
Abstract
Efficacious antitumor vaccines strongly stimulate cancer-specific effector T cells and counteract the activity of tumor-infiltrating immunosuppressive cells. We hypothesised that combining cytokine expression with silencing programmed cell death ligand 1 (PD-L1) could potentiate anticancer immune responses of lentivector vaccines. Thus, we engineered a collection of lentivectors that simultaneously co-expressed an antigen, a PD-L1-silencing shRNA, and various T cell-polarising cytokines, including interferon γ (IFNγ), transforming growth factor β (TGFβ) or interleukins (IL12, IL15, IL23, IL17A, IL6, IL10, IL4). In a syngeneic B16F0 melanoma model and using tyrosinase related protein 1 (TRP1) as a vaccine antigen, we found that simultaneous delivery of IL12 and a PD-L1-silencing shRNA was the only combination that exhibited therapeutically relevant anti-melanoma activities. Mechanistically, we found that delivery of the PD-L1 silencing construct boosted T cell numbers, inhibited in vivo tumor growth and strongly cooperated with IL12 cytokine priming and antitumor activities. Finally, we tested the capacities of our vaccines to counteract tumor-infiltrating myeloid-derived suppressor cell (MDSC) activities ex vivo. Interestingly, the lentivector co-expressing IL12 and the PD-L1 silencing shRNA was the only one that counteracted MDSC suppressive activities, potentially underlying the observed anti-melanoma therapeutic benefit. We conclude that (1) evaluation of vaccines in healthy mice has no significant predictive value for the selection of anticancer treatments; (2) B16 cells expressing xenoantigens as a tumor model are of limited value; and (3) vaccines which inhibit the suppressive effect of MDSC on T cells in our ex vivo assay show promising and relevant antitumor activities.
Keywords::
Abbreviations:
- DC, dendritic cell
- G-MDSC, granulocytic MDSC
- IL, interleukin
- IiOVA, MHC II invariant chain-ovalbumin
- MDSC, myeloid-derived suppressor cell
- M-MDS, monocytic MDSC
- MLR, mixed lymphocyte reaction
- OVA, chicken ovalbumin
- p1, PD-L1-targeted microRNA
- PD-L1, programmed cell death 1 ligand 1
- PD-1, programmed cell death 1
- shRNA, short hairpin RNA
- TAA, tumor associated antigen
- TCR, T cell receptor
- Th, T helper lymphocyte
- TRP1, tyrosinase related protein 1;
- TRP2, tyrosinase related protein 2
- 142 3p, target sequence for the microRNA 142 3p
Introduction
Effective antitumor T cell responses are required for successful cancer immunotherapy. However, activation of cytotoxic T cells specific for tumor-associated antigens (TAAs) in a systemic immunosuppressive environment is a major challenge. Firstly, circulating TAA-specific T cells are usually anergic or present low-affinity T cell receptors (TCRs).Citation1,2 Therefore, their activation threshold is greater than the one for T cells specific for non-self antigens. Secondly, once activated these TAA-specific effector T cells have to resist the highly immunosuppressive tumor environment. There are several cell types that inhibit anticancer immune responses, among which are tumor-infiltrating myeloid-derived suppressor cells (MDSCs). MDSCs have recently become major targets for cancer immunotherapy.Citation3,4
T cells are activated by 3 signals provided by antigen-presenting dendritic cells (DCs). These include: the binding of the antigenic peptide-major histocompatibility complex (MHC) to the T cell receptor (TCR); the co-stimulatory/inhibitory ligand-receptor interactions between DCs and T cells; and cytokine priming that promotes T cell differentiation.Citation5 Manipulation of any, or all 3 of these signals can enhance T cell responses. One way to achieve this is by removing negative co-stimulation provided through binding of programmed death 1 ligand 1 (PD-L1) to the T cell inhibitory receptor programmed cell death 1 (PDCD1, better known as PD-1).Citation6-11 We previously demonstrated that PD-L1 silencing on DCs inhibited TCR down-modulation and hyperactivated CD8+ T cells. The lack of PD-L1/PD-1 binding in the immunological synapse prevented up-regulation of the E3 ubiquitin ligase Cbl-b in CD8+ T cells. Cbl-b expression plays a physiological role in triggering ligand-induced TCR down-modulation after antigen presentation.Citation1,12 As a consequence, T cells undergoing antigen presentation in the absence of PD-L1/PD-1 co-stimulation presented high TCR surface levels, expanded rapidly, increased pro-inflammatory cytokine secretion and accelerated anticancer activities.Citation1,12 Surprisingly, cellular vaccines transferring DCs with silenced PD-L1 did not improve cure rates in a model of lymphoma expressing the xenoantigen ovalbumin (OVA) as a surrogate tumor antigen. We also demonstrated that only the combination of PD-L1 silencing with either ERK inhibition or p38 activation increased the capacity of lentivector vaccines to significantly improve cure rates in a thymoma mouse model.Citation12 Therefore, we concluded that additional signals such as adequate cytokine priming must be provided to T cells to fully activate them.Citation1,5 In addition, activated TAA-specific T cells are strongly inhibited by the suppressive activity of MDSCs, and other immunosuppressive cells, present within the tumor environment.Citation3,13 An efficacious vaccine, therefore, will stimulate TAA-specific T cells while concurrently inhibiting MDSC suppressive activities within the tumor environment.Citation4
In this study we designed lentivector vaccines for melanoma immunotherapy that co-expressed a neoplastic cell antigen, a PD-L1-silencing microRNA and an array of T cell-polarising cytokines. T-cell responses and antitumor activities of each candidate lentivector were prospectively evaluated in detail. Only co-delivery of IL12 with a PD-L1 silencing microRNA showed significant therapeutic activities in a syngeneic melanoma mouse model, using the endogenous TAA tyrosinase related protein 1 (TRP1) as a vaccine antigen. Mechanistically, its efficacy relied on efficient activation of TRP1-specific T cells by IL12 expression and direct inhibition of tumor growth by PD-L1 silencing. In vitro co-delivery of IL12 with a PD-L1 silencing microRNA exhibited highly effective anti-tumor-infiltrating MDSC activities, suggesting another important mechanistic effect of this cancer vaccine. Interestingly, we demonstrated that methods based on the surrogate tumor antigen (OVA) to evaluate vaccine efficacyCitation12,14-17 in healthy or B16-IiOVA tumor-bearing mice were unable to predict therapeutic outcome. Only the treatments that overcame tumor-infiltrating MDSC suppressive activities ex vivo exhibited therapeutically relevant anti-melanoma activities in vivo in mouse models.
Results
Engineering lentivectors to simultaneously deliver a PD-L1 silencing microRNA, a vaccine antigen and cytokines
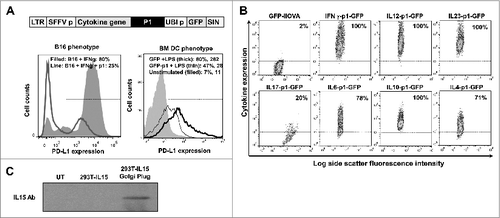
On the basis of prior results, we reasoned that T cells must receive additional signals along with PD-L1 silencing to acquire potent antitumor activities.Citation1,5,18 As proof-of-principle, we engineered a collection of lentivectors that would simultaneously deliver a PD-L1 silencing microRNA (designated here as p1) with a vaccine antigen of interest, in combination with various cytokines. As a starting point, we used a previously described lentivector backbone encoding green fluorescence protein (GFP) as a reporter gene ().Citation12,19 As expected, transduction with the modified lentivector encoding p1 efficiently reduced PD-L1 expression in bone marrow-derived DCs treated with lipopolysaccharide (LPS) afterwards, and in B16F0 melanoma cells treated with interferon γ (IFNγ; ). We used this lentivector backbone to insert different cytokine genes including: pro-inflammatory cytokines IL12, IFNγ, IL15, or IL6; anti-inflammatory and T helper type-2 (Th2) cytokines IL10 or IL4; regulatory T cell (Treg) inducers such as IL10 or transforming growth factor β (TGFβ), and Th17 stimulators such as IL23, IL17 or IL6. These cytokines were selected as they possessed a wide range of properties and functions. The constructed lentivectors efficiently expressed the corresponding cytokines in transduced cells, as assessed by flow cytometry using cytokine-specific antibodies, or immunoblot for IL15 detection ().
Figure 1. Characterization of lentivectors co-expressing an array of cytokines and a PD-L1-targeted shRNA. (A) Lentivector system used to co-express cytokine genes, a PD-L1-targeted microRNA (p1), and green fluorescent protein (GFP). The histograms show PD-L1 expression in B16F0 cells (B16, left) transduced with the lentivectors co-delivering GFP-p1, and treated with IFNγ and bone marrow-derived dendritic cells (BM-DCs, right) transduced with GFP-p1 or only GFP and treated with lipopolysaccharide (LPS). Percentages and mean fluorescent intensities (MFI) for the indicated treatments are shown. Horizontal lines in the histograms represent the gate excluding 95% of non-transduced (GFP−) cells. LTR, long-terminal repeat; SFFVp, spleen focus-forming virus promoter; UBIp, ubiquitin promoter; SIN, self-inactivating LTR. (B) Flow cytometry density-plots showing cytokine expression (detected by intracellular staining with cytokine-specific antibodies) in 293T cells transduced with the indicated lentivectors. Percentages of cytokine-expressing cells are shown within the graphs. Horizontal lines represent exclusion of 95% of non-transduced cells. (C) IL15 expression assessed by inmunoblot of protein prepared from 293T cells transduced with a lentivector encoding IL15. GolgiPlug was added (top) to allow cytokine accumulation prior to cell harvest. UT, untransduced. A bioassay using SMAD-GFP cellsCitation19 was used for TGFβ detection (5.1 ± 1.03 μg TGF-β/mL lentivector stock).
