
Abstract
Transient Receptor Potential Vanilloid 4 (TRPV4) is a mechano- and osmosensitive cation channel that is highly expressed in chondrocytes, the cells in cartilage. A large number of mutations in TRPV4 have been linked to skeletal dysplasias, and the goal of this addendum is to shed light on the mechanisms by which mutations in TRPV4 can cause skeletal dysplasias by focusing on 3 recent publications. These papers suggest that skeletal dysplasia-causing TRPV4 mutations reprogram chondrocytes to increase follistatin production, which inhibits BMP signaling, thus slowing the process of endochondral ossification and leading to skeletal dysplasia. In spite of these important advances in our understanding of the disease mechanism, much remains to be elucidated. Nonetheless, these new data suggest that inhibiting aberrant TRPV4 activity in the cartilage may be a promising direction for therapeutic intervention.
Abbreviations
| TRPV4 | = | transient receptor potential vanilloid 4 |
| BMP | = | bone morphogenetic protein |
| TRP | = | transient receptor potential |
| TRPV | = | transient receptor potential vanilloid |
| SRY | = | sex determining region Y |
| SOX9 | = | SRY-box 9 |
| Col2a1 | = | collagen type II, alpha 1 |
| iPSCs | = | induced pluripotent stem cells |
| RUNX2 | = | runt-related transcription factor 2 |
| FST | = | follistatin |
| BV/TV | = | bone volume per trabecular volume |
| CRE | = | cyclic AMP response element |
| CREB | = | CRE binding protein |
Introduction
The Transient Receptor Potential (TRP) superfamily of ion channels has been implicated in sensation of a variety of physical and chemical stimuli throughout the body. TRP channels are typically non-selective cation channels that preferentially permeate calcium due to cellular calcium gradients.Citation1 Most TRP channels contain 6 transmembrane domains with cytoplasmic termini that may interact with other proteins and the cytoskeleton. The TRP Vanilloid (TRPV) channels have been implicated in thermo-, osmo-, and mechanosensation in a wide variety of tissues. In particular, TRPV4 is a polymodally activated TRP channel that is expressed in a variety of tissues and exhibits diverse functions. In sensory neurons, such as the dorsal root and trigeminal ganglion neurons, TRPV4 has been shown to play a role in mechanosensation, osmosensation and nociception/pain.Citation2,3 TRPV4 activation in keratinocytes of the skin by UVB mediates sunburn pain and tissue damage.Citation4 TRPV4 is also expressed in kidney, where it acts not only as an osmosensor, but also as a mechanosensor that can sense shear flow.Citation5 Furthermore, certain TRPV4 polymorphisms in humans coincide with alterations in systemic osmolarity.Citation6 In knockout mice, the lack of TRPV4 decreases homeostatic water intake behavior and thus increases systemic osmolarity.Citation7 In zebrafish, in situ hybridization has shown high TRPV4 expression in the developing kidney and in developing cartilage.Citation8
TRPV4 in the Skeletal System
Along with diverse roles throughout the body, TRPV4 plays a crucial role in the skeletal system, where it is expressed in several tissues, such as cartilage and bone.Citation9 In bone, TRPV4 plays a key role in bone homeostasis by regulating osteoclast differentiation. Mice lacking TRPV4 have greater bone mass due to impaired bone resorption.Citation10 In cartilage, TRPV4 has been shown to play a crucial role in chondrocyte mechanotransduction and osmosensation.Citation11,12 Blocking TRPV4 in chondrocytes prevents an anabolic response to mechanical loads, while activating TRPV4 mimics the mechanical loading response.Citation12 In cartilage, osmotic fluctuations occur as the tissue is mechanically loaded, and chondrocytes sense hypoosmotic change via TRPV4.Citation11 Furthermore, TRPV4 is involved in the normal maintenance of the joint, as mice lacking this channel develop earlier and more severe osteoarthritis than their wild-type counterparts.Citation13 Not only does TRPV4 play a key role in adult chondrocytes, it is also involved in differentiation of stem cells into a chondrogenic lineage.Citation14,15 TRPV4 expression levels increase as ATDC5 and C3H10T1/2 cells differentiate into chondrocytes. Furthermore, activation of TRPV4 in these cells, in concert with growth factors, leads to increased levels of the chondrogenic transcription factor, SRY (sex determining region Y)-box 9 (SOX9), and synthesis of cartilage-specific matrix molecules.Citation15 TRPV4 may also be involved in the conversion of cartilage to bone as it plays a role in chondrocyte volume regulation, which is integral to chondrocyte hypertrophy during endochondral ossification.Citation16
