
Abstract
Despite a major improvement in the treatment of advanced kidney cancer by the recent introduction of targeted agents such as multi-kinase inhibitors, long-term benefits are still limited and a significant unmet medical need remains for this disease. Cancer immunotherapy has shown its potential by the induction of long-lasting responses in a small subset of patients, however, the unspecific immune interventions with (high dose) cytokines used so far are associated with significant side effects. Specific cancer immunotherapy may circumvent these problems by attacking tumor cells while sparing normal tissue with the use of multi-peptide vaccination being one of the most promising strategies. We here summarize the clinical and translational data from phase I and II trials investigating IMA901. Significant associations of clinical benefit with detectable T cell responses against the IMA901 peptides and encouraging survival data in treated patients has prompted the start of a randomized, controlled phase III trial in 1st line advanced RCC with survival results expected toward the end of 2015. Potential combination strategies with the recently discovered so-called checkpoint inhibitors are also discussed.
Abbreviations
| μg | = | Microgram |
| 5-FU | = | 5 fluorouracil |
| AE | = | Adverse event |
| ccRCC | = | Clear cell renal cell carcinoma |
| CTL | = | Cytotoxic T-lymphocyte |
| CY | = | Cyclophosphamide |
| DC | = | Dendritic cell |
| DCR | = | Disease control rate |
| ECG | = | Electrocardiogram |
| ELISpot | = | Enzyme-linked immunospot assay |
| FDA | = | Food and Drug Administration |
| HBV | = | Hepatitis B virus |
| HLA | = | Human leukocyte antigen |
| i.d. | = | intradermal; intradermally |
| IFN | = | Interferon |
| IL | = | Interleukin |
| mg | = | Milligram |
| MHC | = | Major histocompatibility complex |
| mRNA | = | Messenger ribonucleic acid |
| MDSC | = | Myeloid-derived suppressor cells |
| MSKCC | = | Memorial Sloan Kettering Cancer Center |
| mTOR | = | Mammalian target of rapamycin |
| n | = | Number |
| NCI-CTC | = | National Cancer Institute-Common Toxicity Criteria |
| OS | = | Overall survival |
| PD | = | Progressive disease |
| PFS | = | Progression-free survival |
| PK | = | Pharmacokinetic |
| PR | = | Partial response |
| RCC | = | Renal cell carcinoma |
| RECIST | = | Response Evaluation Criteria in Solid Tumors |
| SAE | = | Serious adverse event |
| s.c. | = | subcutaneous, subcutaneously |
| SD | = | Stable disease |
| TKI | = | Tyrosine-kinase inhibitors |
| TNF | = | Tumor necrosis factor |
| Tregs | = | Regulatory T-cells |
| TUMAP | = | Tumor-associated peptides |
| VEGF | = | Vascular endothelial growth factor |
Introduction
Renal cell carcinoma (RCC)
Renal cell carcinoma (RCC) is the most common kidney cancer accounting for 85% of all kidney cancers.Citation1 RCCs arise from the renal tubular epithelium Citation2 and are mostly adenocarcinomas.Citation3 Kidney cancer belongs to the 10 most common cancers and is responsible for 1.9% of all cancers worldwide.Citation4 The incidence varies widely with higher rates in US and Europe and lower rates in Asian countries.Citation5,6 About twice as many men as women are diagnosed with RCC.Citation4,5,7,8 80% of RCCs are diagnosed in patients aged between 40 and 69 years with an average age in the low 60s.Citation9,10 Worldwide, the incidence of total kidney cancer and simultaneously RCC has steadily increased over the last 2 decades, while the mortality has decreased. Citation5,8,9, Citation11-13 This rise in incidence appears to result from an increase in the frequency of risk factors such as obesity and smoking and in the more frequent usage of diagnostic imaging procedures.Citation11,12,14-16 In the US, an estimated 63,920 people (39,140 male; 24,780 female) will be diagnosed with RCC, and about 13,860 (8,900 male; 4,960 female) will die from this disease during the year 2014.Citation17 The 5-year overall survival rates for patients diagnosed with localized, regional or distant disease between 1999 and 2005 were 90.4%, 62.3% and 10.4%, respectively.Citation8 In Europe, kidney cancer was the 9th most common malignancy in 2008 with 88,400 new cases (56,000 men; 32,400 women) and 39,300 deaths (24,800 men; 14,500 women).Citation18
RCC is a heterogeneous, malignant disease and has traditionally been classified according to mainly histological features, which are generated through examining excised tumor tissues.Citation19 Based on the findings of routinely conducted histological assessments, patients are grouped into 5 major histologic subtypes, which differ in prognosis and response to anti-tumor therapy.Citation20,21 Clear cell renal cell carcinoma (ccRCC) is the most prevalent histologic subtype being assigned to 70–80% of all RCC patients. The prognosis of patients with ccRCCs is less favorable compared with papillary (chromophile) RCCs accounting for 10–15% of RCCs and chromophobe RCCs being responsible for 3–5% of RCC cases.Citation22,23 Rare histologic RCC subtypes, such as collecting duct carcinomas are aggressive diseases associated with poor outcome.Citation24
Treatment of advanced RCC
Nephrectomy, ideally with curative or at least with debulking intent, is the treatment of choice for advanced and metastatic RCC.Citation25 Additionally, metastasectomy should be considered for patients with solitary lung, bone or brain metastases and a potentially resectable primary tumor as well as for patients with a solitary recurrence after a long disease-free interval.Citation26,27 Radiotherapy and chemotherapy is barely effective and should be mainly considered as palliative treatment in RCC patients. Unspecific immunotherapy, mostly consisting of interleukin (IL)‑2 and/or interferon (IFN)‑α with or without 5‑fluorouracil (5-FU) has been the only systemic treatment option for many years leading to clinical improvements in a small proportion of patients, however at the cost of substantial toxicities.Citation25 A significant change in standard of care has been introduced with the approval of targeted therapies including tyrosine kinase inhibitors (TKIs) and inhibitors of the mammalian target of rapamycin (mTOR inhibitors). The TKIs sunitinib (Sutent®) and pazopanib (Votrient®) are well established first-line therapies. Other first-line therapy options include the monoclonal antibody bevacizumab (Avastin®) combined with IFN‑α and the mTOR inhibitor temsirolimus (Torisel®), the latter for poor-risk patients. Selected first-line patients may be treated with high dose IL-2 or sorafenib (Nexavar®). Standard second-line therapies include sorafenib (Nexavar®), everolimus (Afinitor®), axitinib (Inlyta®) and IL-2.Citation28–31 However, despite the significant clinical improvements on the progression of disease seen with these targeted agents, there still remains a significant medical need for additional treatment options as the effect of TKI and mTOR-inhibitors on survival seem to be relatively marginal and almost all of advanced RCC patients still die of their disease. Unspecific immunotherapy with cytokines has shown promise in advanced RCC achieving long-term response and survival in a small fraction of patients albeit with significant side effects. Improved cancer immunotherapy approaches such as (peptide-based) cancer vaccines are therefore of high interest.
Peptide-based cancer vaccines
Active immunotherapy of cancer is based on the premise that a vaccine targeting tumor-associated antigens will raise a cytotoxic immune response to these antigens, destroying malignant cells without harming normal cells. Human leukocyte antigen (HLA) molecules, also known as MHC (major histocompatibility complex) molecules, present peptide fragments derived from internal cellular proteins on the cell surface, thus permitting T cells to discriminate between healthy cells and diseased cells including virus-infected and tumor cells.Citation32 The peptides which are predominantly presented on tumor cells (and not at all or to a far lesser degree on healthy cells) are named TUMAPs (tumor-associated peptides).
