Abstract
Hypoxia-inducible pro-death protein BNIP3 (BCL-2/adenovirus E1B 19-kDa interacting protein 3), provokes mitochondrial permeabilization causing cardiomyocyte death in ischemia-reperfusion injury. Inhibition of autophagy accelerates BNIP3-induced cell death, by preventing removal of damaged mitochondria. We tested the hypothesis that stimulating autophagy will attenuate BNIP3-induced cardiomyocyte death. Neonatal rat cardiac myocytes (NRCMs) were adenovirally transduced with BNIP3 (or LacZ as control; at multiplicity of infection = 100); and autophagy was stimulated with rapamycin (100 nM). Cell death was assessed at 48 h. BNIP3 expression increased autophagosome abundance 8-fold and caused a 3.6-fold increase in cardiomyocyte death as compared with control. Rapamycin treatment of BNIP3-expressing cells led to further increase in autophagosome number without affecting cell death. BNIP3 expression led to accumulation of autophagosome-bound LC3-II and p62, and an increase in autophagosomes, but not autolysosomes (assessed with dual fluorescent mCherry-GFP-LC3 expression). BNIP3, but not the transmembrane deletion variant, interacted with LC3 and colocalized with mitochondria and lysosomes. However, BNIP3 did not target to lysosomes by subcellular fractionation, provoke lysosome permeabilization or alter lysosome pH. Rather, BNIP3-induced autophagy caused a decline in lysosome numbers with decreased expression of the lysosomal protein LAMP-1, indicating lysosome consumption and consequent autophagosome accumulation. Forced expression of transcription factor EB (TFEB) in BNIP3-expressing cells increased lysosome numbers, decreased autophagosomes and increased autolysosomes, prevented p62 accumulation, removed depolarized mitochondria and attenuated BNIP3-induced death. We conclude that BNIP3 expression induced autophagosome accumulation with lysosome consumption in cardiomyocytes. Forced expression of TFEB, a lysosomal biogenesis factor, restored autophagosome processing and attenuated BNIP3-induced cell death.
Keywords: :
Introduction
In myocardial ischemia-reperfusion injury, programmed cell death causes substantial cardiac myocyte loss in addition to accidental necrosis triggered by lack of oxygen and nutrients in the ischemic core.Citation1 The BCL-2 family of proteins is a key regulator of the initiation and execution of programmed cell death pathways.Citation2 BNIP3Citation3,Citation4 a pro-death member of this family,Citation5-Citation7 is transcriptionally upregulated in hypoxic cardiac myocytes,Citation6,Citation8 and causes mitochondrial permeabilizationCitation6,Citation7 and dysfunctionCitation9 leading to cell death, which is an important determinant of cardiac dysfunctionCitation10 and post-infarction remodeling following ischemia-reperfusion injury.Citation11
In the setting of increased BNIP3 expression, as happens with cardiac ischemia-reperfusion injury,Citation6 cardiomyocyte autophagy is upregulated.Citation10,Citation12-Citation14 Autophagy is an evolutionarily conserved lysosomal degradative pathway to remove damaged intracellular constituents that facilitates cellular homeostasis, and promotes cell survival under stress such as nutrient deprivation and hypoxia.Citation15 Induction of autophagy is protective in the ischemic heart,Citation10,Citation12-Citation14 but has been implicated in causing cardiomyocyte death in myocardial reperfusion injury.Citation14 Forced expression of BNIP3 stimulates autophagy in cardiac myocytes,Citation9,Citation10,Citation13,Citation16 with a dose-dependent increase in autophagosome abundance.Citation16 While BNIP3-induced autophagy has been implicated in causing cell death in cancerous cells,Citation17,Citation18 induction of autophagy in the setting of BNIP3 expression is protective in cardiac myocytes, as inhibition of autophagosome formation either pharmacologically [with 3-methyladenine (3MA)Citation9] or with co-expression of dominant negative autophagy-related (Atg) protein 5 (Atg5)Citation9,Citation10 increases BNIP3-induced cardiomyocyte death. Conversely, enhancing autophagosome formation with forced expression of Atg5 and BECN1 appears to attenuate BNIP3-induced cell death in HL-1 cardiac myocytes.Citation10,Citation13 It is not known whether further induction of protective autophagy in BNIP3-expressing cardiac myocytes is limited by the availability of constituents of the autophagic machinery, or is actively suppressed, whereby it is unable to fully protect cells from BNIP3-induced cell death.
BNIP3 permeabilizes cardiac mitochondria,Citation19 promotes mitochondrial fission,Citation20 and/or renders them dysfunctional,Citation9 and the damaged mitochondria are removed via macroautophagy,Citation10,Citation20 to ensure cellular viability. Indeed, BNIP3 has been proposed as a key mediator for autophagic removal of damaged mitochondria under hypoxic stressCitation21,Citation22 and in unstressed cardiac myocytes.Citation23 This process involves coordinated action of multiple Atg proteins to sequester cargo, such as BNIP3-damaged mitochondria that are targeted for destruction within autophagosomes, which then fuse with lysosomes, wherein degradative enzymes break down complex organic matter in an intralysosomal acidic environment, to recycle amino acids, simple sugars and lipids.Citation24 Rapamycin, a potent inducer of cardiomyocyte autophagy,Citation14 also stimulates selective removal of dysfunctional mitochondria in yeastCitation25 and neurons,Citation26 and attenuates apoptotic cell death.Citation26 In this study, we evaluated whether further induction of autophagosome formation with rapamycin treatment attenuates BNIP3-induced cell death. Our results implicate lysosomal consumption as a rate-limiting factor in BNIP3-induced autophagy, whereby a strategy for enhancing flux through the macroautophagy pathway, rather than stimulating autophagosome formation alone, may accelerate removal of BNIP3-damaged mitochondria.
Recent studies have identified a critical role for transcription factor EB (TFEB), in upregulating synthesis of autophagy proteins and stimulating lysosomal biogenesis in a coordinated fashion to facilitate starvation-induced autophagy.Citation27,Citation28 We therefore evaluated whether expression of TFEB will enhance flux through macroautophagy in the setting of increased BNIP3 expression and protect against BNIP3-induced cell death. Our results demonstrate that exogenous expression of TFEB increased lysosome biogenesis, alleviating this rate limiting step in BNIP3-induced autophagy and significantly attenuated BNIP3-induced cardiomyocyte death.