TRPV4 Mutations
The first disease-causing TRPV4 mutations were identified in 2008.Citation17 Since that time, TRPV4 mutations that cause diseases, i.e., “channelopathies,” have been identified at an astonishing rate, including 47 skeletal dysplasia-inducing mutationsCitation18, 20 neuropathy-inducing mutationsCitation19, and one osmoregulatory polymorphism.Citation6 The skeletal dysplasia mutations have a wide range of functional consequences, from relatively mild brachyolmia to severe neonatal lethal metatropic dysplasia.Citation20 Interestingly, these mutations occur at a wide range of sites throughout the TRPV4 protein, with no apparent relationship between mutation site and functional consequences: 4 mutations are in the amino-terminus, 26 are within the ankyrin repeat domains, 18 are within the transmembrane domains, and 9 are in the carboxy-terminus.Citation20 While some mutations cause both neural and skeletal disease,Citation21 it is surprising to find seemingly very different disease phenotypes caused by a single channel,Citation20 further supporting the diverse and tissue-specific function of this channel.
New Insight Into How TRPV4 Mutations Cause Skeletal Dysplasia
Our recent paperCitation22 and 2 other reportsCitation23,24 have begun to unravel the mystery of how these TRPV4 mutations cause skeletal dysplasias. Briefly, Weinstein et al. describe the process of endochondral ossification that is affected by a TRPV4 mutation and leads to skeletal dysplasias.Citation23 Saitta et al. describe how the biochemical pathway driving endochondral ossification is malfunctioning.Citation24 Our study provides a mechanism that explains how the mutations are able to affect that pathway by overproducing an inhibitor of bone morphogenetic proteins (BMPs).Citation22 Importantly, given the tissue-specific function of this channel, these 3 studies have examined the effects of TRPV4 mutations in cartilage and chondrocytes, which provide an appropriate cellular context as the tissues and cells most affected by the mutations. Taken together, these studies provide new insights into how TRPV4 mutations can lead to skeletal dysplasias.
The earliest description of TRPV4 mutation-induced skeletal dysplasias speculated that these malformations might develop because of problems with endochondral ossification.Citation17 Further studies examined cartilage from individuals with severe lethal metatropic dysplasia and found poorly organized growth plate cartilage with few hypertrophic chondrocytes and islands of cartilage left in zones that should be fully mineralized, which also suggested problems with endochondral ossification of the cartilage.Citation25 Weinstein et al. created mice that expressed the TRPV4R594H mutation in order to examine more carefully how the mutation alters the process of endochondral ossification.Citation23 The channelopathy mutation was directed to chondrocytes by the collagen, type II, alpha 1 (Col2a1) promoter, an early and strong chondrocytic promoter. In humans, TRPV4R594H causes a moderately severe skeletal dysplasia, spondylometaphyseal dysplasia, Kozlowski type. The mice not only had major skeletal deformities, but also showed clear evidence of impaired differentiation of hypertrophic chondrocytes, with the most severely affected mice having almost no mineralization in endochondral bones. Taken together, these data suggest that robust expression of a skeletal dysplasia-causing TRPV4 channelopathy mutation, directed early to the chondrocytic lineage, evokes severe skeletal deformities that resulted from dysfunctional endochondral ossification.