HLA molecules occur in many different forms, known as alleles. While hundreds of different alleles exist, some of them are predominantly present in human populations. For example, the allele HLA-A*02 is expressed by 45–50% of the Caucasian population, while the allele HLA-A*24 is expressed by over 60% of the South-East-Asian population. As the interactions between TUMAPs and their corresponding HLA allele are highly specific, only patients expressing the respective HLA allele(s) can benefit from receiving a given TUMAP-based immunotherapy.
A cytotoxic T cell specific for a certain tumor-associated peptide (TUMAP) will recognize a tumor cell through interaction with this HLA molecule-bound TUMAP presented on the cell surface.
There are 2 kinds of TUMAPs:
Class I TUMAPs are oligopeptides (8 to 10 amino acids) which activate CD8+ cytotoxic T lymphocytes (CTLs). Activated CTLs have the potential to directly kill tumor cells that present such TUMAPs by releasing cytolytic substances, or by forcing tumor cells into apoptosis.
Class II TUMAPs are longer peptides (approximately 15 or more amino acids), which activate CD4+ T-helper cells. Activated T-helper cells support CTLs by locally increasing the concentration of certain cytokines and stimulating the production of antibodies by B cells.
Despite the ability of T cells to detect TUMAPs that are naturally presented by most tumor cells, naturally occurring T cells are not activated (or even silenced by the tumor) and, therefore, do not protect the body against cancer. This requires the help of co-stimulatory molecules. These co-stimulatory molecules are exclusively expressed on professional antigen-presenting cells, e.g. dendritic cells (DCs).
Vaccination with TUMAPs is believed to activate the immune system against cancer in 4 stages (). The underlying activation cascade comprises vaccination, priming, proliferation and elimination as described in detail below.
Figure 1. Vaccination with several TUMAPs simultaneously triggers a broad immune attack against the tumor.
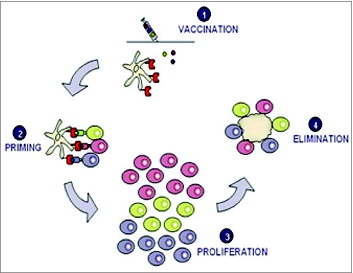
Vaccination: TUMAPs are dissolved in aqueous buffer and administered intradermally together with adjuvants/immunomodulators, e.g., GM-CSF. GM-CSF creates an inflammatory milieu and helps to recruit DCs to the administration site as well as maturate DCs for full expression of co-stimulatory molecules.Citation33
Priming: TUMAPs injected into the skin bind to dermal DCs. TUMAPs are loaded onto HLA molecules that are located on the surface of the DCs without any further requirement of processing. DCs then migrate into the lymph nodes, where they encounter and activate (“prime”) naïve T cells specifically recognizing the TUMAPs used in the vaccine.
Proliferation: Once T cells are primed by DCs, their number increases rapidly (clonal proliferation). Soon thereafter, they leave the lymph nodes and begin searching for tumor cells displaying exactly the same TUMAP by which they were activated in the process of priming.
Elimination: Once CTLs recognize a TUMAP on the HLA molecules of tumor cells, they are able to mount a cytolytic/apoptotic attack against the tumor cells.
The priming of CTLs directed against only one specificity, i.e. a single tumor antigen, is normally not sufficient to eradicate all tumor cells. Tumor cells have the ability to mutate more frequently than other cells and are thus able to evade a CTL attack by changing their protein expression pattern and in this way can no longer be recognized by CTLs (so-called “tumor escape”). Vaccination with multiple TUMAPs allows the priming of different T-cells that can act synergistically by simultaneously recognizing a matching number of independently encoded tumor antigens.
IMA901 vaccine
IMA901 is composed of 9 HLA class I-binding tumor-associated peptides (TUMAPs) with the capacity to activate CD8+ CTLs and one HLA class II-binding TUMAP with the capacity to activate CD4+ helper T-cells. describes the process of identifying the HLA class I and II TUMAPs by using an integrated functional genomics approach combining gene expression analysis, mass spectrometry, and T-cell immunology.Citation34 This methodology served as the basis for selection of the TUMAPs included in IMA901. By analyzing tumor samples from more than 30 RCC patients, this selection procedure ensures that the majority of RCC tumors in a patient population are recognized by the induced T cells.
Figure 2. Selection, Identification and Validation of TUMAPs.
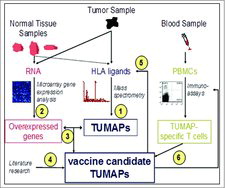
Samples from surgically removed malignant tissue from RCC patients and blood from healthy donors were analyzed as follows:
HLA ligands from the malignant material were identified by mass spectrometry.
Genes that were over-expressed in the malignant RCC tissue compared with a range of normal organs and tissues were identified using genome-wide messenger ribonucleic acid (mRNA) expression analysis by microarrays.
Gene expression data were compared to identify HLA ligands. Peptides that were encoded by selectively expressed or overexpressed genes as detected in step 2 were considered potential candidates for a multi-peptide vaccine.
Literature research was conducted with the intention to identify additional evidence for the relevance of the TUMAP candidates.
At the mRNA level, the relevance of over-expression was verified by redetection of the selected TUMAPs from step 3 on tumor tissue.
Several immunoassays (in vitro T-cell assays) were used to test the reactivity of peripheral T-cells from blood of healthy persons against the TUMAP candidates. It can be speculated, that the IMA901 in vitro immunogenicity data obtained by analyzing PBMCs from HLA-A*02+ healthy individuals as well as from HLA-A*02+ RCC patients would have been similar, as this could be shown for IMA950, a sister product of IMA901 developed by the same technology platform for the treatment of glioblastoma.Citation35
The most relevant results of the multi-step procedure leading to selection of TUMAPs for IMA901 are summarized in for each peptide. “X” indicates that a criterion was fulfilled for the respective TUMAP.
Table 1. Summary of peptide selection
Adjuvants/Immunostimulants
TUMAPs are on their own poorly immunogenic unless the presenting cells (i.e., DCs) are further activated to allow for full T cell priming and stimulation. Therefore, the co-administration of immunological adjuvants or immunomodulators is crucial for eliciting a strong and sustainable immune response against the targeted tumor cells.
GM-CSF
GM-CSF is widely used as an adjuvant in clinical vaccination studies with cellular vaccines, proteins, peptides, or antigen-loaded DCs.Citation36 Autologous and allogenic cancer vaccines genetically modified to generate GM-CSF have been successfully applied in man and are in late-stage clinical development.Citation37 With the FDA approval for Sipuleucel-T (Provenge®), an autologous cancer vaccine modified to produce GM-CSF, the first autologous cellular immunotherapy is available.Citation38 A number of clinical trials mostly using melanoma peptides in combination with GM-CSF have been performed. Some of these pilot trials have demonstrated that a high administration frequency of the peptides, particularly within the first 2 weeks induces effective priming of naive T-cells.Citation39,40 Meta-analysis of published trials suggested, that 40–80 μg GM-CSF applied s.c. or i.d. for 1–5 d at the site of vaccination improves the cellular immune response, while a systemic treatment with ≥ 100 μg does not enhance the efficacy of a peptide vaccine due to expansion of immune-inhibiting myeloid-derived suppressor cells (MDSCs).Citation41
GM-CSF was therefore chosen as the immunological adjuvant in clinical studies with IMA901. GM-CSF is applied locally (i.d.) in the low-dose range (75 μg per injection).
Cyclophosphamide
Regulatory T-cells (Tregs) represent a T-cell population that functionally suppresses immune responses by having an influence on other immune effector cells.Citation42 Results from experiments using mouse tumor models suggest, that Tregs are the main regulators of tumor-directed immunity specifically suppressing cytotoxic effector functions of CTLs.Citation43,44 Several phenotypically distinct Treg populations have been described: The focus of most intensive recent research are CD4+ CD25+ Foxp3high T cells, which also express glucocorticoid-induced tumor necrosis factor (TNF) receptor-related protein at high levels. These Tregs are thought to be key mediators of peripheral immune tolerance. There is evidence from a number of pre-clinical and clinical studies that low-dose cyclophosphamide (CY) administered prior to vaccination is of benefit to cancer patients most likely through inhibition of Tregs and subsequently enhanced immune responses and/or clinical response to vaccination.Citation45-51 In addition, positive immunomodulatory effects of CY on dendritic cells Citation52-55, other antigen presenting cellsCitation52, tumor infiltrating cellsCitation53,55,Citation56 and MDSCsCitation52,57,58 have been shown in in vitro experiments as well as in animal models.