Results
Rapamycin treatment does not attenuate BNIP3-induced cardiomyocyte death
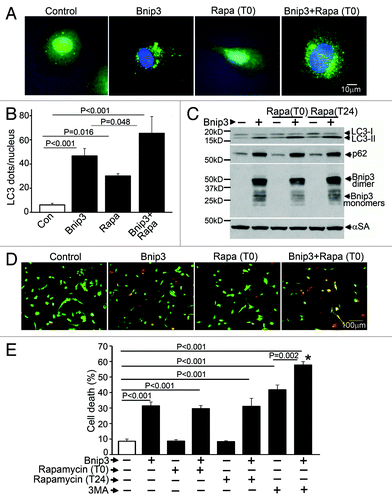
BNIP3-induced autophagy appears protective in cardiac myocytes, as inhibition of autophagy increases cell death.Citation9,Citation10 In myocardial ischemia-reperfusion injury, induction of BNIP3 is a critical determinant of cell death leading to myocardial dysfunction and post-ischemic ventricular remodeling.Citation10,Citation11 Rapamycin is a potent inducer of autophagy in cardiomyocytes;Citation14 rapamycin pretreatment attenuates hypoxic cardiomyocyte death,Citation29 and mTOR inhibition with a related compound, Everolimus, reduces infarct size and attenuates post-infarction remodeling in rats subjected to cardiac ischemia-reperfusion injury.Citation30 Accordingly, to test the hypothesis that further induction of autophagy with rapamycin will attenuate BNIP3-induced cell death, we treated neonatal rat cardiac myocytes (NRCMs) with rapamycin, both simultaneously and 24 h after adenoviral transduction of BNIP3 and assessed cell death at 48 h. BNIP3 expression induced autophagy with a significant increase in punctate GFP-LC3 localization () and increased autophagosome-bound LC3-II abundance (), without a change in LC3 transcription (Fig. S1A). Importantly, levels of p62, a protein that brings ubiquitinated aggregates into autophagosomes and gets consumed during autophagy,Citation31 were elevated in BNIP3-expressing cells (), with a trend toward reduction in p62 transcription (Fig. S1B), suggesting BNIP3-induced impaired clearance of p62. Rapamycin added simultaneously () stimulated autophagy with increased autophagosome abundance (), and increased LC3-II expression with a decline in p62 abundance () as compared with control. Interestingly, while rapamycin increased autophagosome numbers () and LC3-II abundance () in BNIP3-expressing cells, p62 levels did not decline () indicating lack of p62 clearance despite further induction of autophagosome formation by rapamycin. BNIP3 expression provoked a ~3.6-fold increase in cell death as compared with controls (), and further induction of autophagy with rapamycin treatment either simultaneously or 24 h after induction of BNIP3 expression (), did not affect BNIP3-induced cell death (). Inhibition of autophagosome formation with 3MA increased cell death in both control and BNIP3-expressing cells, confirming a beneficial role for autophagy in cardiomyocyte homeostasis and protection from BNIP3-induced cell death, as previously described.Citation9
Figure 1. Rapamycin treatment does not protect against BNIP3-induced cell death. (A) Representative epifluorescence images (630× magnification) demonstrating cellular localization of GFP-LC3 in NRCMs adenovirally transduced with BNIP3 or LacZ (as control) for 48 h, and treated with rapamycin (100 nmol/L) at T (time) = 0 (simultaneously at transduction). Nuclei are blue (DAPI). (B) Quantitation of punctate GFP-LC3 dots in cells from (A) (n = 25–40 nuclei/group). p values are by post-hoc test. (C) Immunoblot demonstrating LC3, p62 and BNIP3 (FLAG) expression in NRCMs adenovirally transduced with BNIP3 or LacZ (Con) for 48 h, and treated with rapamycin (100 nmol/L) at T (time) = 0 or 24 h after transduction. Expression of α-sarcomeric actin (αSA) was assessed as loading control. (D) Representative images (200× magnification) demonstrating live (green) and dead (red) cells treated as in (A). (E) Cell death in NRCMs adenovirally transduced with BNIP3 or LacZ (as control) for 48 h, and treated with rapamycin (100 nmol/L) at T(time) = 0 or 24 h after transduction; or 3 methyl-adenine (7 mmol/L) at t = 24 h. n = 8/group. *p < 0.001 vs Bnip3-expressing cells. All p values are by post-hoc test.
BNIP3 induces autophagosome accumulation in cardiac myocytes
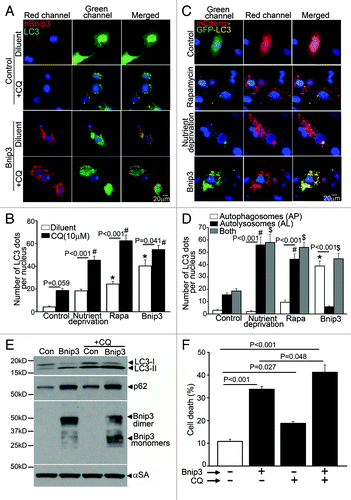
Accumulation of p62 with increased autophagosome abundance in BNIP3-expressing cells () suggested impairment of autophagosome processing. Accordingly, we examined autophagosome abundance in the absence and presence of chloroquine (CQ), a lysosomal acidification inhibitor, which leads to accumulation of autophagosomes due to impairment in autophagosome-lysosome fusion and autophagosome removal.Citation32 Cumulative flux, expressed as a ratio of autophagosome abundance in the presence of CQ to autophagosome numbers in its absence, was partially impaired with BNIP3 expresssion as compared with LacZ-expressing adenovirus-transduced control cells (1.3 vs. 4.4 in the control; ). In contrast, nutrient deprivation and rapamycin led to autophagy induction with better preserved flux as compared with BNIP3-expressing cells (ratio of autophagosome abundance with/without CQ: 2.5 with nutrient deprivation and 2.5 with rapamycin; ). We next examined the relative abundance of autophagosomes and autolysosomes in BNIP3-expressing NRCMs lentivirally transduced with an mCherry-GFP dual tandem tagged LC3. While autophagosomes are evident as dual fluorescent LC3 puncta (red + green = yellow), autolysosomes appear red, as GFP fluorescence is quenched in the acidic intralysosomal environment.Citation33 Control cells demonstrated basal autophagy with a preponderance of autolysosomes (). BNIP3 expression led to increased autophagosome abundance, without an increase in autolysosomes (). This is in contrast to the predominant increase in autolysosomes as compared with controls, observed with autophagy induction due to nutrient deprivation and rapamycin treatment, indicating intact flux through the macroautophagy pathway under these conditions (). Chloroquine treatment of cells transduced with control (LacZ) adenovirus increased autophagosome-bound LC3-II abundance and led to p62 accumulation (), which was associated with a 1.8-fold increase in cell death (), likely secondary to autophagosome accumulation () and lack of homeostatic clearance of autophagic cargo such as damaged mitochondria. In contrast, while CQ treatment of BNIP3-expressing cells caused a further increase in LC3-II abundance (as compared with BNIP3-expressing cells treated with diluent; ); there was no further p62 accumulation (), suggesting impaired autophagosome clearance in BNIP3-expressing cells. In contrast to the effect of CQ on cell death in control cells, the effect of chloroquine treatment on cell death in BNIP3-expressing cells was marginal (1.2 fold; ), correlating with an underlying impairment in clearance of autophagosomes and possibly damaged mitochondria in BNIP3-expressing cells in the absence of CQ treatment. Interestingly, CQ treatment provoked an accumulation of the monomeric, but not the dimeric forms of BNIP3 () and rapamycin treatment preferentially reduced monomeric BNIP3 protein levels (), indicating an underlying impairment in clearance of BNIP3-dimer and likely mitochondria that are permeabilized only by the dimeric forms of BNIP3, during BNIP3-induced autophagy.Citation4,Citation5,Citation34,Citation35
