
Abstract
Cellular senescence is classically considered a tumor suppressive mechanism. In addition to having stably exited the cell cycle, senescent cells secrete inflammatory factors. We recently demonstrated that senescence correlates with accelerated cancer progression in a mouse model of pancreatic ductal adenocarcinoma. Here, we discuss the implications of this study.
Abbreviations
| PanIN | = | pancreatic intraepithelial neoplasia |
| PDAC | = | pancreatic ductal adenocarcinoma |
| SASP | = | senescence-associated secretory phenotype |
Author View
Pancreatic ductal adenocarcinoma (PDAC) is primarily an incurable disease with a median survival of only 6 months, due in large part to the late clinical presentation of the disease. The majority of PDACs arise from activating mutations within the GTPase Kirsten rat sarcoma viral oncogene homolog (KRAS) gene that drive the development and stepwise progression of preneoplastic lesions called pancreatic intraepithelial neoplasms (PanINs). As PanINs progress the cells acquire additional mutations within various tumor suppressor genes including cyclin-dependent kinase inhibitor 2A (CDKN2A) and tumor protein p53 (TP53), and the lesions display increased dysplasia and proliferation.Citation1 It is believed that oncogene activation drives the cells that consitute early PanIN lesions into cellular senescence, thus halting their proliferation and preventing PanIN progression.Citation2 Cellular senescence corresponds to an irreversible exit from the cell cycle that is triggered by various stimuli and prevents the uncontrolled proliferation of damaged cells. Studies in both mouse and human tissues have demonstrated that senescent cells accumulate within preneoplastic lesions including PanIN, but are absent in frank carcinoma. Furthermore, in numerous mouse models of cancer, abrogating the molecular pathways required for entry into cellular senescence correlates with accelerated cancer progression. These observations helped establish the classically accepted role for senescence as a barrier to cancer progression.Citation3
Recently, we demonstrated that mouse embryonic fibroblasts with inactivation of the chromatin-associated SWI-independent transcription regulator family member B (Sin3B) protein are refractory to oncogene-induced senescence. Unlike other proteins required for cellular senescence, including p16INK4A (encoded by CDKN2A) and p53 (encoded by TP53), deletion of Sin3B does not sensitize cells to transformation.Citation4 Therefore, modulating Sin3B levels, as opposed to other regulators of senescence, provides a context within which the effects of cellular senescence in cancer progression can be studied without the confounding effects of accelerated transformation. To better understand the role of cellular senescence in PDAC progression, we genetically engineered mice with pancreatic-specific expression of oncogenic KRasG12D concomitant with deletion of the Sin3B locus. Based on the requirement for Sin3B for entry into cellular senescence and the role of senescence as a barrier to cancer progression, we hypothesized that Sin3B deletion would accelerate KRasG12D-driven PDAC progression in the mouse. Surprisingly, we observed the opposite phenomenon: Sin3B-deleted animals exhibited delayed PanIN and PDAC progression and displayed increased survival compared to their wild-type counterparts.Citation5
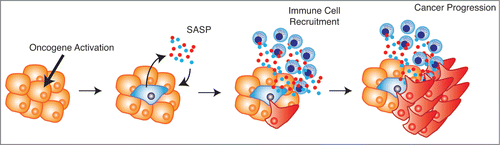
We initially asked whether this unexpected observation was due to a requirement for Sin3B in acinar-to-ductal metaplasia (ADM), an initiating event in at least some PDAC models. Although ADM was delayed in Sin3B-deleted pancreata in vivo, ADM initiation did not require Sin3B in vitro. We hypothesized that this discrepancy reflected a non-cell autonomous effect of Sin3B on the pancreatic microenvironment. Emerging evidence suggests that cellular senescence has both cell autonomous and non-cell autonomous effects, and that senescent cells, although no longer cycling, actively communicate with neighboring cells and the surrounding tissue. Senescent cells produce and secrete numerous factors such as cytokines, chemokines, proteases, and growth factors, collectively referred to as the senescence-associated secretory phenotype (SASP). The SASP reinforces senescence in both an autocrine and a paracrine fashion, and recruits innate and adaptive immune cells.Citation3 Since PDAC relies on an inflammatory microenvironment, we postulated that the SASP, driven by Sin3B-dependent senescence, may paradoxically promote PDAC progression by recruiting immune cells that generate an inflammatory microenvironment. Accordingly, we observed lower expression levels of markers of senescence in Sin3B null pancreata, and the infrequent PanIN lesions within Sin3B-deleted pancreata were negative for established markers of senescence. Furthermore, we detected a drastic reduction in immune cell infiltration in Sin3B-deleted pancreata. We concluded that Sin3B was required for both KRas-induced senescence and inflammation in vivo, and that entry into senescence surprisingly correlated with pancreatic cancer progression.Citation5 While these observations remain correlative, we hypothesize that senescent cells stimulate the generation of an inflammatory microenvironment and promote PDAC progression, questioning the classical view of cellular senescence as merely a tumor suppressive mechanism in vivo ().