The effect of CY on Tregs was investigated in the phase II study with IMA901 in a randomized fashion.
Results and Discussion
Phase I study with IMA901 in advanced RCC
The study IMA901–101 was an open-label, multicenter, first-in-man trial with the primary objective to investigate systemic safety and local tolerability of IMA901 plus GM-CSF in advanced/metastatic RCC patients. Secondary objectives of the trial were immunogenicity and first signs of anti-tumor activity. The study IMA901–101 enrolled 30 patients with stage III/IV RCC regardless of risk categorization, previous treatments and progressing or non-progressing disease. Except for one patient, all had undergone nephrectomy. 47% of all enrolled patients (n=14) had received previous systemic therapies, most frequently consisting of IFN-α and/or IL-2 and often combined with chemotherapy, being most commonly 5-Fluorouracil (5-FU). The remaining 53% of patients (n=16) had received no systemic treatment before the start of vaccination with IMA901 plus GM-CSF.
After positive eligibility screening, patients received up to 8 vaccinations each consisting of 75 μg GM-CSF applied intradermally followed after 15–30 minutes by intradermally applied 4.13 mg IMA901 (413 μg per peptide). In the phase I trial, an additional (non-TUMAP) peptide derived from Hepatitis B virus was added as a marker peptide to better interpret the association of immunological responses and clinical outcome.Citation59 All GM-CSF and IMA901 applications were administered in close proximity at the same injection site in order to re-stimulate (boost) T cells at the same draining lymph node. The vaccination schedule applied is outlined in .
Table 2. Vaccination schedule in IMA901–101Citation59
Safety was assessed according to NCI-CTC criteria, version 3.0 and comprised continuous adverse event (AE) monitoring, assessment of hematology, biochemistry and urinalysis, physical examinations, ECG, vital signs and pregnancy testing at specified time points. Local tolerability was evaluated based on protocol-defined criteria (i.e. rating of injection site related pain, itching, induration, edema and erythema). The immunogenicity of the vaccination therapy was determined by assessing the T-cell response to single TUMAPs contained in IMA901 before, during and after vaccination by applying amplified IFN-γ ELISpot and amplified HLA multimer assays. Tumor response evaluations according to RECIST 1.0 Citation60 were conducted at baseline and during follow up visit taking place about 3 months after study start (2 months of vaccination and 1 month safety follow up) by the local investigator.
All enrolled patients received at least one dose of study medication and were included into the safety population. Two patients were not HLA-A*02 positive and thus, excluded from the efficacy population (n = 28) after the first 2 vaccinations. Twenty-seven patients completed the regular course of the study and received all 8 vaccinations (one patient received only 7 vaccinations). Treatment with IMA901 plus GM-CSF was safe and overall well tolerated. In summary, 21 patients (70%) experienced at least one treatment-emergent adverse event (in total 83 AEs). The most commonly observed AEs independent of relationship were fatigue (16.7%), cough (13.3%), headache (10.0%), pyrexia (10.0%), influenza (10.0%) and anemia (10.0%). The most common AEs considered at least possibly related to study medication were lymphadenitis (6.7%) and injection site pruritus (6.7%). All treatment-emergent AEs with an anticipated relationship to study drug were mild or moderate (maximum severity of NCI-CTC grade 1 or 2) and no patient discontinued study therapy due to AEs. Three SAEs were observed and all resolved and were considered either not related or unlikely related to study drug. One patient was withdrawn from the study after vaccination 7 due to lack of efficacy and died 2 months later due to tumor progression. No indications of autoimmune reactions were observed. Further to AE recording, a more sensitive, protocol-defined scale was used to evaluate local tolerability. Erythema, induration, edema, itching and pain at the injection site were rated 5 minutes and 24 hours post vaccination and at the end-of-study visit as illustrated in .Citation59
Figure 3. Local injection site reactions i.e., erythema, induration, edema, itching and pain 5 minutes and 24 hours post vaccination and about 1 month after last vaccination. (Safety population; N = 30).
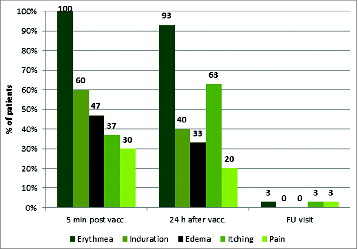
The most frequently observed local injection site reaction was erythema experienced by all patients. Induration and itching were reported by about 60% of patients. Edema and pain were experienced by 47% and 20% of patients, respectively. Interestingly, 24 hours after vaccination erythema, induration and edema decreased, whereby itching and pain increased. Nearly all reactions had resolved at the last study visit. Most of these injection site reactions were not reported as AEs since investigators considered them as not fulfilling the predefined AE criteria (NCI-CTC).
The T-cell responses to class I TUMAPs contained in IMA901 were evaluable for 27 out of 28 HLA-A*02 positive patients. Overall, IMA901 was able to induce a T-cell response to at least one class I TUMAP in 74% of immune response evaluable patients (n = 20) and 30% of patients (n = 8) responded to 2 or more TUMAPs and were classified as multi-TUMAP responders (19 % and 11% of patients had a vaccine-induced T-cell response to 2 or 3 TUMAPs, respectively). 52% (n = 14) of patients developed a T-cell response to the viral HBV-derived marker peptide.Citation59 The onset of immune response appeared after 8 to 15 days of vaccination (after the 3rd or 4th vaccination) in about 70% of patients. The peak of T-cell responses could typically be observed after 6 to 8 vaccinations (in week 4 to 10 after start of treatment). More than 50% of responses were still detected 3 months after the first vaccination. Hence, the vaccination schedule used with intense vaccinations at the beginning and longer intervals later on and, the mode and the route of vaccination seemed to be suitable and were therefore maintained in future trials.Citation59
Tumor response evaluations could be undertaken in all 28 HLA-A*02 positive patients. One patient (3.6%) achieved a partial response (PR), 11 patients (39.3%) had stable disease (SD) and 16 patients (57.1%) had progressive disease (PD) according to the local investigator. Thus, 12 patients (42.9%) had responding or stabilized disease at the time of the last study visit being collectively termed patients with clinical benefit.Citation59 The analysis included 5 patients without measurable disease at baseline. If no new tumor lesions were discovered at the follow-up visit, the patients had been assessed as having stable disease. The tumor response rate achieved was rather modest, however expected, as it seems to reflect the anticipated mode of action of cancer vaccinesCitation61,62. Similar observations were made in other randomized vaccine trialsCitation38,63-66 showing only a moderate improvement in tumor response-dependent clinical trial endpoints such as objective tumor response rate and progression-free survival. Most importantly, clinical benefit was associated with vaccine-induced immune responses as suggested by the cluster of Multi-TUMAP responders (≥ 2 T-cell responses to TUMAPs) on the right hand side of the waterfall plot ().
Figure 4. Waterfall plot and vaccine-induced T-cell responses as indicated by the TUMAPs named above the bar representing the percentage of change in longest diameter of target lesions. The occurrence of new lesions is outlined and patients without measurable disease according to RECIST at baseline are marked with a shadow around the zero. Patient numbers and the treatment line of the vaccination therapy are illustrated.