With the knowledge that improper endochondral ossification is responsible for the skeletal deformities, the next question is what causes the problems with endochondral ossification. A recent paper by Saitta et al.Citation24 has very effectively shown how the endochondral ossification process is disrupted in the presence of a TRPV4 dysplasia-inducing mutation. Using induced pluripotent stem cells (iPSCs) derived from patients with the severe lethal TRPV4I604M mutation, they created stem cell based micromasses and examined their susceptibility to key regulators of chondrocyte hypertrophy. In normal cells, BMP2 drives the production of the key chondrocyte proteins type II collagen, SOX9, and aggrecan; and, during chondrocyte hypertrophy, it drives the production of type X collagen and runt-related transcription factor 2 (RUNX2). In iPSCs with the TRPV4I604M skeletal dysplasia mutation, mRNA for all of these genes was significantly decreased compared to micromasses from control iPSCs. The micromasses with the TRPV4I604M mutation were significantly less sensitive to BMP2.
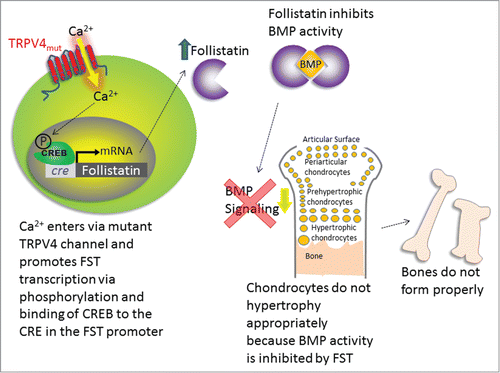
This finding of decreased BMP2 sensitivity fits like the missing puzzle piece into our recent findings of how TRPV4 channelopathy mutations can cause skeletal dysplasias (). Our findings show that Ca2+ entering through the mutant TRPV4 channels activates a maladapted genetic program, which leads to the overproduction of follistatin (FST), a known and potent binding and neutralizing factor for BMP-related ligands. FST overproduction is transcriptionally mediated and critically depends on Ca2+ entering the chondrocyte via the TRPV4 channelopathy mutation.Citation22 Indeed, the expression of a variety of skeletal dysplasia-inducing TRPV4 mutations in chondrocytes results in overexpression of FST. This increase in FST expression is specific to skeletal dysplasia-inducing TRPV4 mutations, in that an arthropathy-inducing TRPV4 mutation does not cause an increase in FST. BMP signaling is key for the transition of chondrocytes to hypertrophic chondrocytes and onto bone,Citation26 and FST attenuates BMP signaling by acting as a “BMP-sponge,” preventing BMPs from binding to their receptors. In separate experiments using a chick embryo model, we also confirmed that excess FST reduced bone ossification in developing limbs. Another piece of the puzzle worth mentioning dates back more than a decade to a study by Devlin et al., which showed that overexpression of noggin, an inhibitor of BMP, caused a significant decrease in bone volume per trabecular volume (BV/TV) (calcified bone fraction) in the mouse skeleton.Citation27 Similarly, we also saw a significant decrease in BV/TV in TRPV4V620I mice, which overexpressed FST. Therefore, with the overexpression of 2 different BMP antagonists, a similar reduction in bone calcification occurs.
Figure 1. Schematic of the mechanisms by which TRPV4 mutations may lead to skeletal dysplasias. Ca2+ enters through the mutant channel, causing phosphorylation of CREB, which binds CRE and causes FST transcription. FST inhibits BMP activity, which prevents chondrocytes from undergoing hypertrophy and forming bone, thus leading to skeletal dysplasia.