All lentivector vaccines induce OVA-specific T-cell responses
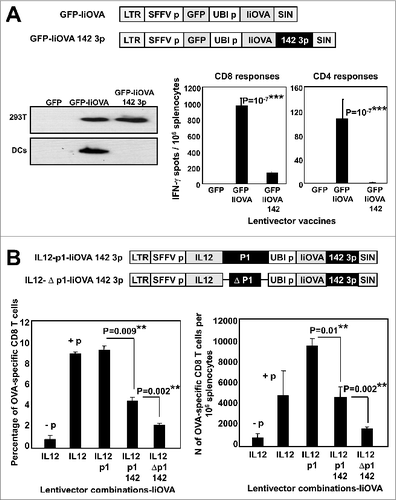
Anticancer vaccines are standardly evaluated first in healthy mice and the strongest T-cell inducers are then selected for testing in tumor models. Accordingly, we wanted to first test our lentivector vaccines in healthy mice. Their capacities to induce CD4+ and CD8+ T-cell responses were characterised in splenocytes after subcutaneous vaccination at the base of the tail. For these experiments we replaced GFP with a IiOVA transgene as a model antigen (). The N-terminal part of the invariant MHC chain (Ii) was fused to OVA (IiOVA) to facilitate presentation of MHC Class II OVA peptide epitopes. Interestingly, OVA-specific T-cell responses were observed in all cases (). However, only expression of IFNγ, IL23, IL12 or IL15 together with p1 resulted in a significant increase in OVA-specific IFNγ+ CD8+ T cells compared to a non-cytokine encoding lentivector control (GFP-IiOVA). The most effective treatment was IFNγ-p1, characterised by a significant expansion of IFNγ+ granzyme B+ CD8+ T cells (). IFNγ and IL23 expression increased the levels of IL17+ CD8+ T cells. Surprisingly, all vaccination groups had low numbers of Foxp3+ Tregs among splenocytes after OVA re-stimulation as compared to the GFP-IiOVA vaccination control. This was even the case when including cytokines in the vectors that are known to induce Tregs ().
Figure 2. T-cell responses in healthy mice immunized with candidate lentivector vaccines. (A) Lentivector constructs used for mouse vaccination, comprising a cytokine from an array of cytokine genes (as indicated) co-expressed with the PD-L1 silencing microRNA (p1) and the model antigen IiOVA. The immunoblot shows IiOVA [detected using a haemagglutinin (HA) tag] and GADPH (loading control) expression in 293T cells transduced with the indicated lentivector constructs (top of the blot). LTR; long terminal repeat, SFFVp; spleen focus forming virus promoter, UBIp; ubiquitin promoter, SIN; self-inactivating LTR. (B) Percentage of OVA-specific IFNγ+ CD8+ T cells (upper left), IL17+ CD8+ T cells (upper right), granzyme B+ IFN−γ+ CD8+ T cells (bottom left), or Foxp3+ CD4+ regulatory T cells (Tregs; bottom right) among splenocytes of mice vaccinated with the indicated lentivector cytokine-p1-IiOVA constructs. Bar charts showing the mean ± S.D. (n = 5 mice per group) of cytofluorimetric analysis of splenocytes immunostained with the indicated antibodies. One representative experiment out of 2 is shown. Relevant statistical comparisons are indicated within the graph, performed by one-way ANOVA, 5 mice per group, 3 replicates; *, P < 0.05. NP, no peptide restimulation; GrzB, granzyme B. The horizontal dotted lines indicate the T-cell levels in mice vaccinated with the GFP-IiOVA lentivector (positive control), and restimulated with OVA peptides. (C) The 2 dot-plots on the left show Foxp3 expression in CD4+ CD25 OT-II transgenic T cells co-cultured for 2 days with immature dendritic cells (DCs) transduced with the lentivectors as indicated; the percentages of Foxp3+ Tregs are indicated. Bar chart (right) indicates inducible Foxp3+ Treg percentages showing the mean ± S.D. (n = 3). Relevant statistical comparisons are indicated within the graph, performed by Student's t-test; the experiment (initially performed in duplicates) was performed in triplicate with similar results achieved.
![Figure 2. T-cell responses in healthy mice immunized with candidate lentivector vaccines. (A) Lentivector constructs used for mouse vaccination, comprising a cytokine from an array of cytokine genes (as indicated) co-expressed with the PD-L1 silencing microRNA (p1) and the model antigen IiOVA. The immunoblot shows IiOVA [detected using a haemagglutinin (HA) tag] and GADPH (loading control) expression in 293T cells transduced with the indicated lentivector constructs (top of the blot). LTR; long terminal repeat, SFFVp; spleen focus forming virus promoter, UBIp; ubiquitin promoter, SIN; self-inactivating LTR. (B) Percentage of OVA-specific IFNγ+ CD8+ T cells (upper left), IL17+ CD8+ T cells (upper right), granzyme B+ IFN−γ+ CD8+ T cells (bottom left), or Foxp3+ CD4+ regulatory T cells (Tregs; bottom right) among splenocytes of mice vaccinated with the indicated lentivector cytokine-p1-IiOVA constructs. Bar charts showing the mean ± S.D. (n = 5 mice per group) of cytofluorimetric analysis of splenocytes immunostained with the indicated antibodies. One representative experiment out of 2 is shown. Relevant statistical comparisons are indicated within the graph, performed by one-way ANOVA, 5 mice per group, 3 replicates; *, P < 0.05. NP, no peptide restimulation; GrzB, granzyme B. The horizontal dotted lines indicate the T-cell levels in mice vaccinated with the GFP-IiOVA lentivector (positive control), and restimulated with OVA peptides. (C) The 2 dot-plots on the left show Foxp3 expression in CD4+ CD25 OT-II transgenic T cells co-cultured for 2 days with immature dendritic cells (DCs) transduced with the lentivectors as indicated; the percentages of Foxp3+ Tregs are indicated. Bar chart (right) indicates inducible Foxp3+ Treg percentages showing the mean ± S.D. (n = 3). Relevant statistical comparisons are indicated within the graph, performed by Student's t-test; the experiment (initially performed in duplicates) was performed in triplicate with similar results achieved.](/cms/asset/1c0697e4-ba7a-4558-a645-ea3f3c6c98bb/koni_a_945378_f0002_b.gif)
PD-L1 expression is required for the differentiation of inducible antigen-specific Tregs.Citation20 Therefore, we tested whether PD-L1 silencing itself was reducing Treg levels in our experimental system. To address this point, immature DCs were transduced with a GFP-IiOVA lentivector and an IiOVA-GFP-expressing lentivector that included the PD-L1 silencing microRNA. Transduced cells were subsequently co-cultured with purified OVA-specific transgenic OT-II CD4+ T cells. As expected, OVA presentation by immature DCs induced CD4+CD25+Foxp3+ Treg differentiation from OT-II cells. In contrast, silencing of PD-L1 alone was sufficient to completely abrogate Foxp3+ Treg differentiation (), which explained the low numbers of Tregs observed by vaccination with the p1-containing lentivectors. Thus, we preferentially (but not exclusively) considered the lentivectors encoding IFNγ, IL12 or IL15 as potential good vaccine candidates, as they were the 3 best inducers of CD8+ T cell responses in healthy vaccinated mice.
Local delivery of a PD-L1 silencing shRNA, particularly to immune cells amplifies T cell quantity and cooperates with cytokine priming
Subcutaneous vaccination with lentivectors transduces conventional DCs, which then migrate to lymph nodes where they trigger T-cell responses.Citation21 However, we have previously shown that lentivectors also transduce cells other than DCs at the vaccination site.Citation19,21-23 To characterise the adjuvant effects of cytokine expression and PD-L1 silencing only on the cells at the injection site other than professional antigen presenting cells, we introduced the target sequence for the haematopoietic endogenous microRNA-142-3p in lentivectors (). This strategy efficiently abrogates transgene expression from lentiviral transduced DCs, and other cells from the haematopoietic lineage such as B and T cells, Langerhans cells and macrophages, as these cells express the miRNA-142-3p and would thus likely degrade mRNA containing the miR-142-3p seed sequence.Citation24-30 We firstly validated this approach by using the GFP-IiOVA lentivector backbone. The 142-3p target abolished GFP and IiOVA expression in DCs and in a large collection of haematopoietic-derived immune cells ( and results not shown). In agreement with other studies, subcutaneous vaccination with the GFP-IiOVA-142-3p construct in mice strongly reduced CD8+ responses (), and completely abrogated CD4+ responses (). These results confirmed that antigen expression in haematopoietic-derived antigen presenting cells is required to trigger antigen-specific T cell responses, as previously described.Citation31 After validation in the GFP-IiOVA backbone, we included the target sequence in the IL12-p1-IiOVA and IFNγ-p1-IiOVA lentivectors (IL12-p1-IiOVA-142-3p, IFNγ-p1-IiOVA-142-3p). We decided to test these 2 cytokine-p1 combinations because the first one is a proto-typical Th1 cytokine, and the second one was the highest inducer of T-cell responses in healthy vaccinated mice ().
Figure 3. Local delivery of a PD-L1-targeted shRNA particularly to immune cells amplifies T-cell responses. (A) GFP-IiOVA lentivectors lacking or containing the miR-142-3p target sequence (as indicated) downstream of the IiOVA gene. Immunoblot shows IiOVA expression detected with an HA (haemagglutinin tag) antibody in 293T and bone marrow-derived dendritic cells (BM-DCs) transduced with the indicated lentivectors. Right; OVA-specific CD8+ and CD4+ responses quantified as a percentage of splenocytes from lentivector immunized mice (n=10 per group) as measured by IFNγ ELISPOT; bars indicate the mean ± S.D. Statistical comparisons were performed by one-way ANOVA; experiments performed in triplicates with similar results achieved. (B) Top, lentivectors containing the miR-142-3p targeting sequence in a IL12-p1-IiOVA backbone, with (p1) or without (Δp1) PD-L1 shRNA. Mice (n = 5 per group) were subcutaneously vaccinated and CD8+ T cell responses evaluated after OVA peptide stimulation 2-weeks later. The left graph shows the fraction of OVA-specific T cells within the CD8+ T cell population, while the right graph shows total numbers of OVA-specific CD8+ T cells among splenocytes; absence (−p) or presence (+p) of OVA peptide stimulation. LTR, long-terminal repeat; SFFVp, spleen focus-forming promoter; UBIp, ubiquitin promoter; SIN, self-inactivating LTR. Relevant statistical comparisons are shown within the graphs, calculated by one-way ANOVA; experiment performed in duplicate with similar results achieved; * P < 0.05, ** P < 0.01, *** P < 0.001.