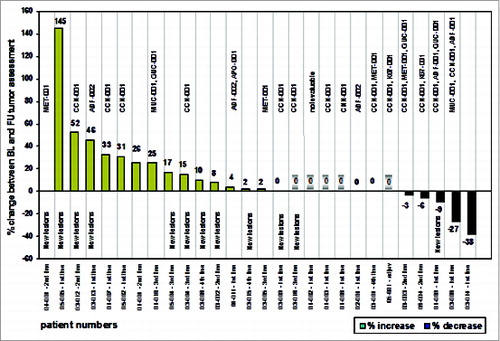
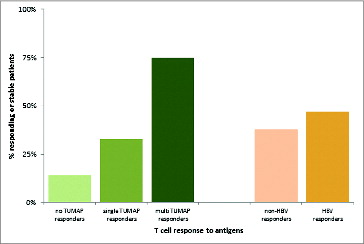
depicts, that 75% of multi-TUMAP responders compared to 26% of non-multi-TUMAP (0–1 T cell responses) responders had stable or responding disease at the follow up visit. In other words, patients with a multiple T-cell response were significantly more likely to experience clinical benefit compared with patients with only 0–1 T-cell responses (Chi squared test, p = 0.0187).Citation59 In contrast, no association was found between T cell responses to the viral marker peptide HBV-001 and clinical benefit (Chi squared test, p = 0.6617)Citation59 suggesting that the observed positive association of clinical outcome with T cell responses against the vaccinated TUMAPs resembles a true therapeutic effect rather than immune responses being just a marker for overall better baseline characteristics, i.e., being a mere prognostic factor. A similar correlation between vaccine-induced immune responses and clinical benefit (responses as well as OS prolongation) was found for other vaccines applied to patients with RCC and other cancers.Citation67-71
Figure 5. Association between vaccine-induced T-cell responses and clinical benefit defined as responding or stable disease after 3 months of vaccination (at the follow up visit). The clinical benefit rates were 14%, 33% and 75% for patients with 0, 1 and ≥ 2 TUMAP responses, respectively. 38% of Non-HBV responders and 47% of HBV responders had their disease controlled at the follow up visit.
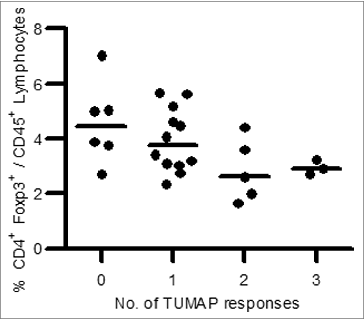
Interestingly, an inverse association between Multi-TUMAP responders and pre-vaccination Tregs frequencies could be detected. Patients with a multiple IMA901 TUMAP response had significantly lower pre-vaccination Treg frequencies () (p = 0.018, Two-sided Wilcoxon scores).Citation59
Figure 6. Association between pre-vaccination Treg frequency and number of induced TUMAP responses (n = 26). Lower pre-vaccination frequencies of Treg cells defined as CD4+Foxp3+ cells among CD45+ lymphocytes (y axis) were associated with the induction of multiple IMA901 TUMAP responses (x axis). Every dot represents one patient and horizontal lines illustrate the median Treg frequencies.
Phase II study with IMA901 in advanced RCC
The IMA901–202 phase II study was designed as a multicenter, open-label, randomized study in patients with RCC to investigate the effect of a systemic treatment with IMA901 plus GM-CSF with or without cyclophosphamide (CY) as an additional immunomodulator. The study population consisted of adult HLA-A*02-positive women or men with advanced clear cell RCC classified as having a favorable or intermediate risk according to the 3-score Memorial Sloan Kettering Cancer Center (MSKCC) risk criteria and having experienced disease progression after previous systemic therapy for advanced disease (tyrosine kinase inhibitors [TKIs] or cytokines). Sixty-eight patients were randomized 1:1 to receive a single infusion of low-dose CY (300 mg/m2) 3 d prior to the first vaccination versus no pre-treatment (33 patients did and 35 patients did not receive pre-treatment with CY). Stratification factors included risk group (favorable or intermediate risk based on the MSKCC criteria) and previous therapy, i.e. TKIs or cytokines. 64 patients were included in the pre-defined efficacy analysis (per protocol population). Patients in both randomization arms received up to 17 vaccinations consisting of 75 μg GM-CSF administered i.d. followed after 10 to 30 minutes by i.d. administration of 4.13 mg IMA901. Tumor assessments were performed by either CT or MRI according to Response Evaluation Criteria in Solid Tumors (RECIST). All images were assessed locally and reviewed centrally by an independent radiologist and oncologist (both blinded for CY pre-treatment). All patients were followed for survival after the final visit. Safety assessments included continuous adverse event (AE) monitoring and physical examinations, vital signs, ECG, assessment of hematology, blood chemistry and urinalysis at scheduled time points.
The primary endpoint of this study was defined as the disease control rate (DCR; percentage of patients with complete or partial response or stable disease according to RECIST) after 26 weeks of treatment. The main secondary endpoints included overall survival (OS), progression-free survival (PFS), safety, immune responses to peptides contained in IMA901 and monitoring of other immune cell populations with a potential influence on T-cell responses such as Tregs.
IMA901/GM-CSF and the single infusion of low-dose CY were safe and overall well tolerated. The most frequently reported AEs independent of relationship were anemia (24%), asthenia (16%), pyrexia (15%), back pain (12%), dyspnea (12%), and fatigue (10%). Most of the AEs which were considered at least possibly related to treatment with IMA901 and/or GM-CSF were injection site reactions with 15 (22%) patients experiencing at least one type of injection site reaction. Except for one patient, local reactions were mild or moderate in severity. Two serious adverse events which were considered related to treatment with IMA901/GM-CSF were reported: one patient experienced a systemic allergic reaction after the twelfth vaccination (caused by CM-CSF as shown in an in vitro basophil degranulation assay) and for another patient a grade 3 localized allergic reaction was reported after the eleventh vaccination, with no similar signs of intolerance after further vaccinations.
Patients randomized to the 2 arms were well balanced with regard to baseline characteristics, risk factors, and pre-treatments. The analysis of the primary efficacy variable showed a DCR of 24.6% (95% CI: 14.5% to 37.3%) at 6 months. For post-cytokine patients (n = 40) DCR at 6 months was 31% compared to 14% in the post-TKI group (n = 24). Progression-free survival was 3.3 months (post-cytokine group 3.4 months, post-TKI group 2 months). As reported for other cancer vaccines objective responses were infrequent, with one complete and 2 partial responses according to investigators’ assessment and one centrally confirmed partial response. Progression-free survival was comparable in both treatment arms ().
Figure 7. OS and PFS of patients treated in the phase II study (per protocol population). (a) PFS of patients pre-treated with cyclophosphamide or without such pre-treatment. (b) OS of patients pre-treated with cyclophosphamide or without such pre-treatment. (c) OS of patients evaluable for immune responses in the following groups: immune responders pre-treated with cyclophosphamide, +Cy ≥ 1; patients pre-treated with cyclophosphamide for whom no immune response was observed, +Cy 0; immune responders without cyclophosphamide pre-treatment, −Cy ≥ 1; patients without cyclophosphamide pre-treatment for whom no immune response was observed, −Cy 0. (d) OS of patients for whom no immune response was observed, immune responders to one TUMAP, immune responders to 2 TUMAPs or immune responders to at least 3 TUMAPs.