The Complex Role of Calcium
Most of the TRPV4 channelopathy mutations that have been described result in an alteration in channel conductance, which results in altered levels of intracellular Ca2+. Most of the dysplasia-causing mutations result in increased basal current density with both increased basal Ca2+ levels and higher Ca2+ levels in response to channel activation.Citation17,22,25,28,29 We and others have looked for correlations between these Ca2+ levels and disease severity, as it seems logical that, if altered channel function is causing disease, the more affected channel activity should cause worse disease. Surprisingly, there appears to be no direct correlation between the current density or Ca2+ concentrations and disease severity.Citation22,28 Loukin et al., however, did find a trend between the relative change in current in response to TRPV4 agonists and disease severity.Citation28 When averaged across broad disease categories, the relative agonist-induced current decreased as disease severity decreased, which may be due to an increase in basal current. However, many individual mutations deviated from this trend (which did not reach statistical significance), suggesting that this measure does not satisfactorily explain the relationship between channel activity and disease severity.
Thus, there appears to be a complex relationship between disease severity and Ca2+. While we did not see a relationship between basal Ca2+ or agonist-induced peak Ca2+ with disease severity, we did find a Ca2+-dependent mechanism that causes increases in FST, which could lead to skeletal dysplasia. This FST increase was prevented when the channel was mutated so that it could not permeate Ca2+, either via a mutation that blocked the channel completely or a mutation that allowed monovalent, but not divalent cations to pass.Citation22 Antagonism of channel activity by a small molecule inhibitor also prevented the FST increase. Furthermore, the FST promoter contains a Cyclic AMP Response Element (CRE). This CRE is thought to be Ca2+ sensitive in that a Ca2+ signal can activate Ca2+/calmodulin-activated kinase that phosphorylates CRE binding protein,Citation30 which then binds CRE and promotes FST transcription. Our results unambiguously demonstrate that the TRPV4 mutation-driven FST increase requires an intact CRE in the promoter of the FST gene.Citation22 Thus, FST upregulation depends on Ca2+ influx via TRPV4 channelopathy mutations. It also critically relies on an intact CRE DNA sequence in the FST promoter, a transcriptional activation mechanism that is known to be Ca2+-dependent. However, our results and the results of others combined do not explain how disease severity in patients correlates with specific mutations of TRPV4 channel proteins.
Conclusion
Although recent studies have made great strides in piecing together the puzzle of TRPV4 channelopathies and skeletal dysplasias, many aspects of the disease process remain to be elucidated. It is likely that different cellular signaling mechanisms may cause different subsets of disease pathology, e.g., some channelopathy mutations are likely de novo targets for phosphorylation. Modeling work could possibly help to clarify the effects of different TRPV4 mutations on the structure of the protein. The TRPV4 amino-terminus has been elucidated structurally, and the TRPV1 cryo-electron microscopy structure is now available. A number of structurally related K-channels have been resolved crystallographically as well, so that, now, structures of TRPV4 channelopathy mutations can be illustrated by homology modeling, yielding a novel angle of insight.
Ultimately, the goal is to first understand how these mutations cause disease, then to develop effective interventions for these skeletal dysplasia patients. In this respect, we established the promising finding that blocking mutant TRPV4 channels with a small molecule TRPV4 inhibitor prevented excessive FST production.Citation22 Given that TRPV4 inhibitors have been used therapeutically in other settings,Citation3,31 it is possible that appropriately timed delivery of a TRPV4 inhibitor may restore normal endochondral ossification toward physiological skeletal development. In addition to chemical agonists, inhibiting TRPV4 via gene therapy approaches, such as dominant-negative TRPV4 or transitory RNAi expression,Citation32 may provide another therapeutic avenue.