Mice were subcutaneously vaccinated with IL12-p1-IiOVA-142-3p, and GFP-IiOVA, IL12-IiOVA and IL12-Δp1-IiOVA-142-3p (without the shRNA) as controls (). Interestingly, after re-stimulation with OVA peptides IL12-IiOVA and IL12-p1-IiOVA vaccination induced the same percentage of OVA-specific T cells within the CD8+ population as assessed by IFNγ expression, indicating equal specificity of the response. The 142-3p target in IL12-p1 significantly reduced this proportion, as this vector only induced expression of transgenes in cells other than DCs at the injection site (). Surprisingly, only removal of p1 from this 142-3p-containing vector to ensure unmodified PD-L1 expression at the injection site decreased CD8+ T-cell responses close to background levels. In contrast, the results were quite different when total numbers of OVA-specific CD8+ T cells within the splenocyte population were quantified, reflecting inequity in the “strength” of the response. Vaccination with IL12-p1-IiOVA was clearly superior to IL12-IiOVA, while vaccination with IL12-p1-IiOVA-142-3p gave the same total number of OVA-specific CD8+ T cells as IL12-IiOVA (). These results suggested that delivery of p1 increased expansion of antigen-specific and non-specific CD8+ T cells. On the other hand, IL12 expression in combination with PD-L1 silencing ensured the highest numbers of antigen-specific CD8+ T cells (). Similar results were observed using the IFNγ-p1-IiOVA backbone for the experiments (results not shown).
Vaccination with lentivectors delivering cytokines and PD-L1 shRNA delays tumor growth using a surrogate tumor antigen in a B16-IiOVA melanoma model
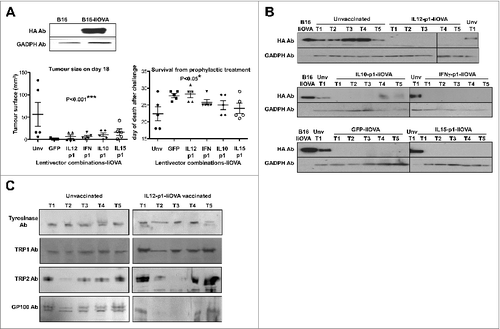
Before testing our lentivector vaccines in a syngeneic B16F0 melanoma model (non-modified cancer cells, only expressing weak TAAs), we used B16-IiOVA cells, which express OVA as a strong surrogate tumor antigen. This model is frequently used to evaluate cancer immunotherapies.Citation12,14-17,19,32,33 We tested a selection of our lentivector vaccines, including IL12-p1-IiOVA and IFNγ-p1-IiOVA, in a prophylactic vaccination protocol. Thus, mice were subcutaneously vaccinated at the base of the tail followed by subcutaneous transfer of B16-IiOVA cells 2-weeks later. Delayed tumor growth and increased survival was observed in all vaccinated groups relative to unvaccinated mice (). Thus, on the basis of the results achieved so far, any of our lentivector vaccines would be a good candidate for cancer immunotherapy. Furthermore, even co-expression of GFP with IiOVA exhibited significant antitumor activities. It has to be taken into account that both GFP and IiOVA are xenoantigens and could have synergistic adjuvant activities. Nevertheless, tumors regressed and we analysed IiOVA as well as common endogenous melanoma tumor-associated antigen (TAA) expression in these tumors. As expected, IiOVA expression had been lost in nearly all escaping tumors (). Interestingly, some of the escaping tumors arising in IL12-p1-IiOVA-vaccinated mice had also lost Trp2 and GP100 expression ().
Figure 4. Lentivector vaccination delays melanoma growth in a B16 model expressing a surrogate tumor antigen. (A–C) C57Bl/6 mice (n = 5 per group) were subcutaneously vaccinated at the base of the tail with 1 × 107 lentiviral particles (as indicated) followed by subcutaneous transfer of 2 × 106 B16-IiOVA cells 2-weeks later. (A) Above, IiOVA (detected by HA-tag) and GADPH (loading control) expression assessed by immunoblot in B16 versus B16-liOVA cell lines. Below, left, tumor sizes on day 18 after B16-IiOVA subcutaneous transfer, when the first control mouse was sacrificed. Below right, time of death after B16-IiOVA transfer. Means ± S.D. are also indicated for each group as intervals. Unv, unvaccinated. (B) Immunoblots of IiOVA and GADPH expression in samples from escaping tumors in each mouse (T1 to T5) vaccinated with the indicated lentivector vaccines (shown on top) and challenged with B16-IiOVA cells. Protein extracts from B16-IiOVA cells and a tumor from unvaccinated, challenged mice (Unv T1) were used as positive controls for IiOVA expression. (C) Expression of the indicated tumor-associated antigens (TAAs) by immunoblot using specific antibodies, from escaping tumors arising in unvaccinated or IL12-p1-IiOVA-vaccinated mice challenged with B16-IiOVA cells. Each tumor was analyzed separately (T1 to T5). Ab, antibody. Statistical comparisons were performed using the non-parametric Kruskal-Wallis test; experiment performed in duplicate with similar results achieved; * P < 0.05, ** P < 0.01, *** P < 0.001.
Prophylactic vaccination with IL12-p1 using endogenous TRP1 as a target antigen enhances survival in a xenoantigen-free syngeneic B16F0 melanoma model
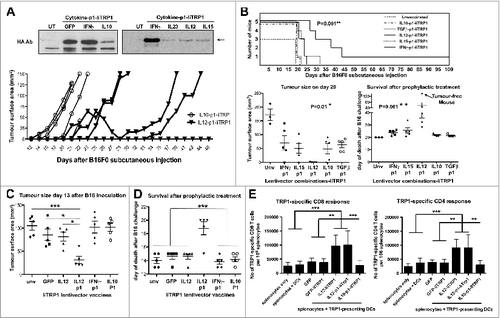
As mentioned above, so far it was unclear which cytokine-p1 combination, if any, would exhibit the strongest antitumor activities. Targeting an immunogenic xenoantigen such as OVA could mask the adjuvant activities of the cytokines in our lentivector constructs (). With this in mind we replaced IiOVA in our constructs with an endogenous TAA in the lentivector vaccines to test them in a syngeneic B16F0 unmodified melanoma model which does not express OVA. Trp1 was chosen as endogenous melanoma TAA because of several reasons. Firstly, it remained highly expressed in escaping tumors (); secondly, lentivector vaccination expressing the wild-type Trp1 antigen (unlike an immunogenic mutated version) does not possess any protective or therapeutic activities.Citation34,35 Therefore, we reasoned that any increase in therapeutic activity observed with our lentivectors, even if a modest one, could be of relevance with a challenging self-antigen as vaccine target. Thus, we delivered Trp1 as a fusion gene between the invariant chain and full-length HA-tagged Trp1 (). These B16F0 cells did not express any xenoantigen that could affect the outcome of the experiments. Mice were vaccinated subcutaneously in the base of the tail with a single lentivector injection followed by B16F0 challenge 2-weeks later. Only vaccination with IL12-p1-IiTrp1 strongly delayed tumor growth and significantly increased survival (). IL12-IiTrp1 vaccination (without the PD-L1 silencing shRNA) also induced slower tumor growth albeit not at the levels of IL12-p1-IiTRP1 (). To confirm the expansion of Trp1-specific T cells in IL12-IiTrp1 and IL12-p1-IiTrp1-vaccinated mice, splenocytes were co-cultured overnight with LPS-treated DCs expressing GFP-IiTrp1. Interestingly, a single vaccination of healthy mice induced strong and significant TRP1-specific CD8 and CD4 systemic T-cell responses (). In contrast, vaccination with the GFP-IiTrp1 lentivector did not raise significant T cell responses, as GFP and wild-type Trp1 are very poor immunogens.Citation34 These results clearly show that our IL12-IiTrp1-based vaccines raise polyclonal Class I and Class II Trp1-specific T cell responses. Moreover, these responses occurred independently of the presence of the PD-L1 silencing shRNA.
Figure 5. Prophylactic vaccination with IL12-p1-IiTrp1 enhances survival in a xenoantigen-free syngeneic B16F0 melanoma model. (A–E) C57Bl/6 mice (n = 5 mice/group) were vaccinated subcutaneously in the base of the tail with 1 × 107 lentiviral particles (as indicated) followed by B16F0 challenge 2-weeks later. (A) Top, IiTrp1 detection by immunoblot using an HA-specific antibody in 293T cells transduced with the indicated lentivectors. IiTrp1 expression levels are noticeably different (arrow). Below, tumor growth in mice vaccinated with the indicated lentivectors on top, followed by transfer of 5 × 105 B16F0 cells per mouse. (B) Top graph, Kaplan-Meier survival plot from (A) but including the indicated lentivector vaccination groups. The graphs below represent tumor size (left) on day 20, and time of death after B16F0 challenge (right). Lentivector vaccines are indicated below each graph. The arrow indicates a long-term protected mouse. C. Tumor size in mice vaccinated with the indicated lentivectors on day 13 after inoculation with B16F0 cells (2 × 106 cells per mouse). (D) Same as in C, but representing the time of death after B16F0 challenge. (E) Quantification of the percentage of TRP1-specific CD4+ and CD8+ T cells in splenocytes from mice vaccinated with the indicated lentivectors. The graphs represent IFNγ-expressing T cells, incubated overnight with dendritic cells (DCs) co-expressing GFP-IiTrp1. Data are presented as the mean ± S.D. CD8+ and CD4+ T-cell responses are represented in the left and right graphs, respectively. Unv, unvaccinated mice; Ab, antibody. Tumor sizes and survival were compared with the non-parametric Kruskal-Wallis test; experiment performed in triplicate with similar results achieved. Expansion of CD4+ and CD8+ T-cell responses between vaccinated groups were analysed by one-way ANOVA; * P < 0.05, ** P < 0.01, *** P < 0.001.