Figure 2. BNIP3 induces autophagosome accumulation in NRCMs. (A) Representative epifluorescence images (630× magnification) demonstrating cellular localization of GFP-LC3 in NRCMs adenovirally transduced with BNIP3 (Red; for FLAG) or LacZ (as control) for 48 h, and treated with chloroquine (10 μM, black bars) or diluent (white bars) for 24 h prior to fixation. Nuclei are blue (DAPI). (B) Quantification of punctate GFP-LC3 dots in cells treated as in (A). and in cells subjected to nutrient deprivation or treated with rapamycin (100 nM) for 4 h in the presence of chloroquine (10 μM, black bars) or diluent (white bars). p values are by post-hoc test. *p < 0.05 vs. diluent-treated control group. #p < 0.05 vs. CQ-treated control group (n = 15–25 nuclei/group). (C) Representative epifluorescence images (630× magnification) demonstrating cellular localization of mCherry-GFP-LC3 in NRCMs adenovirally transduced with Bnip3 or LacZ (as control) for 48 h; subjected to nutrient deprivation or treated with rapamycin (100 nM) for 4 h. (D) Quantitation of autophagosomes (green+red; white bars), autolysosomes (red, black bars) and both (gray bars) in NRCMs treated as in (C) (n = 20–40 nuclei/group). p values are by post-hoc test. *p < 0.05 for autophagosomes vs. control; #p < 0.05 for autolysosomes vs. control and $p < 0.05 for both vs. control (n = 15–25 nuclei/group). (E) Immunoblot demonstrating LC3, p62 and BNIP3 (FLAG) expression in NRCMs adenovirally transduced with BNIP3 or LacZ (Con) for 48 h and treated with chloroquine (10 μmol/L) or diluent for 24 h. Expression of α-sarcomeric actin (αSA) was assessed as loading control. (F) Cell death in NRCMs treated as in E (n = 8–24/group).
The transmembrane domain is essential for the interaction of BNIP3 with LC3
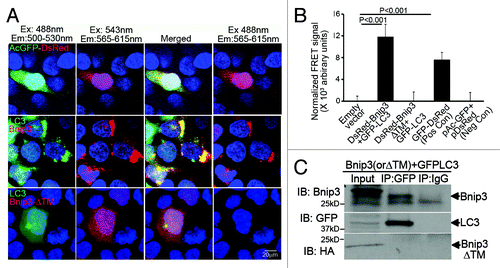
The C-terminal transmembrane domain of BNIP3 is essential for targeting mitochondriaCitation4,Citation5 and the endoplasmic reticulum,Citation36 inducing mitochondrial permeabilization and causing cell death.Citation4-Citation7,Citation34 Recent studies have identified an interaction between BNIP3 and LC3,Citation9 likely via amino acid sequences upstream of BNIP3’s C-terminal transmembrane domain, based on similarities with a related protein NIX/BNIP3L,Citation37,Citation38 suggesting that BNIP3 acts as a ‘receptor’ for targeting mitochondria to autophagosomes.Citation9,Citation10,Citation23 Interestingly, an endogenous hypoxia-inducible splice variant of BNIP3 lacking the transmembrane domain (BNIP3Δex3), but retaining the higher affinity LC3 interaction region (LIR; homologous to LIR labeled W35 in BNIP3LCitation37) was recently identified.Citation39 It is conceivable that at high levels of BNIP3 expression, such as may occur during hypoxia,Citation6,Citation8 the interaction between concomitantly hypoxia-upregulated BNIP3Δex3Citation39 and LC3 protein involving the non-transmembrane segment of the BNIP3 protein, such as observed in silico with BNIP3L/NIX,Citation37,Citation38 leads to sequestration of the available LC3, preventing its role in autophagic removal of damaged mitochondria. To examine this premise, we assessed the interaction of a transmembrane deletion mutant of BNIP3 (BNIP3ΔTM; with all putative LIR regionsCitation37 intact) and LC3, each tagged with a FRET compatible fluorophore partner in HEK 293 cells. As previously demonstrated,Citation4 BNIP3ΔTM demonstrated diffuse cellular localization (;Fig. S2), and did not induce autophagy,Citation10 assessed as increased punctate LC3 localization, as compared with controls (;Fig. S2). Interestingly, induction of autophagy by rapamycin treatment led to markedly increased punctate LC3 localization, but did not alter the subcellular localization of BNIP3ΔTM (Fig. S2), suggesting that the interaction between BNIP3 and LC3 requires activation of autophagy with an intact transmembrane domain. Indeed, assessment of interaction between full-length BNIP3, and BNIP3ΔTM with LC3 by FRET () and co-immunoprecipitation as previously described for BNIP3 and LC3Citation9 (), confirmed an obligate role for the transmembrane domain in the interaction between BNIP3 and LC3, potentially involving additional proteins recruited upon BNIP3-induced mitochondrial permeabilization and dysfunctionCitation9,Citation16,Citation19,Citation23 as observed with a closely related protein, BNIP3L/NIX.Citation37,Citation41 Also, while full-length BNIP3 colocalized both with LC3 and mitochondria in NRCMs, BNIP3ΔTM did not (Fig. S3); indicating that only the full-length BNIP3 protein acts as a receptor to target damaged mitochondria into autophagosomes, as observed with the closely related protein, BNIP3L/NIX.Citation37,Citation41 This suggests that increased BNIP3 expression results in accumulation of autophagosomes containing full-length BNIP3-targeted mitochondria,Citation7,Citation9,Citation10,Citation13,Citation16 with potential deleterious consequences secondary to impaired removal of these damaged organelles.
Figure 3. The transmembrane domain of BNIP3 is required for its interaction with LC3 to target mitochondria into autophagosomes. (A) Representative confocal images (630X) demonstrating FRET interaction between BNIP3, BNIP3ΔTM (both tagged with DsRed monomer) and LC3 (tagged with GFP) in HEK293 cells. Cells transfected with DsRed-GFP dual fluorescent constructCitation40 are shown as positive control. Nuclei are blue (Hoechst dye). Images with acquired at the excitation (Ex) and emission (Em) settings indicated and merged as shown. (B) Quantitation of FRET signal in cells treated as in (A) (n = 8/group). Cells transfected with constructs expressing DsRed-monomer and GFP, separately, are shown as negative control. p values are by post-hoc test. (C) 3T3 fibroblasts were co-transduced with adenoviruses coding for FLAG-BNIP3, or HA-BNIP3ΔTM and GFP-LC3 (100 MOI each for 48 h) and extracts subjected to co-immunoprecipitation employing BNIP3, GFP and IgG control antibodies.