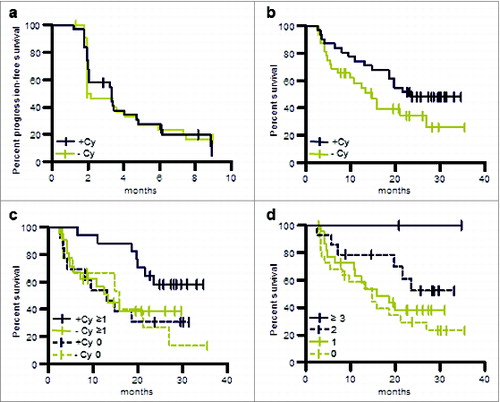
Patients were followed for survival during the study and every 3 months thereafter via standardized questionnaires including information on subsequent systemic treatments for advanced RCC. A strong trend toward improved survival was observed in patients who received pre-treatment with CY (HR = 0.57; p = 0.09) () compared to patients without CY pre-treatment. Survival rates and median survival observed in the IMA901 phase II study appear to be particularly encouraging when compared to data from studies investigating sunitinib and sorafenib in the same patient population. Median survival in the IMA901–202 phase II study was 19.8 months in all second-line patients previously treated with cytokines and had not been reached (> 26 months) at the time of the data cut-off in patients pretreated with CY. This compares favorably to published results in comparable patients with a median OS of 17.8 months for sorafenib Citation72 and 16.4 months for sunitinib. Citation73Neither a difference in baseline characteristics (all relevant parameters were indeed very comparable between IMA901–202 phase II study and the above cited sunitinib and sorafenib studies) nor post-study treatments (only 31% of patients received further systemic therapy after IMA901 treatment) were able to explain the favorable survival data in the IMA901 phase II trial.
Immunogenicity (as measured in peripheral blood, N = 61) of IMA901 class I peptides was comparable to that observed in the phase I trial (phase II: 64% immune responders, 27% multi-TUMAP responders). Patients pre-treated with or without CY showed comparable immune response rates, indicating that CY did not alter the induction of T cell responses but rather had a positive effect on the functionality of such T cells either directly or by affecting the tumor microenvironment. Immune responders (response to ≥ 1 TUMAP) had a significantly longer survival compared to non-responders (p = 0.048). Furthermore, longer survival was observed with an increasing number of responses (p = 0.023, log-rank test for trend in patients with increasing number of responses) (), thereby confirming the hypothesis from the phase I trial results that clinical benefit is associated with the breadth of immune responses. The above mentioned survival benefit of CY-treated patients was observed only in the patients with vaccine-induced immune responses (p = 0.040; HR = 0.38), but not in patients lacking a response to TUMAPs (p = 0.870, HR = 0.92) ().Citation59 This suggests that CY acts as an immune modulator and does not have a single agent activity. The immune modulating effect of CY can be best explained by the observed decrease in regulatory T cells (Tregs): the prospectively defined analysis showed an approximately 20% reduction in regulatory T cells within 3 d (p = 0.013) compared to pre-treatment baseline levels.Citation59
Conclusions and future prospects
In summary, these 2 studies showed that IMA901 plus GM-CSF without or with CY is a safe, immunogenic and a promising vaccine to be taken forward into late stage clinical development.
Support for the presence of sufficient anti-tumor activity of IMA901 was derived from the finding in the phase I study that multiple TUMAP responses, but not HBV responses were associated with stable or responding disease. Moreover, OS in the phase II study compared favorably to historic controls and was significantly longer in TUMAP responders corresponding to the anticipated mode of action of cancer vaccines. Based on these data, a randomized, open-label phase III trial investigating IMA901 plus GM-CSF in combination with sunitinib compared to sunitinib alone in first-line advanced RCC patients was initiated in 2011. The recruitment has meanwhile been completed and survival data are expected toward the end of 2015.
The percentage of patients with observed responses to multiple peptides was lower than hoped for in both the phase I and II study (16 out of 61 immune-evaluable patients in the phase II study and 8 out of 27 immune-evaluable patients in the phase I study). While responses to multiple peptides have been hypothesized as a biomarker for efficacy, it has also been shown in the phase II trial that in patients pre-treated with cyclophosphamide (CY) the observation of at least one TUMAP immune response was associated with longer survival. In fact, patients in the ongoing phase III study will all be pre-treated with CY. Furthermore, the phase III study also incorporates a secondary endpoint, which investigates overall survival benefit in a predefined subgroup of patients with high levels of the serum markers CCL17 and/or Apolipoprotein A1. Both markers were associated with longer overall survival and higher immune response rates in the phase II study.
Emerging immunotherapy approaches, with the potential to prevent the down-regulation of a tumor-directed immune response by blocking co-inhibitory receptors on immune cells or their soluble or membrane-bound ligands the so called checkpoint inhibitors, are rational candidates for combination with IMA901. Preclinical findings Citation74–77 as well as early clinical data in pancreatic, melanoma and prostate cancer Citation78–80 suggest synergistic effects between cancer vaccines and immune checkpoint blockade. Nivolumab, a programmed death-1 (PD-1) inhibitor blocking the PD-1 co-inhibitory receptor on activated T-cells is a promising and in RCC well advanced checkpoint inhibitor, currently being investigated in a phase III trial to assess whether it prolongs survival of second- or third-line RCC patients compared with everolimus. In a previous RCC trial, nivolumab achieved a tumor response rate of 29% with mostly durable responses lasting for 24 or more weeks with manageable toxicities.Citation81 Lambrolizumab, another promising PD-1 inhibitor is currently being tested in first-line advanced RCC patients in a large phase I/II study in combination with pazopanib. A third interesting potential combination partner, MPDL3280A is an antibody targeting PD-L1. MPDL3280A was investigated in advanced RCC in a phase I dose-finding study.Citation82 Tumor responses including complete responses and prolonged disease stabilizations were seen across all dose levels. The PFS rate was 50% at 24 weeks. A randomized phase II study is currently ongoing in initially diagnosed, unresectable advanced or metastatic RCC patients comparing monotherapy with MPDL3280A vs. MPDL3280A in combination with avastin or sunitinib. Some other checkpoint inhibitors, such as pidilizumab (PD-1 inhibitor), MAG-271 (B7-H3 inhibitor), BMS-936559 (PD-L1 inhibitor) and MEDI4736 (PD-L1 inhibitor) are in early RCC clinical trials and could also be synergistic combination partners for IMA901. It can be hypothesized that the addition of IMA901, which is able to induce a multi-clonal and tumor-directed T-cell response could increase the clinical benefit observed with checkpoint inhibitors alone, leading to a considerable prolongation of survival ideally in all patients.
Disclosure of Potential Conflicts of Interest
The authors are employees of Immatics biotechnologies GmbH.
Acknowledgments
The authors would like to express their special thanks to all staff involved at immatics biotechnologies GmbH and to members of the Scientific Advisory Board and Data Safety Monitoring Board who contributed to the development of IMA901. In addition we would like to thank all investigators and patients participating in the clinical studies.
References
- Lynch CF, Cohen MB. Urinary system. Cancer 1995;75(1 Suppl):316-29; PMID:8001003; http://dx.doi.org/10.1002/1097-0142(19950101)75:1+%3c316::AID-CNCR2820751314%3e3.0.CO;2-T
- Pantuck AJ, Zisman A, Belldegrun AS. The changing natural history of renal cell carcinoma. J Urol 2001;166(5):1611-23; PMID:11586189; http://dx.doi.org/10.1016/S0022-5347(05)65640-6
- Chow WH, Devesa SS, Warren JL, Fraumeni JF, Jr. Rising incidence of renal cell cancer in the United States. JAMA 1999;281(17):1628-31; PMID:10235157; http://dx.doi.org/10.1001/jama.281.17.1628
- Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin 2005;55(2):74-108; PMID:15761078; http://dx.doi.org/10.3322/canjclin.55.2.74
- Murai M, Oya M. Renal cell carcinoma: etiology, incidence and epidemiology. Curr Opin Urol 2004;14(4):229-33; PMID:15205579; http://dx.doi.org/10.1097/01.mou.0000135078.04721.f5
- Marugame T, Matsuda T. Comparison of time trends in kidney cancer incidence (1973-97) in East Asia, Europe and USA, from cancer incidence in five continents, Vols IV-VIII. Jpn J Clin Oncol 2008;38(7):508-9; PMID:18664483; http://dx.doi.org/10.1093/jjco/hyn060
- Vaishampayan UN, Do H, Hussain M, Schwartz K. Racial disparity in incidence patterns and outcome of kidney cancer. Urology 2003;62(6):1012-7; PMID:14665346; http://dx.doi.org/10.1016/j.urology.2003.07.010
- Horner MJ, Ries LAG, Krapcho M et al. SEER Cancer Statistics Review, 1975-2006. based on November 2008 SEER data submission. 2009. http://seer.cancer.gov/csr/1975_2006/, National Cancer Institute. Bethesda, MD.