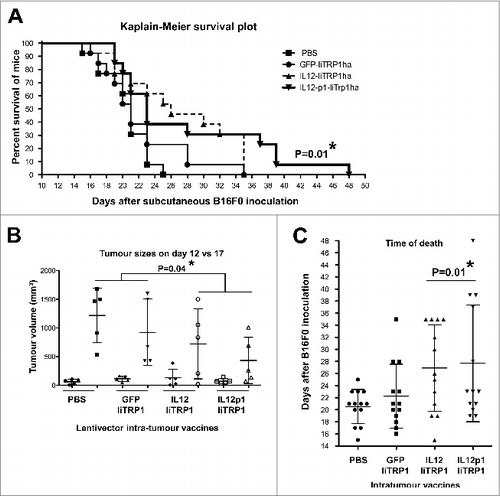
Therapeutic intratumoral vaccination with IL12-p1-IiTrp1 controls tumor growth in a xenoantigen-free syngeneic B16F0 melanoma model
Vaccination with IL12-p1-IiTrp1 exhibited the strongest antitumor activities in prophylactic experiments, which depend on the priming of T-cell responses. In these experiments, the incorporation of the PD-L1-silencing shRNA had a clear enhancing effect. We further tested IL12-p1-IiTrp1 vaccination in therapeutic experiments in mice by intratumoral administration. Of note, in these experiments, TAA-independent mechanisms might be taking place that could also contribute to therapeutic activities. Thus, a single intratumoral injection was carried out with lentivectors GFP-IiTrp1, IL12-IiTrp1, IL12-p1-IiTrp1 or PBS in rapidly growing B16F0 melanoma tumors (about 12 days after B16F0 transfer).Citation1,12 We chose a single injection regimen to prevent the boosting of primary T-cell responses, even though modest therapeutic effects might be achieved in this way. However, as with prophylactic experiments, these responses could highlight relevant activities that might be otherwise missed. Injections were performed into the tumors when the mean tumor volume reached between 50 and 100 mm3. Tumor growth was significantly delayed upon injection of either IL12-IiTrp1 or IL12-p1-IiTrp1 vaccines, and were particularly retarded in the case of IL12-p1-based vaccination (). However, no prominent p1-specific differences were observed. In the therapeutic model (unlike in the prophylactic model) the inclusion of p1 might not be the critical factor for the lentivector activities. Therefore, survival was prolonged after vaccination with IL12-expressing lentivectors, with or without the PD-L1 silencing p1 component, as compared to vaccination with PBS or GFP-IiTrp1 controls ().
Figure 6. Therapeutic intratumor vaccination with IL12-p1-IiTrp1 can control tumor growth in a xenoantigen-free syngeneic B16F0 melanoma model. (A–C) C57Bl/6 mice were subcutaneously injected with 3 × 105 B16F0 cells/mouse (n = 5 per group) at the base of the tail. Mice were vaccinated 12 days later (average tumor size between 50–200 mm3) with 1 × 107 lentivector transducing particles intratumorally injected and tumor size was monitored every 2 days. (A) Kaplan-Meier survival plot. Pooled data from 2 independent experiments are plotted. (B) Tumor volume in mice on the day of vaccine injection (day 12) and on day 17. Data are presented as the mean ± S.D. (C) The time of death after lentivector vaccination. In this case, data from 2 independent experiments were pooled. Data are presented as the mean ± S.D. Tumor sizes and survival between groups were analysed statistically with the non-parametric Kruskal-Wallis test; duplicate experiments were performed with similar results achieved; * P < 0.05.
Delivery of PD-L1 silencing shRNA to tumor cells delays tumor growth
To gain insight into the therapeutic mechanisms of the activity of the IL12-based lentivector vaccines after intratumoral administration, we studied their inhibitory effects over cancer cell growth. For this purpose, B16F0 cells were transduced ex vivo and then injected into mice, assuring that only cancer cells would be transduced. We chose the lentivector expressing GFP-IiOVA as the non-cytokine encoding control, the IL12-p1-IiOVA lentivector that we had found to be the most effective in the syngeneic B16F0 model, the IFNγ-p1-IiOVA lentivector among the strongest T cell stimulators in healthy vaccinated mice and the IL10-p1-IiOVA lentivector as a control encoding an immunosuppressive cytokine. In vitro growth of B16F0 cells was not affected with the exception of IFNγ-p1-IiOVA transduction, which stopped cell growth. Injection of B16F0 melanoma cells expressing GFP-IiOVA enhanced tumor growth (), suggesting that IiOVA expression did not inhibit tumor establishment in vivo. Surprisingly, tumor growth was significantly delayed in all groups vaccinated with cytokine gene-containing lentivectors () while IL12-p1-IiOVA expression in cancer cells was the most effective at attenuating tumor growth.
Figure 7. Delivery of PD-L1 silencing microRNA exclusively to B16F0 melanoma cells delays tumor growth. (A and B) To specifically analyse the effects of vaccination on cancer cell growth, B16F0 cells were transduced ex vivo and then injected into recipient mice (n = 5 mice per group). (A.) Top, tumor growth after subcutaneous transfer of B16F0 cells transduced with GFP-IiOVA (solid symbols) or IL12-p1-IiOVA (open symbols) lentivectors, as indicated. Below, the same as top, but with individual tumor volumes plotted at the indicated time points; bars indicate the mean ± S.D. Pooled data from 2 control experiments are included. (B) Left, tumor growth induced by B16F0 cells previously transduced ex vivo with the lentivectors GFP-IiOVA (solid symbols) and GFP-p1-IiOVA (open symbols) and subcutaneously injected into mice 4 days later. On the right, the same individual tumor volumes plotted at the indicated time points; bars indicate the mean ± S.D. Relevant statistical comparisons are shown within the graphs. Tumor sizes and survival between groups were compared with the non-parametric Kruskal-Wallis test; duplicate experiments were performed with similar results achieved; * P < 0.05, ** P < 0.01, *** P < 0.001.
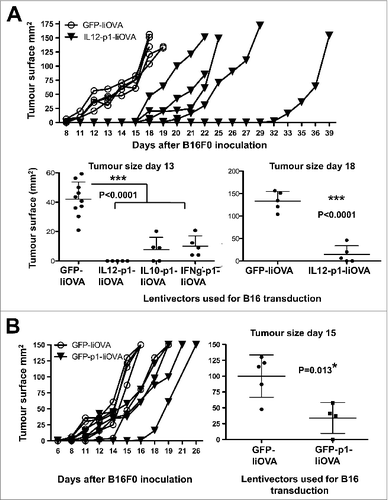
As GFP-IiOVA-expressing B16F0 cells increased the tumor growth rate, we next studied the contribution of PD-L1 silencing alone to cancer cell growth inhibition. B16F0 cells were thus transduced ex vivo with GFP-p1-IiOVA, which encompassed the silencing PD-L1 shRNA and were subsequently transferred to mice. Interestingly, tumor growth was significantly delayed by including p1 in the GFP-IiOVA lentivector backbone (). Thus, PD-L1 silencing in cancer cells was sufficient to delay tumor growth to some degree. However, as all of our lentivector vaccines already contained p1 as well as the targeted antigen, suggesting that the PD-L1 silencing shRNA and IiTrp1 (unlike in the prophylactic experiments) were not significantly contributing to the therapeutic activities of the IL12-containing lentivector after intra-tumor injection. We concluded that intratumoral IL12 expression was the key effector molecule in the therapeutic setting.
Only IL12 lentivector-based vaccines counteract MDSC-T cell suppressive activities
Tumor-infiltrating MDSCs strongly contribute to the highly immunosuppressive tumor environment by inhibiting T-cell responses. We therefore wanted to characterise the in vivo effects of our lentivector vaccines over MDSCs, particularly the IL12-based vaccines. In fact, IL12 expressed by genetically modified CD8+ T cells is capable of converting tumor-associated myeloid cells into effective antigen-presenting cells.Citation36 Therefore, although IL12 exerts multiple activities over a variety of cell types within the tumor, this mechanism can affect myeloid cells, thereby contributing to tumor regression.Citation36 However, after intratumoral injection, most of the transduced cells are B16F0 cancer cells (about 80%), while transduced MDSCs accounted for less than 5% of total tumor cells (data not shown and. Citation37) Purification of sufficient numbers of intra-tumor transduced MDSCs for analyses would be impractical, especially considering the number of different treatments to compare at the same time. To overcome this significant limitation, we developed a method to produce large numbers of MDSCs ex vivo that resemble melanoma-infiltrating MDSCs. To achieve this, we modified melanoma B16F0 cells and a non-neoplastic control cell line (293T cells) with lentivectors expressing GM-CSF. This approach was taken as BM-derived MDSCs using cancer cell-derived conditioning medium were shown to be equivalent to MDSCs found in tumor-bearing mice.Citation13 These cells produced MDSC-conditioning medium mimicking the tumor environment and strongly enhancing MDSC proliferation. As shown in (and manuscript in preparation), culturing bone marrow cells with MDSC conditioning medium expanded high numbers of MDSCs as defined by their phenotype (CD11cneg/low , CD11bhigh, GR1high, CD80high, F4/80neg/low, Flt3neg, CD34neg, CD62Lhigh MHC IIlow, CD86low). These MDSCs were a mixture of monocytic (Ly6C+, Ly6Glow/neg) and granulocytic (Ly6C+, Ly6Ghigh) populations (). B16-MDSCs expressed inducible nitric oxide synthase (iNOS) and arginase-1, a signature of tumor-infiltrating MDSCs Citation3,38 ( and manuscript in preparation). These cells were further compared to spleen and tumor-infiltrating MDSCs from B16F0 melanoma-bearing mice. Their respective phenotypes were analysed according to expression of representative markers widely used in flow cytometric characterisation of MDSCs.Citation13,39 Ex vivo-produced B16-MDSCs phenotypically resembled tumor-infiltrating rather than spleen MDSCs from melanoma-bearing mice (). Ex vivo B16-MDSCs were further sorted into monocytic and granulocytic populations, and both strongly inhibited proliferation and IFNγ production of anti-CD3/anti-CD28-activated CD8+ T cells in standard suppression assays (manuscript in preparation). Further, we used an MDSC-T cell antigen presentation assay to determine whether our treatments could increase the antigen presentation capacities of the ex vivo MDSCs. We tested most of our lentivector vaccines expressing the model antigen IiOVA. Thus, transduced MDSCs were co-cultured with OVA-specific OT-I CD8+ T cells at a ratio of 1:10 (). Strikingly, only transduction with IL12-p1-IiOVA strongly enhanced OT-I activation above controls ().