BNIP3 does not target lysosomes or alter lysosome pH
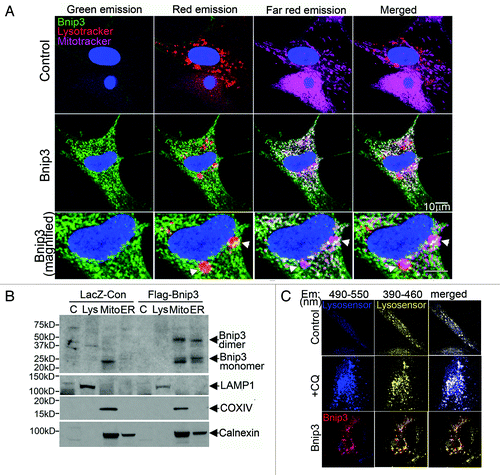
Accumulation of autophagosomes in the setting of increased BNIP3 expression may occur due to inhibition of subsequent autophagosome processing. Additionally, previous studies have suggested direct targeting of BCL-2 family proteins to lysosomes as a potential mechanism for activation of cell death.Citation42,Citation43 Accordingly, we examined whether BNIP3 targets to lysosomes or alters lysosomal integrity. BNIP3 colocalizes together with both lysosomes and mitochondria in NRCMs (, see arrows, bottom panel). This may indicate that BNIP3 targets to the lysosomes independently or via localization on mitochondria engulfed within autophagosomes that have subsequently fused with lysosomes. Accordingly, to examine the organelle-specific localization of BNIP3, we performed subcellular fractionation to isolate fractions enriched in lysosomes, mitochondria and the endoplasmic/sarcoplasmic reticulum in HL-1 cardiac myocytes, a cell line that displays mammalian cardiomyocyte physiology and can be easily expanded as necessary.Citation44 As previously demonstrated,Citation36 BNIP3 protein segregated to subfractions enriched in the mitochondrial marker COX IV, and the endoplasmic reticulum marker CALNEXIN, but not a lysosome marker, LAMP-1 ().Taken together, these data indicate that BNIP3 protein does not target lysosomes independently of being localized to mitochondria, and suggest that autophagosome-lysosome fusion in the setting of BNIP3-induced autophagy is not disrupted. Additionally, BNIP3 did not cause lysosome permeabilization (Fig. S4) or alter lysosome pH in NRCMs ().
Figure 4. BNIP3 does not target to lysosomes or affect lysosomal acidification. (A) Representative confocal images (630×) of NRCMs adenovirally transfected with LacZ (Control, top panel) or BNIP3 (green, middle panel, zoomed-in view in bottom panel), costained for lysosomes (red; LysoTracker red) and mitochondria (pink, MitoTracker deep red), demonstrating colocalization of BNIP3 with mitochondria and lysosomes (white arrowheads). (B) HL-1 cardiac myocytes were adenovirally transduced with LacZ (Control) or FLAG-Bnip3 for 48 h and subcellular fractionation performed to obtain fractions enriched for lysosomes (Lys), mitochondria (mito), endoplasmic reticulum (ER) and cytoplasm (C). Representative immunoblot demonstrating distribution of BNIP3 (FLAG) in fractions enriched for LAMP1 (lysosome marker), COXIV (mitochondrial marker) and calnexin (ER marker). (C) Representative confocal images (630×) of NRCMs adenovirally transduced with LacZ (Control) and BNIP3 (red) for 48 h and stained with lysosensor yellow/blue to assess acidification status of lysosomes. Images were obtained at excitation with 360 nm and emission split between 390–460 nm (blue, corresponding to emission maxima at pH 9.0) and 490–550 nm (yellow, corresponding to emission maxima at pH 3.0). NRCMs treated with chloroquine (10 µmol/L for 1 h) to inhibit lysosomal acidification are shown as controls.
BNIP3-induced autophagososme formation leads to lysosome consumption
We next examined whether BNIP3 affects lysosome abundance. Expression of full-length BNIP3, but not BNIP3ΔTM, provoked a ~20% reduction in lysosome numbers in NRCMs as assessed by uptake of two pH-dependent lysosome probes, LysoTracker red () and LysoTracker green (Fig. S5A), and cellular levels of LAMP-1 (; Fig. S6A), an abundant lysosomal membrane protein. This decline in lysosome numbers closely tracked LC3-II and p62 accumulation, and was observed early (within 24 h of BNIP3 transduction; Fig. S5B and S5C), and at 10-fold lower infective viral dose (MOI = 10; Fig S5D and S5E). BNIP3-induced decline in lysosome abundance was prevented by 3MA-mediated inhibition of BNIP3-induced autophagy (), suggesting lysosomal consumption during macroautophagy as the mechanism for reduced lysosome abundance in BNIP3-expressing cells, without requisite replenishment as observed in starvation- and rapamycin-induced autophagy.Citation28,Citation45 Interestingly, a decline in LAMP-1 levels was also observed in NRCMs subjected to prolonged hypoxia, which provokes maximal accumulation of the BNIP3 proteinCitation8,Citation46 (Fig. S7), suggesting that BNIP3-induced lysosomal consumption may be of pathophysiological significance in ischemic cardiac injury.
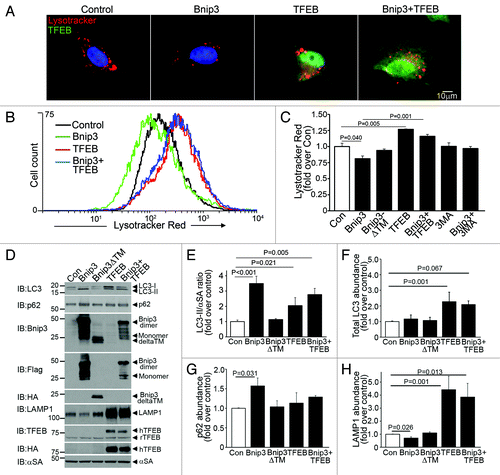
Figure 5. BNIP3-induced autophagy is associated with reduced lysosome abundance, which is restored by co-expression of TFEB. (A) Representative epiflourescence images (630X) demonstrating lysosome distribution (by LysoTracker red staining) in cells adenovirally transduced with Bnip3, TFEB (green, at 100 MOI), BNIP3+TFEB (at 100 MOI each) for 48 h. Nuclei are blue (Hoechst dye). Adenovirus coding for LacZ expression was added as necessary to result in equivalent MOIs (at total 200 MOI per treatment). (B) Flow cytometric analysis of LysoTracker red staining in cells treated as in (A). Control is depicted in black, BNIP3 in green, TFEB in red and BNIP3+TFEB in blue. (C) Assessment of LysotTracker red expression by flow cytometry in NRCMs expressing BNIP3, BNIP3 ΔTM, TFEB, BNIP+TFEB for 48 h; and in BNIP3 expressing cells treated for 24 h with 3MA (7 mmol/L). (D) Representative immunoblots demonstrating LC3, p62, BNIP3 (FLAG), BNIP3ΔTM (HA), TFEB (both rat TFEB and HA tagged human TFEB) and LAMP1 expression, with α-sarcomeric actin (αSA) in NRCMs adenovirally transduced with LacZ (control), BNIP3, BNIP3ΔTM, TFEB and BNIP3+TFEB as in (A) (for 48 h). (E–H) Quantitative assessment of LC3-II/α-sarcomeric actin ratio (E), total LC3 (F), p62 (G) and LAMP1 (H) abundance in NRCMs treated as in D (n = 3–7/group). p values are by post-hoc test.
