; http://dx.doi.org/10.4161/epi.6.12.18296
- Novakovic B, Yuen RK, Gordon L, Penaherrera MS, Sharkey A, Moffett A, Craig JM, Robinson WP, Saffery R. Evidence for widespread changes in promoter methylation profile in human placenta in response to increasing gestational age and environmental/stochastic factors. BMC Genomics 2011; 12:529; PMID:22032438; http://dx.doi.org/10.1186/1471-2164-12-529
- Freathy RM, Mook-Kanamori DO, Sovio U, Prokopenko I, Timpson NJ, Berry DJ, Warrington NM, Widen E, Hottenga JJ, Kaakinen M, et al. Variants in ADCY5 and near CCNL1 are associated with fetal growth and birth weight. Nature Genetics 2010; 42:430-35; PMID:20372150; http://dx.doi.org/10.1038/ng.567
- Piedrahita JA. The role of imprinted genes in fetal growth abnormalities. Birth Defects Research (Part A) 2011; 91:682-92; http://dx.doi.org/10.1002/bdra.20795
- Hattersley AT, Tooke JE. The fetal insulin hypothesis: an alternative explanation of the association of low birth weight with diabetes and vascular disease. Lancet 1999; 353:1789-92; PMID:10348008; http://dx.doi.org/10.1016/S0140-6736(98)07546-1
- Fowden AL, Sibley C, Reik W, Constancia M. Imprinted genes, placental development and fetal growth. Horm Res (Suppl 3) 2006; 65:50-58; PMID:16612114; http://dx.doi.org/10.1159/000091506
- Jacob KJ, Robinson WP, Lefebvre L. Beckwith – Weidemann and Silver-Russell syndromes: opposite developmental imbalances in imprinted regulators of placental function and embryonic growth. Clin Genet 2013; 84:326-34; PMID:23495910; http://dx.doi.org/10.1111/cge.12143
- Smyth GK. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 2004; 3:Article 3; PMID:16646809
- Bejamini Y, Hochberg Y. Controlling the False Discovery Rate: A practical and powerful approach to multiple testing. J Royal Stat Soc, Series B 1995; 57:289-300.
- Lowe R, Rakyan VK. Marmal-aid – a database for Infinium Human Methylation450. BMC Bioinformatics 2013; 14:359; PMID:24330312; http://dx.doi.org/10.1186/1471-2105-14-359
- Quinello C, Silveira-Lessa AL, Ceccon MEJR, Cianciarullo MA, Carneiro Sampaio, Palmeira P. Phenotypic differences in leucocyte populations among healthy preterm and full-term newborns. Scand J Immunol 2014; 80, 57-70; PMID:24724912; http://dx.doi.org/10.1111/sji.12183
- Du P, Zhang X, Huang C-C, Jafari N, Kibbe WA, Hou L, Lin SM. Comparison of b value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics 2010; 11:587-598; PMID:21118553; http://dx.doi.org/10.1186/1471-2105-11-587
- Chen Y-a, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW, Gallinger S, Hudson TJ, Weksberg R. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium Humanmethylation450 microarray. Epigenetics 2013:8:203-209; PMID:23314698; http://dx.doi.org/10.4161/epi.23470
- Koon HB, Ippolito GC, Banham AH, Tucker PW FOXP1: a potential therapeutic target I cancer. Expert Opin Ther Targets 2007; 11:955-65; PMID:17614763; http://dx.doi.org/10.1517/14728222.11.7.955
- Shan J, Wang P, Zhou J, Wu D, Shi H, Huo K. RIOK3 interacts with caspase-10 and negatively regulates the NF-kB signaling pathway. Mol. Cell Biochem, 2009; 332:113-20; PMID:19557502; http://dx.doi.org/10.1007/s11010-009-0180-8
- Prasad S, Ravindra J, Aggarwal BB NF-kB and cancer: how intimate is this relationship. Mol. Cell Biochem 2010; 336:25-37; PMID:19823771; http://dx.doi.org/10.1007/s11010-009-0267-2
- Bhattacharya S, Takada S, Jacobson RH Structural analysis and dimerization potential of the human TAF5 subunit of TFIID. PNAS 2007; 104:1189-94; PMID:17227857; http://dx.doi.org/10.1073/pnas.0610297104