Figure 8. IL12-based lentivector vaccines exclusively strongly counteract B16F0 melanoma-infiltrating MDSC suppression of T-cell activities. (A–E) Characterization of IL-12-based lentiviral vaccines modulation of myeloid-derived suppressor cell (MDSCs) immunosuppressive activities in vitro. A. Characterization of bone marrow-derived myeloid cells cultured in the presence of conditioning media from GM-CSF expressing 293T or dendritic cells (DCs) to phenocopy the tumor microenvironment and generate MDSCs. Bar graph on the left represents the percentage of expression of the indicated markers (bottom) after 8 days in culture with DC medium or 293T-MDSC conditioning medium (CM) as indicated. Statistical comparisons between DC and MDSC markers (n = 5) are shown, analysed using a parametric t-test; more than 10 independent experimental replicates performed with similar results achieved. Histogram on the right shows CD62L expression on conventional DCs, 293T-MDSCs, and B16-MDSCs. (B) Ly6C-Ly6G density plot profiles from conventional DCs, or MDSCs differentiated in 293T- or B16-CM as indicated. Monocyte-(M) or granulocytic-(G) MDSCs are gated within the graphs with their corresponding percentages. M-MDSCs and G-MDSCs, monocytic and granulocytic MDSCs. (C) Phenotype profiling of M-MDSC and G-MDSC populations from different sources (tumor-infiltrating MDSCs, spleen MDSCs from tumor-bearing mice, and ex vivo-produced B16-MDSCs) are shown; Data are presented as the mean ± S.D. with the percentage of expression of the indicated markers on top of each graph. (D) Mixed leukocyte reaction to measure the effect of lentivector vaccine on MDSC immunosuppressive activities. MDSCs transduced with the indicated lentivector were co-cultured with OVA-specific OT-I CD8+ T cells at a ratio of 1:10 (MDSC:T cells). Top flow cytometry density plots show IFNγ/Granzyme B (Grz B) expression profiles from OT-I cells. The bottom graph represents the percentage of IFNγ-producing OT-I cells in the MDSC-T cell assay. Statistical comparisons between groups (n = 5 per group) were performed with the parametric test one-way ANOVA; the experiment was repeated numerous times with similar results achieved. (E) T cell proliferation assay. Figure 8 (See opposite page). Number of CD8+ T cells in standard suppression assays using B16-MDSCs, represented as a bar graph. CD8+ T cells were activated with anti-CD28 anti-CD3 beads. Anti-PD1 antibody, anti-CTLA-4 antibody, and 250 pg IL12 were added as indicated. GFP indicates the addition of control supernatants (non-antibody control) from 293T cells expressing GFP. Statistical comparisons between groups (n = 3 per group) were performed with the parametric test one-way ANOVA; the experiment was repeated independently twice with similar results achieved. (F) The same as in e but plotting the amount of secreted IFNγ after 72 hours of co-culture, as assessed by ELISA. no, no treatment; IC, isotype control; b, anti-CD3/CD28 activation beads. GFP indicates the addition of supernatant from 293T-expressing GFP. Statistical comparisons between groups (n = 3 per group) were performed with the parametric test one-way ANOVA; the experiment was repeated independently twice with similar results achieved; * P < 0.05, ** P < 0.01, *** P < 0.001; data are presented as the mean ± S.D.
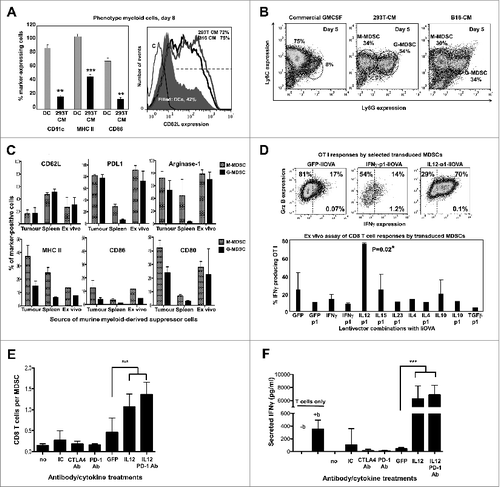
IL12 provided in trans to MDSCs has been shown to increase the expression of conventional maturation and antigen presentation markers such as CD80, CD86 and MHC II.Citation36,40,41 To determine if IL12 produced in cis by MDSCs after lentivector transduction would significantly change their phenotype, the expression of four representative maturation and lineage markers (CD86, MHC II, CD11c and Ly6G) were assayed (Fig. S1). The up-regulation of CD86 and MHC II are indicative of myeloid cell maturation.Citation17,36,40-43 Therefore, ex vivo B16-MDSCs were transduced with lentivectors expressing selected cytokines (IL12, IFNγ, IL10 and TGFβ) and p1 relative to a GFP-expressing control lentivector. Phenotypic characterization of transduced MDSCs was subsequently performed by surface staining and flow cytometry. Interestingly, no dramatic changes in the expression of these makers were observed in MDSCs transduced with lentivectors expressing IL12 (Fig. S1). In contrast, upregulation of CD86, MHC II and Ly6G was observed in response to IFNγ expression, while a tendency to exhibit decreased expression was detected in response to either IL10 or TGFβ (Fig. S1).
Nevertheless, the main targets of intratumoral lentivector injection are cancer cells. Additionally, T cells in the tumor environment are infiltrating effector T cells.Citation44 To assess whether direct transduction of MDSCs in the tumor would be required to counteract their activities over activated T cells, MDSC-activated T cell co-cultures were treated with soluble IL12 and anti-PD1 antibody. Soluble IL12 clearly overcame MDSC-T cell inhibitory activities in a standard MDSC suppression assay using anti-CD3/anti-CD28-activated CD8+ T cells as targets (). The combination of soluble IL12 and anti-PD1 antibody increased CD8+ T-cell responses as compared with IL12 alone (). Therefore, direct transduction of MDSCs would not be strictly required to counteract their suppressive activities within the tumor environment.
Concluding, although these ex vivo assays cannot be directly linked to the outcome of the disease, IL12 (a known and potent antitumor agent) stood out among the other cytokines.
Discussion
Blockade or interference with PD-L1/PD-1 interaction is becoming a very promising therapeutic procedure with good clinical outcomes in a number of human cancers.Citation45-48 In previous work, we characterised in detail the anticancer effects of PD-L1 silencing alone using lentivector vaccines. In brief, we observed that PD-L1 silencing inhibits ligand-induced TCR down-modulation in antigen-specific CD8+ T cells, by preventing the up-regulation of the E3 ubiquitin ligase Cbl-b.Citation1,12 Consequently, there is a rapid expansion of hyperactivated TCRhigh CD8+ T cells which overexpress pro-inflammatory cytokines and are strongly cytotoxic. Interference with PD-L1/PD-1 interactions with blocking antibodies has shown the same mode of action as PD-L1 silencing by lentivector transduction.Citation1,12 Thus, when we observed that PD-L1 silencing significantly attenuated tumor growth but was not sufficient to strongly prolong long-term survival,Citation1,12 we hypothesised that cytokine priming could potentiate antitumor activities of PD-L1 silencing using lentivector vaccines.Citation5
Therefore, we engineered lentiviral constructs to test a variety of cytokines that could potentially induce different T-cell responses together with delivery of a PD-L1 silencing shRNA to select the more efficacious combination. To select a treatment that could predict therapeutic potential, we performed standard characterisation experiments, which included vaccination of healthy mice to evaluate T-cell responses, followed by a melanoma B16-IiOVA model. These experiments were firstly performed with the model OVA antigen, frequently used for these studies. Rather surprisingly, all of our lentivector vaccines showed strong T-cell stimulatory activities even with anti-inflammatory cytokines or Treg-inducing cytokines. In agreement with others, we found that PD-L1 silencing significantly inhibited Treg differentiation, an activity which could counteract immunosuppressive mechanisms.Citation22 Additionally, we also previously showed that PD-L1 silencing hyperactivates T cells leading to increased IFNγ production. Consequently, all lentivector vaccines also increased survival in a transplantable B16-IiOVA melanoma model, which is extensively used in cancer research.Citation15,16,33 We further reasoned that the use of a highly immunogenic xenoantigen could obscure any significant therapeutic effect of our vaccines. Hence, we chose wild-type Trp1 because it does not possess protective or therapeutic activities in lentivector vaccines.Citation34,35 When Trp1 was targeted, one of the vaccines was clearly superior in an unmodified B16F0 melanoma model (which do not express IiOVA) in particular, the lentiviral construct based on the combination of IL12 expression and PD-L1 silencing. The therapeutic effects that we observed were modest, but of significance considering that lentivector-expressed wild-type Trp1 is not immunogenic, and that mice were vaccinated only once. In addition, using unmodified syngeneic B16F0 melanoma cells instead of B16-IiOVA circumvented the expression of strongly immunogenic xenoantigens in cancer cells that could change the outcome of anticancer immune responses.