- Lipworth L, Tarone RE, McLaughlin JK. The epidemiology of renal cell carcinoma. J Urol 2006;176(6 Pt 1):2353-8; PMID:17085101; http://dx.doi.org/10.1016/j.juro.2006.07.130
- Pascual D, Borque A. Epidemiology of kidney cancer. Adv Urol 2008;782381; PMID:19009036
- Nepple KG, Yang L, Grubb RL, III, Strope SA. Population based analysis of the increasing incidence of kidney cancer in the United States: evaluation of age specific trends from 1975 to 2006. J Urol 2012;187(1):32-38; PMID:22088338; http://dx.doi.org/10.1016/j.juro.2011.09.028
- Nepple, Wood. Kidney cancer on the rise. 2011. Genitourinary Cancers Symposium, Orlando, Fla., Feb. Seventeen-19, 2011.
- Levi F, Ferlay J, Galeone C, Lucchini F, Negri E, Boyle P, La Vecchia C. The changing pattern of kidney cancer incidence and mortality in Europe. BJU Int 2008;101(8):949-58; PMID:18241251; http://dx.doi.org/10.1111/j.1464-410X.2008.07451.x
- Mathew A, Devesa SS, Fraumeni JF, Jr., Chow WH. Global increases in kidney cancer incidence, 1973-1992. Eur J Cancer Prev 2002;11(2):171-8; PMID:11984136; http://dx.doi.org/10.1097/00008469-200204000-00010
- Tate R, Iddenden R, Harnden P et al. Increased incidence of renal parenchymal carcinoma in the Northern and Yorkshire region of England, 1978-1997. Eur J Cancer 2003;39(7):961-7; PMID:12706365; http://dx.doi.org/10.1016/S0959-8049(03)00070-4
- Chow WH, Devesa SS. Contemporary epidemiology of renal cell cancer. Cancer J 2008;14(5):288-301; PMID:18836333; http://dx.doi.org/10.1097/PPO.0b013e3181867628
- Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin 2014;64(1):9-29; PMID:24399786; http://dx.doi.org/10.3322/caac.21208
- Ferlay J, Parkin DM, Steliarova-Foucher E. Estimates of cancer incidence and mortality in Europe in 2008. Eur J Cancer 2010;46(4):765-781; PMID:20116997; http://dx.doi.org/10.1016/j.ejca.2009.12.014
- Prasad SR, Humphrey PA, Catena JR, Narra VR, Srigley JR, Cortez AD, Dalrymple NC, Chintapalli KN. Common and uncommon histologic subtypes of renal cell carcinoma: imaging spectrum with pathologic correlation. Radiographics 2006;26(6):1795-1806; PMID:17102051; http://dx.doi.org/10.1148/rg.266065010
- Cheng L, Zhang S, MacLennan GT, Lopez-Beltran A, Montironi R. Molecular and cytogenetic insights into the pathogenesis, classification, differential diagnosis, and prognosis of renal epithelial neoplasms. Hum Pathol 2009;40(1):10-29; PMID:19027455; http://dx.doi.org/10.1016/j.humpath.2008.09.009
- Algaba F, Trias I, Scarpelli M, Boccon-Gibod L, Kirkali Z, van PH. Handling and pathology reporting of renal tumor specimens. Eur Urol 2004;45(4):437-443; PMID:15041106; http://dx.doi.org/10.1016/j.eururo.2003.11.026
- Cheville JC, Lohse CM, Zincke H, Weaver AL, Blute ML. Comparisons of outcome and prognostic features among histologic subtypes of renal cell carcinoma. Am J Surg Pathol 2003; 27(5):612-624; PMID:12717246; http://dx.doi.org/10.1097/00000478-200305000-00005
- Jones TD, Eble JN, Cheng L. Application of molecular diagnostic techniques to renal epithelial neoplasms. Clin Lab Med 2005; 25(2):279-303; PMID:15848737; http://dx.doi.org/10.1016/j.cll.2005.01.011
- Eble JN, Sauter G, Epstein JI, Sesterhenn IA. Pathology and Genetics of Tumours of the Urinary System and Male Genital Organs. IARC, Press, Lyon. In press.
- Staehler M, Rohrmann K, Bachmann A, Zaak D, Stief CG, Siebels M. Therapeutic approaches in metastatic renal cell carcinoma. BJU Int 2005;95(8):1153-61; PMID:15877725; http://dx.doi.org/10.1111/j.1464-410X.2005.05537.x
- Vogl UM, Zehetgruber H, Dominkus M, Hejna M, Zielinski CC, Haitel A, Schmidinger M. Prognostic factors in metastatic renal cell carcinoma: metastasectomy as independent prognostic variable. Br J Cancer 2006; 95(6):691-8; PMID:16940978; http://dx.doi.org/10.1038/sj.bjc.6603327
- Ljungberg B. The role of metastasectomy in renal cell carcinoma in the era of targeted therapy. Curr Urol Rep 2013;14(1):19-25; PMID:23212738; http://dx.doi.org/10.1007/s11934-012-0293-6
- Stadler WM. Maturing of renal cancer therapeutics. J Clin Oncol 2014;32(8):722-724; PMID:24516015; http://dx.doi.org/10.1200/JCO.2013.54.1748
- Motzer RJ, Agarwal N, Beard C, Bolger GB, Boston B, Carducci MA, Choueiri TK, Figlin RA, Fishman M, Hancock SL, et al. NCCN clinical practice guidelines in oncology: kidney cancer. J Natl Compr Canc Netw 2009;7(6):618-30; PMID:19555584
- Ljungberg B, Cowan NC, Hanbury DC, Hora M, Kuczyk MA, Merseburger AS, Patard JJ, Mulders PF, Sinescu IC; European Association of Urology Guideline Group. EAU Guidelines on Renal Cell Carcinoma: The 2010 Update. Eur Urol 2010;58:398-406; PMID:20633979
- Escudier B, Kataja V. Renal cell carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2010;21 Suppl 5:v137-9; PMID:20555064; http://dx.doi.org/10.1093/annonc/mdq206
- Rammensee HG, Falk K, Rotzschke O. Peptides naturally presented by MHC class I molecules. Annu Rev Immunol 1993;11:213-44; PMID:8476560; http://dx.doi.org/10.1146/annurev.iy.11.040193.001241
- Molenkamp BG, Vuylsteke RJ, van Leeuwen PA, Meijer S, Vos W, Wijnands PG, Scheper RJ, de Gruijl TD. Matched skin and sentinel lymph node samples of melanoma patients reveal exclusive migration of mature dendritic cells. Am J Pathol 2005;167(5):1301-7; PMID:16251414; http://dx.doi.org/10.1016/S0002-9440(10)61217-5
- Weinschenk T, Singh H, Mahr A, Fritsche J. Biomarkers for predicting the efficacy of an immunotherapy against cancer, Patent PCT/EP2011/070661, published as WO2012069462. 2012 May 31.
