
Abstract
The present study supports a model in which Pten loss-induced senescence is hindered in prostate tumor cells by non cell-autonomous mechanisms. Indeed, paracrine signaling by tumor-infiltrating CD11b+Gr-1+ myeloid cells triggers senescence evasion in prostate lesions of Pten-null mice, eventually promoting tumor progression.
Cellular senescence is a stable growth arrest that constrains cell cycle progression in case proliferation becomes aberrant, thus limiting tumorigenesis.Citation1 Oncogenic mutations and loss of tumor-suppressor genes that cause uncontrolled cell-cycle progression are required events in tumor development. Nevertheless, many of these genetic alterations retain an intrinsic tumor-suppressive potential that gives rise to oncogene-induced senescence (OIS).Citation2 This type of cellular senescence response represents a complex combination of events occurring in vivo, as demonstrated in several mouse models.Citation3 In addition, tumor cell senescence can also be promoted by anticancer drugs that either enhance DNA damage, or alternatively, target genes involved in the regulation of pro-senescence pathways.Citation4 Interestingly, tumor-infiltrating T helper 1 cells can also drive senescence in cancers in a cytokine-induced manner by secreting interferon γ (IFNγ) and tumor necrosis α (TNFα) into the tumor microenvironmentCitation5 (). These lines of evidence support the idea that a strong interplay occurs between cancer cells, senescence and tumor-infiltrating immune populations thus opening the possibility to tune such interplay for cancer treatment.
Figure 1. A model of tumor heterogeneity and senescence evasion. Whereas both intrinsic and extrinsic mechanisms induce cellular senescence in the tumor tissue that constrains neoplastic cell growth and limits tumorigenesis, multiple events trigger senescence evasion in tumor cells, thus breaking down the barrier to cell proliferation and cancer progression.
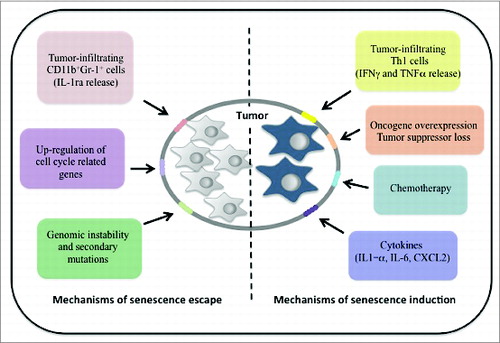
Notably, senescence is enriched in pre-malignant lesions, but it generally disappears in malignant tissues,Citation6 suggesting that intrinsic or extrinsic factors determine the propensity of cells to evade the senescence response, thereby eventually promoting tumor progression. Senescence by-pass also occurs during anticancer chemotherapy, accounting for tumor progression and relapse.Citation4 Remarkably, although mounting evidence has previously demonstrated that senescence induction can be overcome by accumulation of secondary mutations in tumor cells, the possibility that extrinsic factors in the tumor microenvironment could also be involved in senescence evasion has not been taken into accountCitation7 (). This is of particular relevance in Pten loss-induced cellular senescence (PICS) that, contrary to OIS, is not preceded by a hyper-proliferative phase and accumulation of DNA damage in vitro, suggesting that senescence evasion in this context is unlikely to occur by accumulation of additional mutations.Citation8 Nevertheless, experiments in transgenic Pten-null and K-RAS+/G12 mice in which Pten deletion is conditionally driven in prostate and K-RAS+/G12 is overexpressed in lung respectively, have suggested that senescence is evaded at early stages of tumorigenesis in vivo.Citation9 This is despite the fact that in the Pten-/- prostate cancer model, complete inactivation of the tumor-suppressor gene Pten in the mouse prostate epithelium induces a strong senescence response at the onset of tumor formation, accompanied by the presence of a proliferative cellular compartment.Citation8 As such heterogeneity can be observed at the earliest stages of tumorigenesis, we reasoned that a cause other than genetic mutations may trigger the senescence escape in such tumors, and that senescence evasion could be attributed to a cell extrinsic mechanism. In a recent manuscript, we definitively provided the first evidence that cellular senescence in tumor cells can be evaded by non cell-autonomous mechanisms and that tumor-infiltrating immune cells can oppose senescence in a paracrine manner in vivo. As Pten-null tumors are strongly infiltrated by CD11b+Gr1+ myeloid cells, we speculated that such a population could be the source of factors that hinder the senescence response. Notably, immunofluorescence analysis showed that the spatial localization of the myeloid compartment was associated with the highest expression of proliferation markers from the prostatic epithelia. Interestingly, when Pten null mice were adoptively transferred with green fluorescence protein-tagged (UBC-GFP) myeloid precursors, we were able to detect an impressive recruitment of the Gr1+ myeloid subset from the peripheral blood to the tumor, again confirming the localization of the immune cells in close proximity to the Pten null proliferating epithelia. Importantly, such recruitment proved the capability of the Pten null tumor to tune its secretome in order to attract these myeloid cells from the periphery and generate immunotolerance at the tumor site. Furthermore, unpublished pieces of evidence from our laboratory indicate that Pten null tumors are infiltrated by diverse immunosuppressive subsets, such as regulatory