Figure 1. Senescence-associated cytokines establish an inflammatory microenvironment and promote cancer progression. Upon oncogene activation cells become senescent. The senescent cells produce and secrete the senescence-associated secretory phenotype (SASP), which reinforces senescence within the lesion and recruits immune cells to the surrounding tissue. The immune cells, along with the SASP, generate an inflammatory microenvironment, which in certain contexts fuels cancer progression.
Recent studies identified a role for the SASP in liver cancer development. However, although one study observed that the SASP inhibited tumor development,Citation6 the other reported that the SASP promoted tumor progression.Citation7 Although the reason for this difference is not immediately clear, it could be due to the different models used by the 2 groups, adding further complexity to the role of senescence and the SASP. To better define its role in cancer progression, factors that are required for the SASP but are not involved in cell cycle exit should be identified. In our model, interleukin-1 α (IL-1α) levels correlated with senescence and the inflammatory response in both mouse and human tissue. Several groups have previously identified IL-1α as an upstream regulator of the SASP both in vitro and in vivo.Citation8,9 Moreover, Chia and colleagues recently described that oncogenic KRas induces IL-1α expression in PDAC, which in turn stimulates NF-kβ activity, previously described to be required for the SASP.Citation10 These studies, in combination with our observations, point to a central role of IL-1α in stimulating an inflammatory microenvironment through the SASP. We believe that dissecting the contribution of IL-1α in various cancers, such as PDAC and hepatocellular carcinoma, will provide an avenue to directly investigate the role of the SASP in cancer progression. If the SASP promotes cancer progression in at least some contexts, IL-1α inhibition may represent a novel treatment option.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to all members of the David laboratory for helpful discussions.
Funding
This work was funded by the American Cancer Society (115014-RSG-08-054-01-GMC to GD), the National Institute of Health (5R01CA148639 and 5R21CA-155736 to GD), the Irma T. Hirschl Charitable Trust (GD), the Samuel Waxman Cancer Research Foundation (GD) and a Feinberg NYU individual grant (GD). DJC was supported by a predoctoral NIH training grant T32CA009161 (D. Levy).
References
- Hezel A, Kimmelman A, Stanger B, Bardeesy N, DePinho R. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev 2006; 20:1218-49; PMID:16702400; http://dx.doi.org/10.1101/gad.1415606
- Caldwell ME, DeNicola GM, Martins CP, Jacobetz MA. Cellular features of senescence during the evolution of human and murine ductal pancreatic cancer. Oncogene 2012; 31(12):1599-1608; PMID:21860420; http://dx.doi.org/10.1038/onc.2011.350
- Pérez-Mancera PA, Young AR, Narita M. Inside and out: the activities of senescence in cancer. Nat Rev Cancer 2014; 14(8):547-58; PMID:25030953; http://dx.doi.org/10.1038/nrc3773
- Grandinetti KB, Jelinic P, DiMauro T, Pellegrino J, Fernández Rodríguez R, Finnerty PM, Ruoff R, Bardeesy N, Logan SK, David G. Sin3B expression is required for cellular senescence and is up-regulated upon oncogenic stress. Cancer Res 2009; 69:6430-7; PMID:19654306; http://dx.doi.org/10.1158/0008-5472.CAN-09-0537
- Rielland M, Cantor DJ, Graveline R, Hajdu C, Mara L, Diaz B de D, Miller G, David G. Senescence-associated SIN3B promotes inflammation and pancreatic cancer progression. J Clin Invest 2014; 124:2125-35; PMID:24691445; http://dx.doi.org/10.1172/JCI72619
- Lujambio A, Akkari L, Simon J, Grace D, Tschaharganeh D, Bolden J, Zhao Z, Thapar V, Joyce J, Krizhanovsky V, et al. Non-cell-autonomous tumor suppression by p53. Cell 2013; 153:449-60; PMID:23562644; http://dx.doi.org/10.1016/j.cell.2013.03.020
- Yoshimoto S, Loo T, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013; 499:97-101; PMID:23803760; http://dx.doi.org/10.1038/nature12347
- Orjalo A, Bhaumik D, Gengler B, Scott G, Campisi J. Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6IL-8 cytokine network. Proc Natl Acad Sci U S A 2009; 106:17031-6; PMID:19805069; http://dx.doi.org/10.1073/pnas.`0905299106
- Acosta J, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton J, Athineos D, Kang T-W, Lasitschka F, Andrulis M, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol 2013; 15:978-90; PMID:23770676; http://dx.doi.org/10.1038/ncb2784
- Ling J, Kang Y, Zhao R, Xia Q, Lee D-FF, Chang Z, Li J, Peng B, Fleming JB, Wang H, et al. KrasG12D-induced IKK2βNF-κB activation by IL-1α and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell 2012; 21:105-20; PMID:22264792; http://dx.doi.org/10.1016/j.ccr.2011.12.006