- Dutoit V, Herold-Mende C, Hilf N, Schoor O, Beckhove P, Bucher J, Dorsch K, Flohr S, Fritsche J, Lewandrowski P, et al. Exploiting the glioblastoma peptidome to discover novel tumour-associated antigens for immunotherapy. Brain 2012;135(Pt 4):1042-54; PMID:22418738; http://dx.doi.org/10.1093/brain/aws042
- Berinstein NL. Enhancing cancer vaccines with immunomodulators. Vaccine 2007;25 Suppl 2:B72-88; PMID:17916464; http://dx.doi.org/10.1016/j.vaccine.2007.06.043
- Hege KM, Jooss K, Pardoll D. GM-CSF gene-modifed cancer cell immunotherapies: of mice and men. Int Rev Immunol 2006;25(5-6):321-352; PMID:17169779; http://dx.doi.org/10.1080/08830180600992498
- Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med 2010;363(5):411-22; PMID:20818862; http://dx.doi.org/10.1056/NEJMoa1001294
- Biskamp M, Jaeger E, Karbach J et al. Phase I study of intensive course immunization with NY-ESO-1 peptides in HLA-A2+ patients with NY-ESO-1+ cancer. ASCO Conference 2003. Proc Am Soc Clin Oncol (ASCO 2003) 22[706].2003
- Gaudernack G, Buanes T, Meo M et al. Clinical trials of a peptide based vaccine targeting telomerase. Proc Am Soc Clin Oncol (ASCO 2003) 22[666].2003.
- Parmiani G, Castelli C, Pilla L, Santinami M, Colombo MP, Rivoltini L. Opposite immune functions of GM-CSF administered as vaccine adjuvant in cancer patients. Ann Oncol 2007;18(2):226-32; PMID:17116643; http://dx.doi.org/10.1093/annonc/mdl158
- Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 2004;10(9):942-9; PMID:15322536; http://dx.doi.org/10.1038/nm1093
- Chen W, Cai MY, Wei DP, Wang X. Pivotal molecules of MHC I pathway in human primary hepatocellular carcinoma. World J Gastroenterol 2005;11(21):3297-9; PMID:15929187
- Turk MJ, Guevara-Patino JA, Rizzuto GA, Engelhorn ME, Sakaguchi S, Houghton AN. Concomitant tumor immunity to a poorly immunogenic melanoma is prevented by regulatory T cells. J Exp Med 2004;200(6):771-82; PMID:15381730; http://dx.doi.org/10.1084/jem.20041130
- Berd D, Maguire HC, Jr., Mastrangelo MJ. Potentiation of human cell-mediated and humoral immunity by low-dose cyclophosphamide. Cancer Res 1984;44(11):5439-5443; PMID:6488195
- Faries MB, Hsueh EC, Gupta RK et al. Immunomodulation of responses to active immunization with a polyvalent immunotherapeutic: cyclophosphamide, H2 blockade, IL-2/IFNa and GM-CSF. J Clin Oncol, 2005 ASCO Annual Meeting Proceedings Vol 23, No.16S, Part I of II (June 1 Supplement)[2564].2005.
- Ghiringhelli F, Menard C, Puig PE, , Ladoire S, Roux S, Martin F, Solary E, Le Cesne A, Zitvogel L, Chauffert B, et al. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother 2007;56(5):641-8; PMID:16960692; http://dx.doi.org/10.1007/s00262-006-0225-8
- Greten TF, Ormandy LA, Fikuart A, Höchst B, Henschen S, Hörning M, Manns MP, Korangy F. Low-dose cyclophosphamide treatment impairs regulatory T cells and unmasks AFP-specific CD4+ T-cell responses in patients with advanced HCC. J Immunother 2010;33(2):211-8; PMID:20139774; http://dx.doi.org/10.1097/CJI.0b013e3181bb499f
- Laheru D, Lutz E, Burke J, Biedrzycki B, Solt S, Onners B, Tartakovsky I, Nemunaitis J, Le D, Sugar E, et al. Allogeneic granulocyte macrophage colony-stimulating factor-secreting tumor immunotherapy alone or in sequence with cyclophosphamide for metastatic pancreatic cancer: a pilot study of safety, feasibility, and immune activation. Clin Cancer Res 2008;14(5):1455-63; PMID:18316569; http://dx.doi.org/10.1158/1078-0432.CCR-07-0371
- Livingston PO, Cunningham-Rundles S, Marfleet G, Gnecco C, Wong GY, Schiffman G, Enker WE, Hoffman MK, et al. Inhibition of suppressor-cell activity by cyclophosphamide in patients with malignant melanoma. J Biol Response Mod 1987;6(4):392-403; PMID:2957470
- Mitchell MS, Kan-Mitchell J, Kempf RA, Harel W, Shau HY, Lind S. Active specific immunotherapy for melanoma: phase I trial of allogeneic lysates and a novel adjuvant. Cancer Res 1988;48(20):5883-93; PMID:3262416
- Salem ML, Kadima AN, El-Naggar SA, Rubinstein MP, Chen Y, Gillanders WE, Cole DJ. Defining the ability of cyclophosphamide preconditioning to enhance the antigen-specific CD8+ T-cell response to peptide vaccination: creation of a beneficial host microenvironment involving type I IFNs and myeloid cells. J Immunother 2007;30(1):40-53; PMID:17198082; http://dx.doi.org/10.1097/01.cji.0000211311.28739.e3
- Radojcic V, Bezak KB, Skarica M, Pletneva MA, Yoshimura K, Schulick RD, Luznik L. Cyclophosphamide resets dendritic cell homeostasis and enhances antitumor immunity through effects that extend beyond regulatory T cell elimination. Cancer Immunol Immunother 2010;59(1):137-48; PMID:19590872; http://dx.doi.org/10.1007/s00262-009-0734-3
- Nakahara T, Uchi H, Lesokhin AM, Avogadri F, Rizzuto GA, Hirschhorn-Cymerman D, Panageas KS, Merghoub T, Wolchok JD, Houghton AN. Cyclophosphamide enhances immunity by modulating the balance of dendritic cell subsets in lymphoid organs. Blood 2010;115(22):4384-92; PMID:20154220; http://dx.doi.org/10.1182/blood-2009-11-251231
- Liu Y, Laszlo C, Liu Y, Liu W, Chen X, Evans SC, Wu S. Regulation of G(1) arrest and apoptosis in hypoxia by PERK and GCN2-mediated eIF2alpha phosphorylation. Neoplasia 2010;12(1):61-68; PMID:20072654
- Ibe S, Qin Z, Schuler T, Preiss S, Blankenstein T. Tumor rejection by disturbing tumor stroma cell interactions. J Exp Med 2001;194(11):1549-59; PMID:11733570; http://dx.doi.org/10.1084/jem.194.11.1549
- Pelaez B, Campillo JA, Lopez-Asenjo JA, Subiza JL. Cyclophosphamide induces the development of early myeloid cells suppressing tumor cell growth by a nitric oxide-dependent mechanism. J Immunol 2001;166(11):6608-15; PMID:11359814; http://dx.doi.org/10.4049/jimmunol.166.11.6608
- Fuentes D, Avellanet J, Garcia A, Iglesias N, Gabri MR, Alonso DF, Vazquez AM, Perez R, Montero E. Combined therapeutic effect of a monoclonal anti-idiotype tumor vaccine against NeuGc-containing gangliosides with chemotherapy in a breast carcinoma model. Breast Cancer Res Treat 2010;120(2):379-389; PMID:19377876; http://dx.doi.org/10.1007/s10549-009-0399-9
- Walter S, Weinschenk T, Stenzl A, Zdrojowy R, Pluzanska A, Szczylik C, Staehler M, Brugger W, Dietrich PY, Mendrzyk R, et al. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat Med 2012;18:1254-61; PMID:22842478; http://dx.doi.org/10.1038/nm.2883
- Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 2000;92(3):205-16; PMID:10655437; http://dx.doi.org/10.1093/jnci/92.3.205
- FDA. Guidance for Industry: Clinical Considerations for Therapeutic Cancer Vaccines. 2011.