Enhancing lysosomal biogenesis with transcription factor EB expression rescues BNIP3- induced cell death
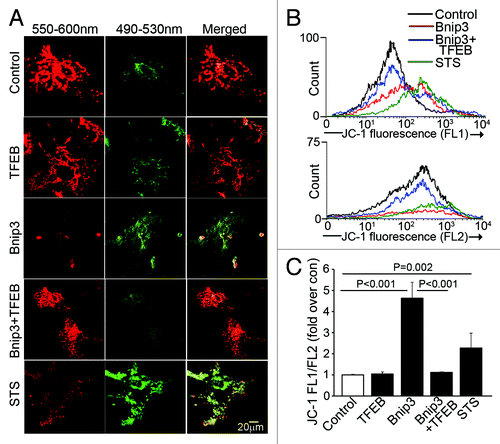
To determine whether restoring lysosome abundance would promote autophagosome clearance in BNIP3-expressing NRCMs, we expressed the transcription factor EB (TFEB), which was recently described to promote biogenesis of lysosomesCitation27,Citation28 and autophagy proteins.Citation28 Adenovirally transduced TFEB localized to the nucleus () and expression of TFEB increased lysosome abundance (;Fig. S5A) with increased abundance of LC3 (; Fig. S6A) and LAMP-1 (; Fig. S6A), induced autophagosome formation as evidenced by punctate GFP-LC3 localization (Fig. S8) and increased the ratio of LC3-II/α-sarcomeric actin (), as previously described.Citation28 Importantly, TFEB expression restored lysosome abundance in BNIP3-expressing cells (; Figs. S5AandS6A), with a reduction in the ratio of LC3-II to LC3-I (;Fig. S6B) and reduced p62 accumulation (; Fig. S6A) as compared with NRCMs treated with BNIP3 alone. Interestingly, while lower expression of TFEB (MOI = 10) did not alter BNIP3 protein abundance (Fig. S6A), higher expression of TFEB (MOI = 100) was associated with a decline in BNIP3 protein levels (;Fig. S9) which was prevented by inhibition of autophagy with 3MA (Fig. S9), suggesting enhanced clearance of BNIP3 by TFEB-induced autophagy. Indeed, exogenous expression of TFEB restored autophagosome processing in BNIP3-expressing cells, with a decline in prevalence of autophagosomes and a commensurate increase in autolysosomes () as compared with cells expressing BNIP3 alone. As shown previously,Citation9,Citation10 BNIP3ΔTM did not stimulate cardiomyocyte autophagy () or increase death (). Enhancement of autophagosome clearance by TFEB was associated with a dose-dependent attenuation in BNIP3-induced cell death () and in BNIP3-induced TUNEL positivity (). This was likely secondary to enhanced removal of damaged mitochondria by a global induction of the autophagic machinery driven by TFEB in the setting of BNIP3 expression, as BNIP3 provoked an increase in green fluorescent JC-1 monomers (, top) with a reduction in red fluorescent JC-1 J-aggregates (, bottom) and markedly increased green to red fluorescence ratio (vs controls, ), indicating increased depolarized mitochondria and reduced numbers of normally polarized mitochondria,Citation47 and suggesting accumulation of damaged mitochondriaCitation48 (), which was reversed by co-expression of TFEB ().
Figure 6. Forced expression of TFEB restores autophagosome processing and attenuates cell death in BNIP3-expressing NRCMs. (A) Representative epifluorescence images (630× magnification) demonstrating cellular localization of mCherry-GFP-LC3 in NRCMs adenovirally transduced with LacZ (as control), BNIP3, TFEB and BNIP3+TFEB for 48 h. Nuclei are blue (DAPI). Adenovirus coding for LacZ expression was added as necessary to result in equivalent MOIs (at total 200 MOI per treatment). (B) Quantitation of autophagosomes (green+red; white bars), autolysosomes (red, black bars) and both (gray bars) LC3 in NRCMs treated as in (A) (n = 25–40 nuclei/group). *p < 0.05 vs. autophagosomes, $p < 0.05 vs autolysosomes and #p < 0.05 vs. both in control group by post-hoc test. (C) Assessment of cell death in NRCMs transduced with LacZ (control), BNIP3, BNIP3ΔTM, TFEB (at MOIs = 10 and 100) and BNIP3+TFEB (at MOIs = 10 and 100) for 48 h. Adenovirus coding for LacZ expression was added as necessary to result in equivalent MOIs (at total 200 MOI per treatment). *p < 0.05 vs. control and #p < 0.05 vs. BNIP3 by post-hoc test. (D) Representative epifluorescence images (200× magnification) demonstrating TUNEL staining (green) in NRCMs treated as in (A). Nuclei are blue (DAPI). (E) Quantitative assessment of TUNEL positivity in NRCMs treated as in (A). Adenovirus coding for LacZ expression was added as necessary to result in equivalent MOIs (at total 200 MOI per treatment) for both (D and E). Treatment with staurosporine (1 μmol/L for 24 h) was employed as positive control (n = 5–9 experiments/group). p values are by post-hoc test.
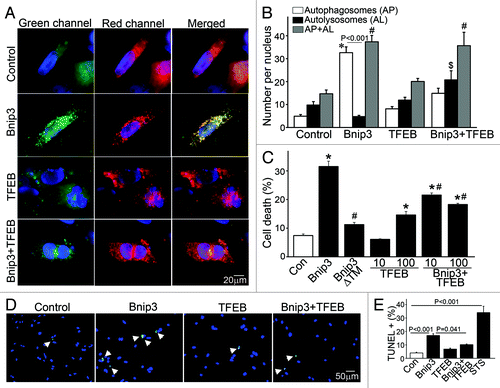
Figure 7. Forced expression of TFEB accelerates clearance of BNIP3-permeabilized mitochondria. (A) Representative confocal images of NRCMs adnovirally transduced with BNIP3, TFEB, BNIP3+TFEB (each at MOI = 100); and LacZ control (added to make total MOI = 200 in each group) for 48 h, demonstrating expression of JC-1 mitochondrial stain. Emission wavelengths employed for imaging the red and green fluorescence are depicted. Staurosporine (STS; 5 μmol/L for 2 h) was added as positive control. (B) Flow cytometric analysis of JC-1 expression with representative traces demonstrating green fluorescence (FL1 channel, B, top) and red fluorescence (FL2 channel, B, bottom). Control is depicted in black, BNIP3 in red, BNIP3+TFEB in blue and staurosporine in green. (C) Quantitation of ratio or FL1/FL2 fluorescence of JC-1 expression in NRCMs treated as in (A). p values depicted are by post-hoc test after one-way ANOVA (n = 3–6/group).