- Pijnappel WWM, Esch D, Baltissen MPA, Wu G, Mischerikow N, Bergsma AJ, van der Wal E, Han DW, vom Bruch H, Moritz S, et al. A central role for TFIID in the pluripotent transcription circuitry. Nature 2013; 495:516-19; PMID:23503660; http://dx.doi.org/10.1038/nature11970
- Parsa A, Kao L, Xie D, Astor BC, Li M, Hsu C-y, Feldman HI, Parekh RS, Kusek JW, Greene TH et al. APOL1 risk variants, race, and progression of chronic kidney disease. N Engl J Med 2013; 369:2183-96; PMID:24206458; http://dx.doi.org/10.1056/NEJMoa1310345
- Freedman BI, Langefeld CD, Murea M, Ma L, Otvos JD, Turner J, Antinozzi PA, Divers J, Hicks PJ, Bowden DW et al. Apolipoprotein L1 nephropathy risk variants associate with HDL subfraction concentration in African Americans. Nephrol Dial Transplant 2011; 26:3805-3810; PMID:21931123; http://dx.doi.org/10.1093/ndt/gfr542
- Gordon L, Joo JE, Powell JE, Ollikainen M, Novakovic B, Li X, Andronikos R, Cruickshank MN, Conneely KN, Smith AK et al. Neonatal DNA methylation profile in human twins is specified by a complex interplay between intrauterine environmental and genetic factors, subject to tissue-specific influence. Genome Res 2012; 22:1395-1406; PMID:22800725; http://dx.doi.org/10.1101/gr.136598.111
- Sookian S and Pirola CJ. Meta-analysis of the influence of I148M variant of patain-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of non-alcoholic fatty liver disease. Hepatology 2011; 53:1883-1894; PMID:21381068; http://dx.doi.org/10.1002/hep.24283
- Xiong Y, Fan C, Kong L, Dong L, Zhu N, Zhang J, Wang L, Qin T, Shen Y, Chen M. Annexin II receptor induces apoptosis independent of Annexin II. Apoptosis 2013; 18:925-39; PMID:23640736; http://dx.doi.org/10.1007/s10495-013-0846-2
- Barg E, Gasiorowski K, Brokos B, Swiedrych A, Skorkowska K. A high frequency of apoptosis was found in cultures of lymphocytes isolated from the venous blood of children born with a low birth weight. Cell Mol Biol Lett 2004; 9:135-43; PMID:15048157
- Hussain N. Epigenetic influences that modulate infant growth, development, and disease. Antioxidants & Redox Signalling 2012; 17:224-36; http://dx.doi.org/10.1089/ars.2011.4484
- Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 2007; 447:425-32; PMID:17522676; http://dx.doi.org/10.1038/nature05918
- Engel S, Joubert BR, Wu MC, Olshan AF, Haberg SE, Ueland PM, Nystad W, Nilsen RM, Vollset SE, Peddada SD et al. Neonatal genome wide methylation patterns in relation to birth weight in the Norwegian mother and child cohort. Am J Epidemiol 2014; 179:834-42; PMID:24561991; http://dx.doi.org/10.1093/aje/kwt433
- Thompson RF, Fazzari MJ, Niu H, Barzilai N, Simmons RA, Greally JM Experimental intrauterine growth restriction induces alterations in DNA methylation and gene expression in pancreatic islets of rats. J Biol Chem 2010; 285:15111-18; PMID:20194508; http://dx.doi.org/10.1074/jbc.M109.095133
- Morgan HD, Sutherland HE, Martin DIK, Whitelaw E. Epigenetic inheritance at the agouti locus in the mouse. Nat Genet 1999; 23:314-18; PMID:10545949; http://dx.doi.org/10.1038/15490
- Ng S-F, Lin RCY, Laybutt DR, Barres R, Owens JA, Morris MJ. Chronic high-fat diet in fathers programs β-cell dysfunction in female rat offspring. Nature 2010; 467:963-67; PMID:20962845; http://dx.doi.org/10.1038/nature09491
- Hillman S, Peebles DM, Williams DJ. Paternal metabolic and cardiovascular risk factors for fetal growth restriction. Diabetes Care 2013; 36:1675-80; PMID:23315598; http://dx.doi.org/10.2337/dc12-1280
- Freathy RM, Bennett AJ, Ring SM, Shields B, Groves CJ, Timpso, NJ, Weedon MN, Zeggini E, Lindgren CM, Lango H. Type 2 diabetes risk alleles are associated with reduced size at birth. Diabetes 2009; 58:1428-33; PMID:19228808; http://dx.doi.org/10.2337/db08-1739