In conclusion, 6 years after the initial description of a TRPV4 channelopathy mutation, our study,Citation22 combined with others,Citation23,24 has yielded new insights into the mechanisms by which TRPV4 mutations cause skeletal dysplasias. We have shown that TRPV4 channelopathies cause skeletal dysplasias by inducing a Ca2+-dependent upregulation of FST in chondrocytes, which inhibits BMP signaling in the developing skeleton, thus preventing chondrocytes from undergoing normal hypertrophy, inhibiting endochondral ossification, and ultimately resulting in skeletal dysplasia.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Leddy, H. A., McNulty, A. L., Lee, S. H., Rothfusz, N. E., Gloss, B., Kirby, M. L., Hutson, M. R., Cohn, D. H., Guilak, F., and Liedtke, W. Follistatin in chondrocytes: the link between TRPV4 channelopathies and skeletal malformations. FASEB Journal Published online before print February 27, 2014, doi: 10.1096/fj.13-245936 fj.13-245936
Funding
This work was supported in part by the US. National Institutes of Health grants AR48182 (FG), AR48852 (FG), AG15768 (FG), AR50245 (FG), AG46927 (FG), and DE018549 (WL).
References
- Kuipers AJ, Middelbeek J, van Leeuwen FN. Mechanoregulation of cytoskeletal dynamics by Trp channels. Eur J Cell Biol 2012; 91:834-46; PMID:22727433; http://dx.doi.org/10.1016/j.ejcb.2012.05.006
- Alessandri-Haber N, Yeh JJ, Boyd AE, Parada CA, Chen X, Reichling DB, Levine JD. Hypotonicity Induces Trpv4-Mediated Nociception in Rat. Neuron 2003; 39:497-511; PMID:12895423; http://dx.doi.org/10.1016/S0896-6273(03)00462-8
- Chen Y, Williams SH, McNulty AL, Hong JH, Lee SH, Rothfusz NE, Parekh PK, Moore C, Gereau RWt, Taylor AB, et al. Temporomandibular joint pain: a critical role for Trpv4 in the trigeminal ganglion. Pain 2013; 154:1295-304; PMID:23726674; http://dx.doi.org/10.1016/j.pain.2013.04.004
- Moore C, Cevikbas F, Pasolli HA, Chen Y, Kong W, Kempkes C, Parekh P, Lee SH, Kontchou NA, Yeh I, et al. Uvb radiation generates sunburn pain and affects skin by activating epidermal Trpv4 ion channels and triggering endothelin-1 signaling. Proc Natl Acad Sci U S A 2013; 110:E3225-34; PMID:23929777; http://dx.doi.org/10.1073/pnas.1312933110
- Pochynyuk O, Zaika O, O’Neil RG, Mamenko M. Novel insights into Trpv4 function in the kidney. Pflugers Arch 2013; 465:177-86; PMID:23207579; http://dx.doi.org/10.1007/s00424-012-1190-z
- Tian W, Fu Y, Garcia-Elias A, Fernandez-Fernandez JM, Vicente R, Kramer PL, Klein RF, Hitzemann R, Orwoll ES, Wilmot B, et al. A loss-of-function nonsynonymous polymorphism in the osmoregulatory Trpv4 gene is associated with human hyponatremia. Proc Natl Acad Sci U S A 2009; 106:14034-9; PMID:19666518; http://dx.doi.org/10.1073/pnas.0904084106
- Liedtke W, Friedman JM. Abnormal osmotic regulation in Trpv4– mice. Proc Natl Acad Sci USA 2003; 100:13698-703; PMID:14581612; http://dx.doi.org/10.1073/pnas.1735416100
- Mangos S, Liu Y, Drummond I. Dynamic expression of the osmosensory channel Trpv4 in multiple developing organs in zebrafish. Gene Expr Patterns 2007; 7:480-4; PMID:17161658; http://dx.doi.org/10.1016/j.modgep.2006.10.011