Discussion
In this study, we demonstrated that BNIP3-induced autophagy in cardiac myocytes was rate limited by lysosome consumption, which led to upstream autophagosome accumulation. Expression of transcription factor EB (TFEB) stimulated lysosome biogenesis and restored processing of autophagosomes. The resultant enhanced flux through the macroautophagy pathway attenuated BNIP3-induced cell death.
The primary stimulus for autophagy induction with increased BNIP3 expression, as occurs with hypoxic insult,Citation6,Citation8 appears to be BNIP3 targeting to the organelle, in particular the mitochondria.Citation4,Citation9,Citation10,Citation36 Indeed, our data confirm an obligate role for the transmembrane domain of BNIP3 protein, which is essential for organelle targeting,Citation4,Citation36 in inducing autophagy. BNIP3 induces mitochondrial damage by multiple mechanisms, such as mitochondrial outer membrane permeabilization in concert with Bax and Bak,Citation49 mitochondrial permeability transition by a novel cyclophilin D-independent mechanism,Citation19 mitochondrial fragmentation in concert with OpaCitation19,Citation50 and Drp1,Citation20 and mitochondrial energetic dysfunction via protease-mediated cleavage of oxidative phosphorylation/electron transport chain proteins.Citation9,Citation23 The obligate role of the transmembrane doman in the interaction of BNIP3 with LC3 proteins (see ) suggests that mitochondrially-localized BNIP3 interacts with autophagosome-bound LC3-II as a receptor to facilitate autophagic removal of these damaged mitochondria.Citation9,Citation10,Citation23
Recent studies have employed bafilomycin A1 to inhibit lysosome acidification and assess cumulative flux through the macroautophagy pathway in BNIP3-expressing cells, and suggest that autophagic flux is intact in this setting.Citation9 While a BNIP3-induced increase in autophagosomes is incontrovertible, the ratio of autophagosome abundance with bafilomycin A1 treatment as compared with no treatment was low in BNIP3-expressing cells when compared with a similar ratio in controls (1.6 in BNIP3-expressing cells vs. 5 in controls).Citation9 These data are comparable to our results with CQ (which also inhibits lysosomal acidification; see ), and taken together, suggest a partial impairment in autophagosome processing in BNIP3-expressing cells. Our data also suggest that under conditions of high levels of BNIP3 expression as may occur with myocardial ischemia-reperfusion injury,Citation6,Citation8 autophagosome processing is impaired, as indicated by accumulation of p62, a protein that gets consumed in the autophagic process,Citation31 and accumulation of autophagosomes without a commensurate increase in autolysosomes. Impaired autophagosome clearance prevents removal of BNIP3-damaged mitochondria, similar to that observed with preventing autophagosome formation (e.g., with 3MA, or dominant negative Atg5Citation9,Citation10) or autophagosome processing (with CQ, see ; or bafilomycin A1 treatmentCitation9), and impairs the role for autophagy in preventing BNIP3-mediated programmed cell death.
Previous studies have demonstrated that inducing autophagosome formation with transfection of Atg5Citation10 or Becn1Citation13 attenuates BNIP3-induced cell death. We observed that stimulating autophagosome formation with rapamycin does not attenuate BNIP3-induced cell death (). The observed differences may be attributable to level of BNIP3 expression, whereby autophagosome accumulation is only observed at high levels (as obtained with adenoviral transduction of Bnip3), but not at lower levels typically achieved with transfection based-methods in cardiac myocytes. Alternatively, the previous studiesCitation10,Citation13 may indicate a specific impairment in ATG5 and/or BECN1 levels or function in the context of BNIP3-induced autophagy, whereby restoring either one attenuates BNIP3-induced cell death. Exogenous administration of TFEB is not known to stimulate synthesis of ATG5 and BECN1,Citation27,Citation28 but could potentially have overcome this limitation via synthesis of alternative proteins in the autophagic pathway, resulting in the observed benefits. Also, while our data suggest that autophagosome-lysosome fusion is not completely disrupted in BNIP3-expressing cells, it may be impaired, and the observed beneficial effects of TFEB expression may relate at least in part to normalization or acceleration of this process. These hypotheses require further investigation.
We found that BNIP3 expression triggers a decline in lysosome abundance in cardiac myocytes. This appears to be due to lysosome consumption in the autophagy process, as it was prevented by inhibition of autophagosome formation with 3MA, and did not occur with the transmembrane deletion variant BNIP3ΔTM which does not induce autophagy. Importantly, BNIP3 did not target to or permeabilize lysosomes whereby it could provoke a lysosomal pathway for cell death.Citation51 Indeed, enhancing lysosomal biogenesis in that setting would have increased BNIP3-induced cell death, akin to adding fuel to the fire. The decline in lysosome numbers with BNIP3-induced autophagy indicates a lack of recruitment of, or an active suppression of mechanisms to enhance lysosomal biogenesis endogenously as occurs in starvation-induced autophagy.Citation28 Notably, rapamycin treatment, which results in an initial depletion of lysosomes followed by a rapid restoration of lysosome abundance,Citation45 did not attenuate BNIP3-induced cell death, suggesting that the observed beneficial effects of TFEB relate to its ability to coordinately upregulate the entire autophagic machinery.Citation28 It is interesting to speculate that active suppression of autophagy by limiting the process at various steps is a mechanism inherent to the programmed cell death process, which prevents the pro-survival function of lysosome-mediated autophagy, and requires further study.