We next sought to determine the mechanisms of action of the IL12-p1 vaccine. We found that local delivery of PD-L1 silencing shRNA amplified T-cell responses, and further, that it directly contributed to inhibition of cancer cell growth. Murine T and B cells are highly refractory to lentivector transduction in vivo,Citation12,49,50 so the potential consequences of PD-L1 silencing in T or B cells could not be evaluated. In a prophylactic setting, p1 further enhanced the antitumor activities of IL12 alone. However, the presence of p1 and the targeted antigen could not explain the antitumor effects of IL12-based lentivector vaccines after intratumoral administration. Therefore, we also studied the capacities of our lentivector vaccines to overcome the immunosuppressive activity of tumor-infiltrating MDSCs. Unfortunately, current ex vivo MDSC production protocols generate low number of cells,Citation4 and as such MDSCs are frequently isolated from a large number of tumor-bearing mice.Citation4,51,52 More importantly, MDSCs purified from spleens of tumor-bearing mice (a widely used source in most published work) are not representative of tumor-infiltrating MDSCs.Citation39 As the numbers of transduced tumor-infiltrating MDSCs were very low, we developed a protocol to differentiate and expand melanoma-infiltrating MDSCs ex vivo from mouse bone marrow. Of note, so far as we are aware, we developed one of the most efficient protocols to date to obtain myeloid cells phenocopying tumor-infiltrating MDSCs ex vivo, which routinely gives around 40-to-60 million MDSCs per mouse within five days (manuscript in preparation). We extensively characterized these ex vivo-generated B16-MDSCs and found that we could substitute them for bona fide melanoma-infiltrating MDSCs in experimental procedures (manuscript in preparation). Intriguingly, only IL12-based vaccines significantly interfered with MDSC suppressive activities on T cells. It is well known that IL12 exerts a wide range of effects within the tumor, affecting not only immune cells but also stromal and endothelial cells.Citation36,53 Curiously, only the vaccines that overcame the suppressive activities of ex vivo MDSCs showed relevant therapeutic activities in vivo. Our data would certainly agree with other published work in which IL12 can convert intratumoral myeloid cells into efficient antigen-presenting cells.Citation36
This is the first time that a single vaccination lentivector simultaneously delivers IL12, a PD-L1-targeted shRNA and a tumor antigen. Therefore, PD-L1 silencing takes place simultaneously to antigen and cytokine expression at the time of immunization. As this design is inherently immunostimulatory, no adjuvant formulation is required. In addition, TAA-specific polyclonal Class I and Class II T-cell responses are effectively achieved. Prime-boosting vaccination strategies, weekly intratumoral injection and targeting more than one TAA will surely improve the efficacy of the IL12-p1 vaccine. Although ex vivo MDSC-T cell assays cannot be directly related to the in vivo disease progression, the results from this study suggest that the early testing of treatments using ex vivo produced tumor-infiltrating MDSCs could simplify the selection of relevant therapeutic treatments. These cell numbers would also make feasible their use in high-throughput screening of multiple compounds.
In conclusion, we have shown that (1) evaluation of vaccines in healthy mice has no significant predictive value for the selection of anti-cancer treatments; (2) the use of B16-IiOVA cells as a tumor model to test anticancer immunotherapeutic regimens has very limited value for the selection of effective anticancer treatments; (3), prospective evaluation of candidate cancer treatments using ex vivo differentiated MDSCs may highlight treatments with significant therapeutic potential; and (4), in the current study, IL12-encoding combined with PD-L1 silencing lentivector vaccines demonstrated promising anti-melanoma activities.
Materials and Methods
Cells and mice
293T, B16F0 cells and bone marrow-derived dendritic cells (BM-DCs) were grown as previously described.Citation17 The mouse strains C57BL/6 mice, OT-I and OT-II mice containing OVA-specific CD8+ and CD4+ T cells.Citation12,19 293T-GM-CSF and B16F0-GM-CSF were generated by transduction using lentivectors co-expressing mouse GM-CSF and puromycin resistance and selected with 3 μg/mL puromycin (SIGMA, cat #P8833). MDSCs were expanded from BM cells extracted from femurs and tibias of C57BL/6 mice, following a similar protocol as for DC differentiation Citation17,19 but including 75% conditioning medium (CM) from the GM-CSF-producing cells. B16F0 cells were obtained from UCL´s cancer cell repository. 293T cells were purchased from the American Type Cell Culture Collection (ATCC) and generously donated by Prof. Mary Collins (UCL). Passage 4 cells were thawed and used for the experiments. Approval for animal studies was obtained from the University College London Animal Ethics Committee.
Immunoblot
Immunoblot was performed as previously described,Citation17 using HA-specific rabbit antibodies (Sigma, cat #H6908), and mouse anti-tyrosinase, TRP1, TRP2 and Gp100-specific antibodies (Abcam, cat #ab738, #ab3312, and #ab137078). Mouse anti-iNOS antibodies were purchased from Cell Signaling (cat #2982), and anti-GADPH from Calbiochem (cat #CB1001-500UG). Peroxidase-conjugated anti-mouse and anti-rabbit antibodies were purchased from DAKO (cat #Z0259, Z0196).
Plasmids, lentivector production, titration and cell transduction
Dual lentivectors co-expressing 2 genes and a microRNA have been previously described.Citation12,19 Mouse cytokine genes were inserted under the transcriptional control of the spleen focus-forming virus (SFFV) promoter following standard cloning techniques. For IL12 and IL23 containing constructs, their 2 component cytokine chains were fused using the following linker sequence: GGCAGTACTTCGGGCAGTGGTAAGCCTGGTAGTGGTGAGGGTAGTACTAAGGGT. The second transgene was expressed from the human ubiquitin promoter (UBI) and corresponded to GFP, HA-tagged IiOVA Citation17,19 or HA-tagged IiTrp1. This last transgene was engineered by standard cloning techniques as a fusion between the N-terminus of the MHC II invariant chain (Ii) with the HA-tagged mouse tyrosinase related protein (Trp1) gene. The PD-L1-specific short-hairpin RNA (shRNA) designated p1, was 5′-AAGGTATATTGCTGTTGACAGTGAGCGCAACCGAAATGATACACAATTCTAGTGAAGCCACAGATGTAGAATTGTGTATCATTTCGGTTTTGCCTACTGCCTCG-3′. When indicated, p1 was removed by NotI digestion.Citation12,19, The target sequence for the haematopoietic endogenous microRNA142-3pCitation31 was included in the indicated pDUAL plasmids and integrated at the XhoI restriction site as follows: 5′-CGGCCGCGACTCTAGAGTCGACTCCA TAAAGTAGGAAACACTACACGATTCCATAAAGTAGGAAACACTACAACCGGTTCCATAAAGTAGGAAACACTACATCACTCCATAAAGTAGGAAACACTACACTAGAGTCGACCTGCAGGCGGCCGCGAATTCACTAGTGATTGGCCGCAGTCGACCTGCAGGCATGCAAGCTTGATATC-3′. Lentivectors were produced and titrated as described.Citation54,55 BMDCs and MDSCs were transduced using previously described protocols.Citation17
Cell staining and flow cytometry
Surface and intracellular staining were performed as described previouslyCitation17 using the following fluorophore (or biotinylated) antibodies.Citation17 From BioLegend: Alexa Fluor 488-Ly6C (#128021), PE-CD3 (100205), PE-CD86 (105007), APC-CD80 (104713), APC-I-A/I-F (107613), Biotin-ICAM I (116103), Biotin-H2kb (116503), Alexa Fluor 488-IFN-γ (#505815), Biotin-PD-L1 (#124305), PE-Cy7-Ly6G (#127618), PE-IL6 (#504503), PE-CD34 (#128609), PE-CD14 (#123309), PE-CD3 (#100205), PE-F4/80 (#123109), Biotin-IL12/IL23 (#505301), Biotin-IL15 (#515103), PE-IL17A (#506903), PE-Cy7-streptavidin (#405206), AF647-Foxp3 (#320014). From eBioscience: PE-CD4 (#12-0042-82), FITC-IA/IE (#107605); APC-IL10 (#17-7101-81). From Raybiotech: FITC-Class I H-2Kb (#129-10449). From BD Bioscience Pharmingen: v500-CD4 (#560783), PE-CD8alpha (#553032), APC-CD11b (#557686), Biotin-IL12 (#554476), AF647-Ki67 (558615). From Southern Biotech: PE-Cy7-CD25 (#1595-17). From LSBio: PE-IL4 (#35203). From Invitrogen: APC-CD11c (#MCD11C05), APC-granzyme B (#GRB05). From AbD Serotec: PE-CD62L (#MCA1259PE); From R&D Systems: anti-human/mouse PE-Arginase 1 (#IC5868P). For indirect staining, the following streptavidin conjugated antibodies were used as the detection reagent: APC-streptatividin (BioLegend, #405207), PE-streptavidin (Invitrogen, #S866), FITC-streptavidin (Invitrogen, #SA1001).
When tumor sizes were between 50 mm2 and 140 mm2, single cell suspensions were obtained using the GentleMACS dissociator system (Miltenyi Biotec, 130-093-235). Tumor and spleen cells were stained by incubating with the above fluorophore conjugated antibodies. Cytofluorimetric analysis was performed on a LSR Fortessa (BD Biosciences) flow cytometer. CD45+ CD11b+ populations were gated and analysed as indicated.
In vitro Suppression Assays
Two types of in vitro suppression assays were performed. The first one tested the capacity of MDSCs to dampen a mixed lymphocyte reaction (MLR), and the second one tested their ability to suppress anti-CD3/anti-CD28-dependent T-cell proliferation. T lymphocytes were isolated from the spleen of C57BL/6 or BALB/c mice using CD8α and CD4+ T cell Isolation Kits II (Miltenyi Biotec, #130-095-236, #130-095-248). If required, CD8+ T lymphocytes were labeled with CFSE (Invitrogen, C34554). Cells were plated at 10Citation5 cells in 100 μL per well in a 96 well plate. Subsequently, the cells were either left unstimulated or were stimulated with a 1/800 dilution of anti-CD3/anti-CD28 coated beads (Invitrogen, #111.61D). MDSCs were either used in bulk or sorted using the MDSC Isolation Kit (Miltenyi Biotec, #130-094-538). When indicated, supernatants containing IL12 were added at 250 pg per well while monoclonal antibodies against CTLA-4, PD1, and control hamster IgG (BioXCell, #UC10-4F10-11, #BE0033-2, #BE0091) were added at 10 μg/mL per well. CFSE dilution was evaluated 3 days later by flow cytometry as a measure of T-cell proliferation. Data was collected using the FACSCanto Flow Cytometer (BD Biosciences) and analyzed with FACSDiva or FlowJo software. Supernatants were collected and screened for IFNγ content using a standard ELISA kit (eBioScience, #88-7314-22).