- Hoos A. Evolution of end points for cancer immunotherapy trials. Ann Oncol 2012;23 Suppl 8:viii47-52; PMID:22918928; http://dx.doi.org/10.1093/annonc/mds263
- Higano CS, Schellhammer PF, Small EJ, Burch PA, Nemunaitis J, Yuh L, Provost N, Frohlich MW. Integrated data from 2 randomized, double-blind, placebo-controlled, phase 3 trials of active cellular immunotherapy with sipuleucel-T in advanced prostate cancer. Cancer 2009;115(16):3670-79; PMID:19536890; http://dx.doi.org/10.1002/cncr.24429
- Small EJ, Schellhammer PF, Higano CS, Redfern CH, Nemunaitis JJ, Valone FH, Verjee SS, Jones LA, Hershberg RM. Placebo-controlled phase III trial of immunologic therapy with sipuleucel-T (APC8015) in patients with metastatic, asymptomatic hormone refractory prostate cancer. J Clin Oncol 2006;24(19):3089-94; PMID:16809734; http://dx.doi.org/10.1200/JCO.2005.04.5252
- Butts C, Socinski MA, Mitchell PL, Thatcher N, Havel L, Krzakowski M, Nawrocki S, Ciuleanu TE, Bosquée L, Trigo JM, et al. Tecemotide (L-BLP25) versus placebo after chemoradiotherapy for stage III non-small-cell lung cancer (START): a randomised, double-blind, phase 3 trial. Lancet Oncol 2014;15(1):59-68; PMID:24331154; http://dx.doi.org/10.1016/S1470-2045(13)70510-2
- Kantoff PW, Schuetz TJ, Blumenstein BA, Glode LM, Bilhartz DL, Wyand M, Manson K, Panicali DL, Laus R, Schlom J, et al. Overall survival analysis of a phase II randomized controlled trial of a Poxviral-based PSA-targeted immunotherapy in metastatic castration-resistant prostate cancer. J Clin Oncol 2010;28(7):1099-105; PMID:20100959; http://dx.doi.org/10.1200/JCO.2009.25.0597
- Ullenhag GJ, Frodin JE, Jeddi-Tehrani M, Strigård K, Eriksson E, Samanci A, Choudhury A, Nilsson B, Rossmann ED, Mosolits S, et al. Durable carcinoembryonic antigen (CEA)-specific humoral and cellular immune responses in colorectal carcinoma patients vaccinated with recombinant CEA and granulocyte/macrophage colony-stimulating factor. Clin Cancer Res 2004;10(10):3273-81; PMID:15161680; http://dx.doi.org/10.1158/1078-0432.CCR-03-0706
- Staff C, Magnusson CG, Hojjat-Farsangi M, Mosolits S, Liljefors M, Frödin JE, Wahrén B, Mellstedt H, Ullenhag GJ. Induction of IgM, IgA and IgE antibodies in colorectal cancer patients vaccinated with a recombinant CEA protein. J Clin Immunol 2012;32:855-65; PMID:22382876
- Mosolits S, Ullenhag G, Mellstedt H. Therapeutic vaccination in patients with gastrointestinal malignancies. A review of immunological and clinical results. Ann Oncol 2005;16(6):847-62; PMID:15829493; http://dx.doi.org/10.1093/annonc/mdi192
- Yu JS, Liu G, Ying H, Yong WH, Black KL, Wheeler CJ. Vaccination with tumor lysate-pulsed dendritic cells elicits antigen-specific, cytotoxic T-cells in patients with malignant glioma. Cancer Res 2004;64(14):4973-79; PMID:15256471; http://dx.doi.org/10.1158/0008-5472.CAN-03-3505
- Wierecky J, Muller MR, Wirths S, Halder-Oehler E, Dörfel D, Schmidt SM, Häntschel M, Brugger W, Schröder S, Horger MS, et al. Immunologic and clinical responses after vaccinations with peptide-pulsed dendritic cells in metastatic renal cancer patients. Cancer Res 2006;66(11):5910-18; PMID:16740731; http://dx.doi.org/10.1158/0008-5472.CAN-05-3905
- Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Staehler M, Negrier S, Chevreau C, Desai AA, Rolland F, et al. Sorafenib for treatment of renal cell carcinoma: Final efficacy and safety results of the phase III treatment approaches in renal cancer global evaluation trial. J Clin Oncol 2009;27(20):3312-18; PMID:19451442; http://dx.doi.org/10.1200/JCO.2008.19.5511
- Motzer RJ, Michaelson MD, Redman BG, Hudes GR, Wilding G, Figlin RA, Ginsberg MS, Kim ST, Baum CM, DePrimo SE, et al. Activity of SU11248, a multitargeted inhibitor of vascular endothelial growth factor receptor and platelet-derived growth factor receptor, in patients with metastatic renal cell carcinoma. J Clin Oncol 2006;24(1):16-24; PMID:16330672; http://dx.doi.org/10.1200/JCO.2005.02.2574
- Hodge JW, Chakraborty M, Kudo-Saito C, Garnett CT, Schlom J. Multiple costimulatory modalities enhance CTL avidity. J Immunol 2005;174(10):5994-6004; PMID:15879092; http://dx.doi.org/10.4049/jimmunol.174.10.5994
- Karyampudi L, Lamichhane P, Scheid AD, Kalli KR, Shreeder B, Krempski JW, Behrens MD, Knutson KL. Accumulation of memory precursor CD8 T cells in regressing tumors following combination therapy with vaccine and anti-PD-1 antibody. Cancer Res 2014; 74:2974-85; PMID:24728077; http://dx.doi.org/10.1158/0008-5472.CAN-13-2564
- Fu J, Malm IJ, Kadayakkara DK, Levitsky H, Pardoll D, Kim YJ. Preclinical evidence that PD-1 blockade cooperates with cancer vaccine TEGVAX to elicit regression of established tumors. Cancer Res 2014;74:4042-52; PMID:24812273; http://dx.doi.org/10.1158/0008-5472
- Mkrtichyan M, Najjar YG, Raulfs EC, Abdalla MY, Samara R, Rotem-Yehudar R, Cook L, Khleif SN. Anti-PD-1 synergizes with cyclophosphamide to induce potent anti-tumor vaccine effects through novel mechanisms. Eur J Immunol 2011;41(10):2977-86; PMID:21710477; http://dx.doi.org/10.1002/eji.201141639
- Madan RA, Mohebtash M, Arlen PM, Vergati M, Rauckhorst M, Steinberg SM, Tsang KY, Poole DJ, Parnes HL, Wright JJ, et al. Ipilimumab and a poxviral vaccine targeting prostate-specific antigen in metastatic castration-resistant prostate cancer: a phase 1 dose-escalation trial. Lancet Oncol 2012;13(5):501-8; PMID:22326924; http://dx.doi.org/10.1016/S1470-2045(12)70006-2
- Le DT, Lutz E, Uram JN, Sugar EA, Onners B, Solt S, Zheng L, Diaz LA Jr, Donehower RC, Jaffee EM, et al. Evaluation of ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. J Immunother 2013;36(7):382-9; PMID:23924790; http://dx.doi.org/10.1097/CJI.0b013e31829fb7a2
- Ribas A, Comin-Anduix B, Chmielowski B, Jalil J, de la Rocha P, McCannel TA, Ochoa MT, Seja E, Villanueva A, Oseguera DK, et al. Dendritic cell vaccination combined with CTLA4 blockade in patients with metastatic melanoma. Clin Cancer Res 2009;15(19):6267-76; PMID:19789309; http://dx.doi.org/10.1158/1078-0432.CCR-09-1254
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366(26):2443-54; PMID:22658127; http://dx.doi.org/10.1056/NEJMoa1200690
- Cho D, Sosman JA, Sznol M et al. Clinical activity, safety, and biomarkers of MPDL3280A, an engineered PD-L1 antibody in patients with metastatic renal cell carcinoma (mRCC). J Clin Oncol 31[(15_suppl), Abstract 4505]. 2013.