Materials and Methods
Cardiac myocyte culture
Neonatal rat cardiac myocyte (NRCM) cultures were prepared as described.Citation14 Briefly, hearts were removed from 1-d-old Sprague-Dawley rats, the atria and great vessels were trimmed off, and tissue was finely minced followed by sequential digestion with 0.5 mg/ml collagenase (WAKO, LK03303). Ventricular cardiomyocytes were separated from fibroblasts by differential plating and were cultured in gelatin-coated 12-well tissue culture plates (0.4 hearts/well) in media containing Dulbecco's modified Eagle's medium, 10% horse serum, 5% fetal calf serum, 100 μmol/L bromodeoxyuridine, penicillin, streptomycin and L-glutamine. Nutrient deprivation was induced as previously described.Citation14 HL-1 cardiac myocytes were a kind gift from Dr. William Claycomb, Louisiana State University, New Orleans, and were cultured as described.Citation44
Generation of viral constructs
The coding sequence for rat Map1lc3β (microtubule-associated protein 1 light chain 3β) was cloned in frame with GFP in pAcGFP-C vector (Clontech, 632470). The GFP-LC3 coding sequence was then cloned downstream of mCherry in pLVx-mCherry-C vector (Clontech, 632561) and lentiviral particles coding for expression of dual fluorescent tandem tagged mCherry-GFP-LC3 were generated per the manufacturer’s instructions at the Hope Center Viral Vectors Core at Washington University School of Medicine. Transduction of lentiviruses was facilitated with polybrene, at 8 μg/ml (Sigma, H9268). Coding sequences for human BNIP3, BNIP3ΔTM (see Supplementary methods for details) and human TFEB, with N-terminal FLAG (DYKDDDDK; on BNIP3) and HA (YPYDVPDYA; on BNIP3ΔTM and TFEB) tags were cloned into pENTR-TOPO vector (Invitrogen, 45.0218) and recombinant adenoviral constructs generated with Clonase mediated recombination (Virapower, Invitrogen, K493000). Adenoviruses were generated in HEK293A cells and titered per the manufacturer’s instructions.
Assessment of cell death
Cell death assays were performed in 96-well plates and chamber slide (Nunc, Fisher, 177429) format with the Live-Dead Cytotoxicity Viability kit for Mammalian cells (Invitrogen, L3224) per manufacturer’s instructions as described.Citation52 Quantitative assessment of fluorescence was performed with BioTek Synergy-2 microplate reader equipped with the appropriate filter sets (Green: Excitation 485 ± 10 nm, Emission 528 ± 10 nm; Red: Excitation 540 ± 7.5 nm, Emission 620 ± 20 nm) at Chemical Genetics Screening Core at Washington University School of Medicine. TUNEL staining was performed as described.Citation52
Immunofluorescence imaging
Imaging for GFP-LC3 and mCherry-GFP-LC3 localization, and immunofluorescence for FLAG and HA epitopes (with Alexa Fluor 488 and 594 tagged secondary antibodies from Invitrogen, A21202 and A21207) were performed on 4% paraformaldehyde fixed cells using Axioscap upright microscope; AxioCam HRC camera and Plan Neofluar objective (63X, NA1.25, oil) (Zeiss) fitted with appropriate filter cubes. Images were acquired and analyzed using Zeiss Axiovision software. Confocal imaging was performed on a Zeiss LSM 510 NLO Meta using an Achroplan 63X (NA 0.95) water objective and Zeiss LSM software. Punctate fluorescent tagged LC3 dots were counted and expressed as number per nucleus using Image J software (NIH) as described.Citation53 Organelle imaging was performed using LysoTracker red (L7528) and green (L7526), MitoTracker deep red FM (M22426), Hoechst dye (H3570), JC-1 (T3168; all from Invitrogen) and DAPI (Vector Labs; H-1200) per manufacturer’s protocols. Acridine orange (Biotium, 40039) staining was performed as described.Citation54 Assessment of lysosomal pH was performed with pH-sensitive dye- Lysosensor yellow/blue DND-160 (Invitrogen, L7545) following manufacturer’s instructions.
Subcellular fractionation
HL-1 cells (7 × 107 cells per group) were adenovirally transduced with LacZ (control) and Bnip3 [at MOI (multiplicity of infection) = 100] for 48 h and subjected to subcellular fractionation on a discontinuous nycodenz gradient using procedures modified from protocols previously described.Citation55,Citation56 Briefly, cells were homogenized in a medium containing 0.25 M sucrose, 1 mM Na2EDTA, 10 mM HEPES adjusted to pH 7.4 with NaOH. Homogenates were cleared off unbroken cells with a brief 120 g spin and a mitochondria+lysosome rich fraction was sedimented at 20,000 g for 20 min. This fraction was layered on a discontinuous nycodenz (Optiprep, Sigma, D1556) gradient (19%, 27% and 30%) and subjected to ultracentrifugation at 110,000 g for 2 h in a swinging bucket rotor. Three-milliliter fractions were collected from the gradient; and lysosomes, and mitochondria with mitochondria-associated-membranes were recovered from the top and 3rd (from top) fractions, respectively. The supernatant from the 20,000 g spin was subjected to ultracentrifugation at 100,000 g for 1 h to recover endoplasmic reticulum rich fraction; with resultant supernatant concentrated as cytosol using Ultracel-10K protein filters (Millipore, UFC801024).
Assessment of FRET interaction
Constructs coding for DsRed-BNIP3 or DsRed-BNIP3ΔTM (see supplementary methods for details) were co-transfected with GFP-LC3 in HEK293 cells. Construct encoding for DsRed-monomer-GFP fusion proteinCitation40 was employed as positive control. Normalized FRET was assessed by confocal microscopy and BioTek Synergy-2 microplate reader equipped with the appropriate filter sets as described.Citation40
Flow cytometry
NRCMs were incubated with LysoTracker Red (1 μmol/L for 15 min at 37° in 5% CO2) or JC-1 (10 μg/ml for 10 min at 37°C in 5% CO2) and subjected to flow cytometry on FACScan instrument (Becton-Dickinson) as described.Citation57 Cyflogic software (CyFlo) was employed to analyze 20,000 events per run.
Co-immunoprecipitation studies
NIH 3T3 fibroblasts were transduced with adenoviruses to co-express BNIP3 or BNIP3ΔTM and GFP-LC3 (all at 100 MOI); and crude extracts prepared as described. One and a half mg of total protein was incubated with anti-GFP (Abcam, ab0290) and normal rabbit IgG; and immunoprecipitation performed using Dynabeads® protein G (Invitrogen, 100.07D).
Immunoblotting
Immunoblotting was performed on cardiac and cellular extracts using previously described techniques.Citation11 Antibodies employed were as follows: FLAG (Sigma, F3165); HA (Sigma, H6908), Bnip3 (Abcam, Ab10433), GFP (Clontech, 632375), LAMP1 (Abcam, AB24170); LC3 (Novus Biologicals, NB100-2220); p62 (Abcam, ab5416); and α-sarcomeric actin (Abcam, ab52219). Image J software was employed for quantitative analysis. Protein abundance was normalized to α-sarcomeric action expression and reported as fold change vs. control. Chemicals employed were obtained as follows: rapamycin (EMD4Biosciences, 553212); chloroquine (Sigma, CC6628); 3-methyladenine (EMD4Biosciences, 189490); and staurosporine (Sigma, S6942).