Vaccination, T cell responses and tumor experiments
All vaccination experiments were repeated independently at least twice unless stated otherwise, using groups of 5 mice, as described.Citation17,21,22 IFNγ ELISPOT assays were performed as previously described.Citation17 Five naïve C57BL/6 mice per group were vaccinated subcutaneously at the base of the tail with 5 × 106 −1 × 107 lentivector transducing particles. Splenocytes were isolated and cultured overnight in the presence of Class I and II OVA peptides.Citation19 Th1/2/17 and CTL responses were assessed by flow cytometry using the appropriate antibody combinations, as previously described.Citation12,17,19,21 T cell responses were also evaluated by ELISPOT assay when indicated, as previously described.Citation12
To carry out therapeutic experiments (first inducing tumor growth, followed by lentivector vaccinations), 3 × 105 B16F0 cells/mouse were firstly subcutaneously transferred at the base of the tail and tumors were allowed to grow. When tumors reached sizes between 50 and 200 mm3, 1 × 107 lentivector transducing particles (as assessed by q-PCR)Citation54,55 were intratumorally injected and tumor size was monitored every 2 days. Mice were sacrificed when tumor volume was above 2500 mm3 (humane endpoint). For prophylactic experiments (first lentivector vaccination, followed by subcutaneous transfer of tumor cells, both at the base of the tail), mice were vaccinated with a dose of 107 lentivector transducing particles per mouse. Two weeks later, 2 × 106 B16 tumor cells per mouse were subcutaneously transferred. Long-term survivors were re-challenged with tumor cells to prove immunity.
To quantify Trp1-specific T-cell responses following lentivector vaccination using this TAA antigen, 3 × 105 BM-DCs were transduced with a GFP-IiTrp1 lentivector for 2 days, treated with 100 ng/mL LPS for 4 hours, and co-cultured overnight with 3 × 106 splenocytes from vaccinated mice. GolgiPlug (BD Biosciences, San Jose, California, United States) was added for 4 hours before intracellular staining of T cells.
Statistical analyses
GraphPad Prism, Salstat and SPSS software packages were used for plotting data and statistical analyses. No data were considered an outlier. 5 mice per group were used following the results of Mead's resource equation for power analysis. In vivo treatments were repeated twice as a minimum, unless stated otherwise. ELISPOT data and mean fluorescence intensities from surface or intracellular staining from multiple groups were analysed by one-way ANOVA followed by a Tukey´s a posteriori test, as previously described.Citation17,19 Survival data from tumor experiments were compared using the Log-Rank test, as previously described.Citation17 Tumor sizes and lifespans were compared with the non-parametric U test of Mann-Whitney or Kruskall Wallis, as previously described.Citation12 In some cases, data were pooled to increase the statistical sensitivity. Percentages of T cells in MLRs were normally distributed and analysed with one-way ANOVA followed by Tukey´s test. Three independent reactions per group were used for these experiments. Significant, very significant and highly significant differences were indicated in the figures with *, ** or *** when the associated probability was P < 0.05 (2-tailed, alpha=0.05, n ≥ 5), P < 0.01 (2-tailed alpha=0.01, n ≥ 5) and P < 0.001 (2-tailed alpha=0.001, n ≥ 5), respectively.
Disclosure of Potential Conflicts of Interest
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. David Escors and Therese Liechtenstein are inventors of the MDSC production system, and the therapeutic lentivector constructs described in this work. A patent on this method has been filed. The other authors disclose no conflicts of interest.
945378_fig_s1.docx
Download MS Word (10.2 KB)945378_Figure_S1.tiff
Download TIFF Image (271.2 KB)Acknowledgments
We sincerely thank Frederick Arce, Grazyna Kochan and Maria Gato for their useful discussions and suggestions.
Additional information
Funding
References
- Karwacz K, Arce F, Bricogne C, Kochan G, Escors D. PD-L1 co-stimulation, ligand-induced TCR down-modulation and anti-tumor immunotherapy. Oncoimmunology 2012; 1:86-8; PMID:22318430; http://dx.doi.org/10.4161/onci.1.1.17824
- Campos-Perez J, Rice J, Escors D, Collins M, Paterson A, Savelyeva N, Stevenson FK. DNA fusion vaccine designs to induce tumor-lytic CD8+ T-cell attack via the immunodominant cysteine-containing epitope of NY-ESO 1. Int J Cancer 2013; 133:1400-7; PMID:23494538; http://dx.doi.org/10.1002/ijc.28156
- Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 2009; 9:162-74; PMID:19197294; http://dx.doi.org/10.1038/nri2506
- Escors D, Liechtenstein T, Perez-Janices N, Schwarze J, Dufait I, Goyvaerts C, Lanna A, Arce F, Blanco-Luquin I, Kochan G et al. Assessing T-cell responses in anticancer immunotherapy: dendritic cells or myeloid-derived suppressor cells? Oncoimmunology 2013; 12:e26148.
- Liechtenstein T, Dufait I, Lanna A, Breckpot K, Escors D. Modulating co-stimulation during antigen presentation to enhance cancer immunotherapy. Immunol, Endocr Metab Agent Med Chem 2012; 12:224-35; http://dx.doi.org/10.2174/187152212802001875
- Latchman YE, Liang SC, Wu Y, Chernova T, Sobel RA, Klemm M, Kuchroo VK, Freeman GJ, Sharpe AH. PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proc Natl Acad Sci U S A 2004; 101:10691-6; PMID:15249675; http://dx.doi.org/10.1073/pnas.0307252101
- Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev 2010; 236:219-42; PMID:20636820; http://dx.doi.org/10.1111/j.1600-065X.2010.00923.x
- Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol 2007; 8:239-45; PMID:17304234; http://dx.doi.org/10.1038/ni1443
- Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity 2007; 27:111-22; PMID:17629517
- Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol 2005; 23:515-48; PMID:15771580; http://dx.doi.org/10.1146/annurev.immunol.23.021704.115611
- Wang L, Pino-Lagos K, de Vries VC, Guleria I, Sayegh MH, Noelle RJ. Programmed death 1 ligand signaling regulates the generation of adaptive Foxp3+CD4 +regulatory T cells. Proc Natl Acad Sci U S A 2008; 105:9331-6; PMID:18599457; http://dx.doi.org/10.1073/pnas.0710441105
- Karwacz K, Bricogne C, Macdonald D, Arce F, Bennett CL, Collins M, Escors D. PD-L1 co-stimulation contributes to ligand-induced T cell receptor down-modulation on CD8(+) T cells. EMBO Mol Med 2011; 3:581-92; PMID:21739608; http://dx.doi.org/10.1002/emmm.201100165
- Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol 2008; 181:5791-802; PMID:18832739; http://dx.doi.org/10.4049/jimmunol.181.8.5791
- Bouwer AL, Saunderson SC, Caldwell FJ, Damani TT, Pelham SJ, Dunn AC, Jack RW, Stoitzner P, McLellan AD. NK cells are required for dendritic cell-based immunotherapy at the time of tumor challenge. J Immunol 2014; 192:2514-21; PMID:24477907; http://dx.doi.org/10.4049/jimmunol.1202797
- Fraser CK, Lousberg EL, Guerin LR, Hughes TP, Brown MP, Diener KR, Diener KR, Hayball JD. Dasatinib alters the metastatic phenotype of B16-OVA melanoma in vivo. Cancer Biol Ther 2010; 10:715-27; PMID:20676039; http://dx.doi.org/10.4161/cbt.10.7.12926
- He Y, Zhang J, Mi Z, Robbins P, Falo LD, Jr. Immunization with lentiviral vector-transduced dendritic cells induces strong and long-lasting T cell responses and therapeutic immunity. J Immunol 2005; 174:3808-17; PMID:15749922; http://dx.doi.org/10.4049/jimmunol.174.6.3808
- Escors D, Lopes L, Lin R, Hiscott J, Akira S, Davis RJ, Collins MK. Targeting dendritic cell signalling to regulate the response to immunisation. Blood 2008; 111:3050-61; PMID:18180378; http://dx.doi.org/10.1182/blood-2007-11-122408
- Liechtenstein T, Dufait I, Bricogne C, lanna A, Pen J, Breckpot K, Escors D. PD-L1/PD-1 co-stimulation, a brake for T cell activation and a T cell differentiation signal. J Clin Cell Immunol 2012; S12: 006:6; PMID:23525238
- Arce F, Breckpot K, Stephenson H, Karwacz K, Ehrenstein MR, Collins M, Escors D. Selective ERK activation differentiates mouse and human tolerogenic dendritic cells, expands antigen-specific regulatory T cells, and suppresses experimental inflammatory arthritis. Arthritis Rheum 2011; 63:84-95; PMID:20967853; http://dx.doi.org/10.1002/art.30099
- Vacchelli E, Vitale I, Tartour E, Eggermont A, Sautes-Fridman C, Galon J, Zitvogel L, Kroemer G, Galluzzi L. Trial Watch: Anticancer radioimmunotherapy. Oncoimmunology 2013; 2:e25595; PMID:24319634
- Goold HD, Escors D, Conlan TJ, Chakraverty R, Bennett CL. Conventional dendritic cells are required for the activation of helper-dependent CD8 T cell responses to a model antigen after cutaneous vaccination with lentiviral vectors. J Immunol 2011; 186:4565-72; PMID:21389256; http://dx.doi.org/10.4049/jimmunol.1002529
- Breckpot K, Escors D, Arce F, Lopes L, Karwacz K, Van Lint S, Keyaerts M, Collins M. HIV-1 lentiviral vector immunogenicity is mediated by Toll-like receptor 3 (TLR3) and TLR7. J Virol 2010; 84:5627-36; PMID:20237085; http://dx.doi.org/10.1128/JVI.00014-10
- Esslinger C, Chapatte L, Finke D, Miconnet I, Guillaume P, Levy F, MacDonald HR. In vivo administration of a lentiviral vaccine targets DCs and induces efficient CD8(+) T cell responses. J Clin Invest 2003; 111:1673-81; PMID:12782670; http://dx.doi.org/10.1172/JCI200317098
- Matsui H, Hegadorn C, Ozelo M, Burnett E, Tuttle A, Labelle A, McCray PB Jr, Naldini L, Brown B, Hough C, Lillicrap D. A microRNA-regulated and GP64-pseudotyped lentiviral vector mediates stable expression of FVIII in a murine model of Hemophilia A. Mol Ther 2011; 19:723-30; PMID:21285959; http://dx.doi.org/10.1038/mt.2010.290
- Goudy KS, Annoni A, Naldini L, Roncarolo MG. Manipulating Immune Tolerance with Micro-RNA Regulated Gene Therapy. Front Microbiol 2011; 2:221; PMID:22144977; http://dx.doi.org/10.3389/fmicb.2011.00221
- Gentner B, Visigalli I, Hiramatsu H, Lechman E, Ungari S, Giustacchini A, Schira G, Amendola M, Quattrini A, et al. Identification of hematopoietic stem cell-specific miRNAs enables gene therapy of globoid cell leukodystrophy. Sci. trans med 2010; 2:58-84; http://dx.doi.org/10.1126/scitranslmed.3001522
- Brown BD, Gentner B, Cantore A, Colleoni S, Amendola M, Zingale A, Baccarini A, Lazzari G, Galli C, Naldini L. Endogenous microRNA can be broadly exploited to regulate transgene expression according to tissue, lineage and differentiation state. Nat Biotechnol 2007; 25:1457-67; PMID:18026085; http://dx.doi.org/10.1038/nbt1372
- Annoni A, Brown BD, Cantore A, Sergi LS, Naldini L, Roncarolo MG. In vivo delivery of a microRNA-regulated transgene induces antigen-specific regulatory T cells and promotes immunologic tolerance. Blood 2009; 114:5152-61; PMID:19794140
- Brown BD, Cantore A, Annoni A, Sergi LS, Lombardo A, Della Valle P, D'Angelo A, Naldini L. A microRNA-regulated lentiviral vector mediates stable correction of hemophilia B mice. Blood 2007; 110:4144-52; PMID:17726165; http://dx.doi.org/10.1182/blood-2007-03-078493
- Brown BD, Sitia G, Annoni A, Hauben E, Sergi Sergi L, Zingale A, Roncarolo MG, Guidotti LG, Naldini L. In vivo administration of lentiviral vectors triggers a type I interferon response that restricts hepatocyte gene transfer and promotes vector clearance. Blood 2006; 109:2797-805.