- Jacob KJ, Robinson WP, Lefebvre L. Beckwith-Wiedemann and Silver-Russell syndromes: opposite developmental imbalances in imprinted regulators of placental function and embryonic growth. Clin Genet 2013; 84:326-34; PMID:23495910; http://dx.doi.org/10.1111/cge.12143
- Hung T-H, Chen S-F, Lo L-M, Li M-J, Yeh Y-L, Hsieh T-T. Increased autophagy in placentas of intrauterine growth restricted pregnancies. Plos One 2012; 7:e40957; PMID:22815878; http://dx.doi.org/10.1371/journal.pone.0040957
- Burton GJ, Yung H-W, Cindrova-Davies T, Charnock-Jones DS. Placental endoplasmic reticulum stress and oxidative stress in the pathophysiology of unexplained intrauterine growth restriction and early onset preeclampsia. Placenta 2009; 30:Supp 43-48; http://dx.doi.org/10.1016/j.placenta.2008.11.003
- Whitehead CL, Walker SP, Lappas M, Tong S. Circulating RNA coding genes regulating apoptosis in maternal blood in severe early onset fetal growth restriction and pre-eclampsia. J Perinatol 2013; 33:600-4; PMID:23429544; http://dx.doi.org/10.1038/jp.2013.16
- Banister CE, Koestler DC, Maccani MA, Padbury JF, Houseman E.A, Marsit CJ. Infant growth restriction is associated with distinct patterns of DNA methylation in human placentas. Epigenetics 2011; 6:920-7; PMID:21758004; http://dx.doi.org/10.4161/epi.6.7.16079
- Veerbeek JH, Nikkels PG, Torrance HL, Gravesteijn J, Post Uiterweer ED, Derks JB, Koenen SV, Visser GH, Van Rijn BB, Franx A. Placental pathology in early intrauterine growth restriction associated with maternal hypertension. Placenta 2014; 35:696-701; PMID:25052232; http://dx.doi.org/10.1016/j.placenta.2014.06.375
- Blair JD, Yuen RKC, Lim BK, McFadden DE, von Dadelszen P, Robinson WP. Widespread DNA hypomethylation at gene enhancer regions in placentas associated with early onset pre-eclampsia. Mol Hum Reprod 2013; 19:697-708; PMID:23770704; http://dx.doi.org/10.1093/molehr/gat044
- Schrey S, Kingdom J, Baczyk D, Fitzgerald B, Keating S, Ryan G, Drewlo S. Leptin is differentially expressed and epigenetically regulated across monochorionic twin placenta with discordant fetal growth. Mol Hum Reprod 2013; 19:764-72; PMID:23832168; http://dx.doi.org/10.1093/molehr/gat048
- Gordon L, Joo J-HE, Andronikos R, Ollikainen M, Wallace EM, Umstad MP, Permezel M, Oshlack A, Morley R, Carlin JB et al. Expression discordance of monozygotic twins at birth. Effect of intrauterine environment and a possible mechanism for fetal programming. Epigenetics 2011; 6:579-592; PMID:21358273; http://dx.doi.org/10.4161/epi.6.5.15072
- Schlinzig T, Johansson S, Gunnar A, Ekstrom TJ, Norman M. Epigenetic modulation at birth – altered DNA-methylation in white blood cells after caesarean section. Acta Paediatr 2009; 98:1096-99; PMID:19638013; http://dx.doi.org/10.1111/j.1651-2227.2009.01371.x
- Baschat AA, Gembruch U, Reiss I, Gortner L, Harman CR, Weiner CP. Neonatal nucleated red blood cell counts in growth restricted foetuses: relationship to arterial and venous Doppler studies. Am J Obstet Gynecol 1999; 181:190-95; PMID:10411818; http://dx.doi.org/10.1016/S0002-9378(99)70458-8
- Rolfo A, Maconi M, Cardaropoli S, Biolcati M, Danise P, Todros T. Nucleated red blood cells in term foetuses: reference values using an automated analyser. Neonatology 2007; 92:205-8; PMID:17476121; http://dx.doi.org/10.1159/000102096
- Sandoval J, Heyn H, Moran S, Serra-Musach J, Pujana MA, Bibikova M, Esteller M. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics 2011; 6:692-702; PMID:21593595; http://dx.doi.org/10.4161/epi.6.6.16196