- Guilak F, Leddy HA, Liedtke W. Transient receptor potential vanilloid 4: the sixth sense of the musculoskeletal system? Ann N Y Acad Sci 2010; 1192:404-9; PMID:20392266; http://dx.doi.org/10.1111/j.1749-6632.2010.05389.x
- Masuyama R, Vriens J, Voets T, Karashima Y, Owsianik G, Vennekens R, Lieben L, Torrekens S, Moermans K, Vanden Bosch A, et al. Trpv4-mediated calcium influx regulates terminal differentiation of osteoclasts. Cell Metab 2008; 8:257-65; PMID:18762026; http://dx.doi.org/10.1016/j.cmet.2008.08.002
- Phan MN, Leddy HA, Votta BJ, Kumar S, Levy DS, Lipshutz DB, Lee SH, Liedtke W, Guilak F. Functional characterization of Trpv4 as an osmotically sensitive ion channel in porcine articular chondrocytes. Arthritis Rheum 2009; 60:3028-37; PMID:19790068; http://dx.doi.org/10.1002/art.24799
- O’Conor CJ, Leddy HA, Benefield HC, Liedtke WB, Guilak F. Trpv4-mediated mechanotransduction regulates the metabolic response of chondrocytes to dynamic loading. Proc Natl Acad Sci U S A 2014; 111:1316-21; http://dx.doi.org/10.1073/pnas.1319569111
- Clark AL, Votta BJ, Kumar S, Liedtke W, Guilak F. Chondroprotective role of the osmotically sensitive ion channel transient receptor potential vanilloid 4: age- and sex-dependent progression of osteoarthritis in Trpv4-deficient mice. Arthritis Rheum 2010; 62:2973-83; PMID:20583100; http://dx.doi.org/10.1002/art.27624
- O’Conor CJ, Griffin TM, Liedtke W, Guilak F. Increased susceptibility of Trpv4-deficient mice to obesity and obesity-induced osteoarthritis with very high-fat diet. Ann Rheum Dis 2013; 72:300-4; http://dx.doi.org/10.1136/annrheumdis-2012-202272
- Muramatsu S, Wakabayashi M, Ohno T, Amano K, Ooishi R, Sugahara T, Shiojiri S, Tashiro K, Suzuki Y, Nishimura R, et al. Functional gene screening system identified Trpv4 as a regulator of chondrogenic differentiation. J Biol Chem 2007; 282:32158-67; PMID:17804410; http://dx.doi.org/10.1074/jbc.M706158200
- Lewis R, Feetham CH, Barrett-Jolley R. Cell volume regulation in chondrocytes. Cell Physiol Biochem 2011; 28:1111-22; PMID:22179000; http://dx.doi.org/10.1159/000335847
- Rock MJ, Prenen J, Funari VA, Funari TL, Merriman B, Nelson SF, Lachman RS, Wilcox WR, Reyno S, Quadrelli R, et al. Gain-of-function mutations in Trpv4 cause autosomal dominant brachyolmia. Nat Genet 2008; 40:999-1003; PMID:18587396; http://dx.doi.org/10.1038/ng.166
- Nishimura G, Lausch E, Savarirayan R, Shiba M, Spranger J, Zabel B, Ikegawa S, Superti-Furga A, Unger S. Trpv4-associated skeletal dysplasias. Am J Med Genet C Semin Med Genet 2012; 160C:190-204; PMID:22791502; http://dx.doi.org/10.1002/ajmg.c.31335
- McEntagart M. Trpv4 axonal neuropathy spectrum disorder. J Clin Neurosci 2012; 19:927-33; PMID:22617546; http://dx.doi.org/10.1016/j.jocn.2011.12.003
- Nilius B, Voets T. The puzzle of Trpv4 channelopathies. EMBO Rep 2013; 14:152-63; PMID:23306656; http://dx.doi.org/10.1038/embor.2012.219
- Cho TJ, Matsumoto K, Fano V, Dai J, Kim OH, Chae JH, Yoo WJ, Tanaka Y, Matsui Y, Takigami I, et al. Trpv4-pathy manifesting both skeletal dysplasia and peripheral neuropathy: a report of three patients. Am J Med Genet A 2012; 158A:795-802; PMID:22419508; http://dx.doi.org/10.1002/ajmg.a.35268