- Brown BD, Venneri MA, Zingale A, Sergi Sergi L, Naldini L. Endogenous microRNA regulation suppresses transgene expression in hematopoietic lineages and enables stable gene transfer. Nat Med 2006; 12:585-91; PMID:16633348; http://dx.doi.org/10.1038/nm1398
- Karwacz K, Mukherjee S, Apolonia L, Blundell MP, Bouma G, Escors D, Collins MK, Thrasher AJ. Nonintegrating lentivector vaccines stimulate prolonged T-cell and antibody responses and are effective in tumor therapy. J Virol 2009; 83:3094-103; PMID:19176629; http://dx.doi.org/10.1128/JVI.02519-08
- Aranda F, Llopiz D, Diaz-Valdes N, Riezu-Boj JI, Bezunartea J, Ruiz M, Martínez M, Durantez M, Mansilla C, Prieto J. et al. Adjuvant combination and antigen targeting as a strategy to induce polyfunctional and high-avidity T-cell responses against poorly immunogenic tumors. Cancer Res 2011; 71:3214-24; PMID:21402711
- Liu Y, Peng Y, Mi M, Guevara-Patino J, Munn DH, Fu N, He Y. Lentivector immunization stimulates potent CD8 T cell responses against melanoma self-antigen tyrosinase-related protein 1 and generates antitumor immunity in mice. J Immunol 2009; 182:5960-9; PMID:19414747
- Guevara-Patino JA, Engelhorn ME, Turk MJ, Liu C, Duan F, Rizzuto G, Cohen AD, Merghoub T, Wolchok JD, Houghton AN. Optimization of a self antigen for presentation of multiple epitopes in cancer immunity. J Clin Invest 2006; 116:1382-90; PMID:16614758; http://dx.doi.org/10.1172/JCI25591
- Kerkar SP, Goldszmid RS, Muranski P, Chinnasamy D, Yu Z, Reger RN, Leonardi AJ, Morgan RA, Wang E, Marincola FM, et al. IL-12 triggers a programmatic change in dysfunctional myeloid-derived cells within mouse tumors. J Clin Invest 2011; 121:4746-57; PMID:22056381; http://dx.doi.org/10.1172/JCI58814
- Emeagi PU, Van Lint S, Goyvaerts C, Maenhout S, Cauwels A, McNeish IA, Bos T, Heirman C, Thielemans K, Aerts JL, Breckpot K. Proinflammatory characteristics of SMAC/DIABLO-induced cell death in antitumor therapy. Cancer Res 2012; 72:1342-52; PMID:22379024; http://dx.doi.org/10.1158/0008-5472.CAN-11-2400
- Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, De Baetselier P, Van Ginderachter JA. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood 2008; 111:4233-44; PMID:18272812; http://dx.doi.org/10.1182/blood-2007-07-099226
- Maenhout SK, Van Lint S, Emeagi PU, Thielemans K, Aerts JL. Enhanced suppressive capacity of tumor-infiltrating myeloid-derived suppressor cells compared to their peripheral counterparts. Int J Cancer 2013; 134:1077-90; PMID:23983191; http://dx.doi.org/10.1002/ijc.28449
- Thaci B, Ahmed AU, Ulasov IV, Wainwright DA, Nigam P, Auffinger B, Tobias AL, Han Y, Zhang L, Moon KS, et al. Depletion of myeloid-derived suppressor cells during interleukin-12 immunogene therapy does not confer a survival advantage in experimental malignant glioma. Cancer Gene ther 2014; 21:38-44; PMID:24434573; http://dx.doi.org/10.1038/cgt.2013.81
- Steding CE, Wu ST, Zhang Y, Jeng MH, Elzey BD, Kao C. The role of interleukin-12 on modulating myeloid-derived suppressor cells, increasing overall survival and reducing metastasis. Immunology 2011; 133:221-38; PMID:21453419; http://dx.doi.org/10.1111/j.1365-2567.2011.03429.x
- Breckpot K, Escors D. Dendritic cells for active anti-cancer immunotherapy: targeting activation pathways through genetic modification. Endocr, Metab Immun Disord Drug Targets 2009; 9:328-43; http://dx.doi.org/10.2174/187153009789839156
- Kochan G, Escors D, Breckpot K, Guerrero-Setas D. Role of non-classical MHC class I molecules in cancer immunosuppression. Oncoimmunology 2013; 2:e26491.
- Zhou Q, Xiao H, Liu Y, Peng Y, Hong Y, Yagita H, Chandler P, Munn DH, Mellor A, Fu N, et al. Blockade of programmed death-1 pathway rescues the effector function of tumor-infiltrating T cells and enhances the antitumor efficacy of lentivector immunization. J Immunol 2010; 185:5082-92; PMID:20926790; http://dx.doi.org/10.4049/jimmunol.1001821
- Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012; 366:2455-65; PMID:22658128; http://dx.doi.org/10.1056/NEJMoa1200694
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012; 366:2443-54; PMID:22658127; http://dx.doi.org/10.1056/NEJMoa1200690
- Vacchelli E, Eggermont A, Galon J, Sautes-Fridman C, Zitvogel L, Kroemer G, Galluzzi L. Trial watch: Monoclonal antibodies in cancer therapy. Oncoimmunology 2013; 2:e22789.
- Aranda F, Vacchelli E, Eggermont A, Galon J, Fridman WH, Zitvogel L, Kroemer G, Galluzzi L. Trial watch: Immunostimulatory monoclonal antibodies in cancer therapy. Oncoimmunology 2014; 3:e27297
- Arce F, Rowe HM, Chain B, Lopes L, Collins MK. Lentiviral vectors transduce proliferating dendritic cell precursors leading to persistent antigen presentation and immunization. Mol Ther 2009; 17:1643-50; PMID:19584812; http://dx.doi.org/10.1038/mt.2009.149
- Escors D, Breckpot K. Lentiviral vectors in gene therapy: their current status and future potential. Arch Immunol Ther Ex 2010; 58:107-19; PMID:20143172; http://dx.doi.org/10.1007/s00005-010-0063-4
- Youn JI, Kumar V, Collazo M, Nefedova Y, Condamine T, Cheng P, Villagra A, Antonia S, McCaffrey JC, Fishman M, et al. Epigenetic silencing of retinoblastoma gene regulates pathologic differentiation of myeloid cells in cancer. Nat Immunol 2013; 14:211-20; PMID:23354483; http://dx.doi.org/10.1038/ni.2526
- Lutz MB, Kukutsch NA, Menges M, Rossner S, Schuler G. Culture of bone marrow cells in GM-CSF plus high doses of lipopolysaccharide generates exclusively immature dendritic cells which induce alloantigen-specific CD4 T cell anergy in vitro. Eur J Immunol 2000; 30:1048-52; PMID:10760792; http://dx.doi.org/10.1002/(SICI)1521-4141(200004)30:4%3c1048::AID-IMMU1048%3e3.0.CO;2-W
- Kerkar SP, Leonardi AJ, van Panhuys N, Zhang L, Yu Z, Crompton JG, Pan JH, Palmer DC, Morgan RA, Rosenberg SA. et al. Collapse of the tumor stroma is triggered by IL-12 induction of Fas. Mol Ther 2013; 21:1369-77; PMID:23568260; http://dx.doi.org/10.1038/mt.2013.58
- Selden C, Mellor N, Rees M, Laurson J, Kirwan M, Escors D, Collins M, Hodgson H. Growth factors improve gene expression after lentiviral transduction in human adult and fetal hepatocytes. J Gene Med 2007; 9:67-76; PMID:17310477; http://dx.doi.org/10.1002/jgm.1000
- Kochan G, Escors D, Gonzalez JM, Casasnovas JM, Esteban M. Membrane cell fusion activity of the vaccinia virus A17-A27 protein complex. Cell Microbiol 2008; 10:149-64; PMID:17708756