T cells and M2-polarized macrophages, supporting the notion that a specific genetic background may have a strong impact on the surrounding immunological network. Strikingly, conditioned media from Gr-1+ cells cultured in vitro were capable of impairing cell senescence in Pten null MEFs (mouse embryonic fibroblasts), demonstrating that Gr-1+ cells can oppose cell senescence in a paracrine manner. Furthermore, cytokine profiles obtained from myeloid cells infiltrating the tumor (as compared to the prostate epithelia) underscored a strong expression of the interleukin IL-1 receptor antagonist (IL-1ra), pointing to the fact that this cytokine may be involved in hindering the senescence response in target cells. Indeed, IL-1ra has been previously reported to impair oncogene-induced senescence in vitro by blocking IL-1α mediated signaling.Citation10 Interestingly, Pten null mice adoptively transferred with IL-1ra-null bone marrow precursors, displayed an almost complete normalization of glands affected by prostatic intraepithelial neoplasia, which was associated with decreased cell proliferation and increased senescence response. Notably, IL-1α signaling was strongly enhanced in the tumor context and peritoneal administration of IL-1α in mice was shown to powerfully potentiate the senescence response in the prostatic epithelia, thus confirming the pro-senescence potential of this cytokine in such a genetic background.Citation9
Importantly, our study unveiled a completely novel mechanism through which senescence may be modulated in vivo. Strikingly, our data indicate that senescence evasion by tumor cells can also occur in a non cell-autonomous manner, thus overcoming prior evidences that commonly ascribed senescence evasion to cellular intrinsic factors.Citation7 Our findings support a model where Pten loss-induced senescence is buffered in vivo by the presence of myeloid cells in the tumor microenvironment. As a consequence Pten-null cells continue to proliferate and accumulate secondary mutations, thus allowing tumor invasiveness.
Our data provide insights into a novel mechanism of senescence escape that could be harnessed therapeutically. Importantly, administration of an antagonist against the chemokine receptor CXCR2, previously described to hinder Gr-1+ cells migration, in combination with the chemotherapeutic agent docetaxel, blocked tumor growth and potentiated the efficacy of chemotherapy-induced senescence. Notably, these findings implicate tumor-infiltrating myeloid cells as drivers of chemoresistance in prostate cancer, thus suggesting that these immune cells play an additional role in tumor progression. Further studies are therefore essential in order to evaluate whether myeloid infiltration may be considered a biological marker to be taken into account when planning chemotherapeutic intervention for the treatment of prostate cancer. Furthermore, whether, or how, different tumor types, heterogeneous genetic backgrounds, or diverse metabolic states may uniquely shape their immune microenvironment, remains to be elucidated. Consequently, a comprehensive study of the relation existing between tumor diversity and recruitment of immune subsets may provide a new starting point to develop personalized therapeutic strategies against cancer.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer 2010; 10:51-7; PMID:20029423; http://dx.doi.org/10.1038/nrc2772
- Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature 2004; 432:307-15; PMID:15549092; http://dx.doi.org/10.1038/nature03098
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997; 88:593-602; PMID:9054499; http://dx.doi.org/10.1016/S0092-8674(00)81902-9
- Ewald JA, Desotelle JA, Wilding G, Jarrard DF. Therapy-induced senescence in cancer. J Natl Cancer Inst 2010; 102:1536-46; PMID:20858887; http://dx.doi.org/10.1093/jnci/djq364
- Braumuller H, Wieder T, Brenner E, Assmann S, Hahn M, Alkhaled M, Schilbach K, Essmann F, Kneilling M, Griessinger C, et al. T-helper-1-cell cytokines drive cancer into senescence. Nature 2013; 494:361-5; PMID:23376950; http://dx.doi.org/10.1038/nature11824
- Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005; 436:725-30; PMID:16079851; http://dx.doi.org/10.1038/nature03918
- Saab R. Senescence and pre-malignancy: how do tumors progress? Semin Cancer Biol 2011; 21:385-91; PMID:21982725; http://dx.doi.org/10.1016/j.semcancer.2011.09.013
- Alimonti A, Nardella C, Chen Z, Clohessy JG, Carracedo A, Trotman LC, Cheng K, Varmeh S, Kozma SC, Thomas G, et al. A novel type of cellular senescence that can be enhanced in mouse models and human tumor xenografts to suppress prostate tumorigenesis. J Cli Invest 2010; 120:681-93; PMID:20197621; http://dx.doi.org/10.1172/JCI40535
- Di Mitri D, Toso A, Chen JJ, Sarti M, Pinton S, Jost TR, D'Antuono R, Montani E, Garcia-Escudero R, Guccini I, et al. Tumour-infiltrating Gr-1+ myeloid cells antagonize senescence in cancer. Nature 2014; 515:134-7; PMID:25156255; http://dx.doi.org/10.1038/nature13638
- Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, Athineos D, Kang TW, Lasitschka F, Andrulis M, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol 2013; 15:978-90; PMID:23770676; http://dx.doi.org/10.1038/ncb2784