- Leddy HA, McNulty AL, Lee SH, Rothfusz NE, Gloss B, Kirby ML, Hutson MR, Cohn DH, Guilak F, Liedtke W. Follistatin in chondrocytes: the link between Trpv4 channelopathies and skeletal malformations. FASEB J 2014; 28:2525-37; PMID:24577120; http://dx.doi.org/10.1096/fj.13-245936
- Weinstein MM, Tompson SW, Chen Y, Lee B, Cohn DH. Mice expressing mutant Trpv4 recapitulate the human Trpv4 disorders. J Bone Miner Res 2014; 29:1815-22; DOI: 10.1002jbmr.2220; PMID:24644033; http://dx.doi.org/10.1002jbmr.2220
- Saitta B, Passarini J, Sareen D, Ornelas L, Sahabian A, Argade S, Krakow D, Cohn DH, Svendsen CN, Rimoin DL. Patient-derived skeletal dysplasia ipscs display abnormal chondrogenic marker expression and regulation by Bmp2 and Tgfbeta1. Stem Cells Dev 2014; 23:1464-78; PMID:24559391; http://dx.doi.org/10.1089scd.2014.0014
- Camacho N, Krakow D, Johnykutty S, Katzman PJ, Pepkowitz S, Vriens J, Nilius B, Boyce BF, Cohn DH. Dominant Trpv4 mutations in nonlethal and lethal metatropic dysplasia. Am J Med Genet A 2010; 152A:1169-77; PMID:20425821; http://dx.doi.org/10.1002/ajmg.a.33392
- Yoon BS, Pogue R, Ovchinnikov DA, Yoshii I, Mishina Y, Behringer RR, Lyons KM. Bmps regulate multiple aspects of growth-plate chondrogenesis through opposing actions on Fgf pathways. Development 2006; 133:4667-78; PMID:17065231; http://dx.doi.org/10.1242/dev.02680
- Devlin RD, Du Z, Pereira RC, Kimble RB, Economides AN, Jorgetti V, Canalis E. Skeletal overexpression of noggin results in osteopenia and reduced bone formation. Endocrinology 2003; 144:1972-8; PMID:12697704; http://dx.doi.org/10.1210/en.2002-220918
- Loukin S, Su Z, Kung C. Increased basal activity is a key determinant in the severity of human skeletal dysplasia caused by Trpv4 mutations. PloS one 2011; 6:e19533; PMID:21573172; http://dx.doi.org/10.1371/journal.pone.0019533
- Krakow D, Vriens J, Camacho N, Luong P, Deixler H, Funari TL, Bacino CA, Irons MB, Holm IA, Sadler L, et al. Mutations in the gene encoding the calcium-permeable ion channel Trpv4 produce spondylometaphyseal dysplasia, kozlowski type and metatropic dysplasia. Am J Hum Genet 2009; 84:307-15; PMID:19232556; http://dx.doi.org/10.1016/j.ajhg.2009.01.021
- West AE, Chen WG, Dalva MB, Dolmetsch RE, Kornhauser JM, Shaywitz AJ, Takasu MA, Tao X, Greenberg ME. Calcium regulation of neuronal gene expression. Proc Natl Acad Sci USA 2001; 98:11024-31; PMID:11572963; http://dx.doi.org/10.1073/pnas.191352298
- Thorneloe KS, Cheung M, Bao W, Alsaid H, Lenhard S, Jian MY, Costell M, Maniscalco-Hauk K, Krawiec JA, Olzinski A, et al. An orally active Trpv4 channel blocker prevents and resolves pulmonary edema induced by heart failure. Sci Transl Med 2012; 4:159ra48; http://dx.doi.org/10.1126/scitranslmed.3004276
- Li J, Kanju P, Patterson M, Chew WL, Cho SH, Gilmour I, Oliver T, Yasuda R, Ghio A, Simon SA, et al. Trpv4-mediated calcium influx into human bronchial epithelia upon exposure to diesel exhaust particles. Environ Health Perspect 2011; 119:784-93; PMID:21245013; http://dx.doi.org/10.1289/ehp.1002807