Statistical analysis
Results are expressed as mean ± SEM. Statistical differences were assessed with the unpaired 2-tailed Student’s t-test for two experimental groups and one-way ANOVA for multiple groups with SPSS software. Bonferroni’s post-hoc testing was employed after ANOVA for testing for significant differences between groups. A two-tailed p value of less than 0.05 was considered statistically significant.
| Abbreviations: | ||
| BNIP3 | = | BCL2/adenovirus E1B 19 kd-interacting protein |
| FRET | = | Forster resonance energy transfer |
| NRCM | = | neonatal rat cardiac myocyte |
| CQ | = | chloroquine |
| 3MA | = | 3-methyladenine |
Additional material
Download Zip (3.2 MB)Acknowledgments
We thank Toni Gonzales for secretarial assistance and express our sincere gratitude to Douglas L. Mann, M.D., chief of cardiology at Washington University School of Medicine, for his helpful comments and support.
This study was supported by the NIH (HL107594), the Department of Veterans Affairs (1 I01 BX000448-01) and the American Heart Association (0735135N Scientist Development Grant) all to A.D. The Hope Center Viral Vectors Core and Alafi Neuroimaging Core at Washington University are supported by P30 NS057105 and P01 NS032636 from the NIH.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Whelan RS, Kaplinskiy V, Kitsis RN. Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu Rev Physiol 2010; 72:19 - 44; http://dx.doi.org/10.1146/annurev.physiol.010908.163111; PMID: 20148665
- Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer 2002; 2:647 - 56; http://dx.doi.org/10.1038/nrc883; PMID: 12209154
- Boyd JM, Malstrom S, Subramanian T, Venkatesh LK, Schaeper U, Elangovan B, et al. Adenovirus E1B 19 kDa and Bcl-2 proteins interact with a common set of cellular proteins. Cell 1994; 79:341 - 51; http://dx.doi.org/10.1016/0092-8674(94)90202-X; PMID: 7954800
- Chen G, Ray R, Dubik D, Shi L, Cizeau J, Bleackley RC, et al. The E1B 19K/Bcl-2-binding protein Nip3 is a dimeric mitochondrial protein that activates apoptosis. J Exp Med 1997; 186:1975 - 83; http://dx.doi.org/10.1084/jem.186.12.1975; PMID: 9396766
- Ray R, Chen G, Vande VC, Cizeau J, Park JH, Reed JC, et al. BNIP3 heterodimerizes with Bcl-2/Bcl-X(L) and induces cell death independent of a Bcl-2 homology 3 (BH3) domain at both mitochondrial and nonmitochondrial sites. J Biol Chem 2000; 275:1439 - 48; http://dx.doi.org/10.1074/jbc.275.2.1439; PMID: 10625696
- Regula KM, Ens K, Kirshenbaum LA. Inducible expression of BNIP3 provokes mitochondrial defects and hypoxia-mediated cell death of ventricular myocytes. Circ Res 2002; 91:226 - 31; http://dx.doi.org/10.1161/01.RES.0000029232.42227.16; PMID: 12169648
- Vande Velde C, Cizeau J, Dubik D, Alimonti J, Brown T, Israels S, et al. BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol Cell Biol 2000; 20:5454 - 68; http://dx.doi.org/10.1128/MCB.20.15.5454-5468.2000; PMID: 10891486
- Kubasiak LA, Hernandez OM, Bishopric NH, Webster KA. Hypoxia and acidosis activate cardiac myocyte death through the Bcl-2 family protein BNIP3. Proc Natl Acad Sci USA 2002; 99:12825 - 30; http://dx.doi.org/10.1073/pnas.202474099; PMID: 12226479
- Rikka S, Quinsay MN, Thomas RL, Kubli DA, Zhang X, Murphy AN, et al. Bnip3 impairs mitochondrial bioenergetics and stimulates mitochondrial turnover. Cell Death Differ 2011; 18:721 - 31; http://dx.doi.org/10.1038/cdd.2010.146; PMID: 21278801
- Hamacher-Brady A, Brady NR, Logue SE, Sayen MR, Jinno M, Kirshenbaum LA, et al. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ 2007; 14:146 - 57; http://dx.doi.org/10.1038/sj.cdd.4401936; PMID: 16645637
- Diwan A, Krenz M, Syed FM, Wansapura J, Ren X, Koesters AG, et al. Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of Bnip3 restrains postinfarction remodeling in mice. J Clin Invest 2007; 117:2825 - 33; http://dx.doi.org/10.1172/JCI32490; PMID: 17909626
- Hamacher-Brady A, Brady NR, Gottlieb RA. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J Biol Chem 2006; 281:29776 - 87; http://dx.doi.org/10.1074/jbc.M603783200; PMID: 16882669
- Hamacher-Brady A, Brady NR, Gottlieb RA, Gustafsson AB. Autophagy as a protective response to Bnip3-mediated apoptotic signaling in the heart. Autophagy 2006; 2:307 - 9; PMID: 16874059
- Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res 2007; 100:914 - 22; http://dx.doi.org/10.1161/01.RES.0000261924.76669.36; PMID: 17332429
- Galluzzi L, Morselli E, Vicencio JM, Kepp O, Joza N, Tajeddine N, et al. Life, death and burial: multifaceted impact of autophagy. Biochem Soc Trans 2008; 36:786 - 90; http://dx.doi.org/10.1042/BST0360786; PMID: 18793137
- Quinsay MN, Thomas RL, Lee Y, Gustafsson AB. Bnip3-mediated mitochondrial autophagy is independent of the mitochondrial permeability transition pore. Autophagy 2010; 6:855 - 62; http://dx.doi.org/10.4161/auto.6.7.13005; PMID: 20668412
- Azad MB, Chen Y, Henson ES, Cizeau J, McMillan-Ward E, Israels SJ, et al. Hypoxia induces autophagic cell death in apoptosis-competent cells through a mechanism involving BNIP3. Autophagy 2008; 4:195 - 204; PMID: 18059169
- Ghavami S, Asoodeh A, Klonisch T, Halayko AJ, Kadkhoda K, Kroczak TJ, et al. Brevinin-2R(1) semi-selectively kills cancer cells by a distinct mechanism, which involves the lysosomal-mitochondrial death pathway. J Cell Mol Med 2008; 12:1005 - 22; http://dx.doi.org/10.1111/j.1582-4934.2008.00129.x; PMID: 18494941
- Quinsay MN, Lee Y, Rikka S, Sayen MR, Molkentin JD, Gottlieb RA, et al. Bnip3 mediates permeabilization of mitochondria and release of cytochrome c via a novel mechanism. J Mol Cell Cardiol 2010; 48:1146 - 56; http://dx.doi.org/10.1016/j.yjmcc.2009.12.004; PMID: 20025887
- Lee Y, Lee HY, Hanna RA, Gustafsson AB. Mitochondrial Autophagy by Bnip3 Involves Drp1-Mediated Mitochondrial Fission and Recruitment of Parkin in Cardiac Myocytes. Am J Physiol Heart Circ Physiol 2011; http://dx.doi.org/10.1152/ajpheart.00368.2011; PMID: 21890690
- Band M, Joel A, Hernandez A, Avivi A. Hypoxia-induced BNIP3 expression and mitophagy: in vivo comparison of the rat and the hypoxia-tolerant mole rat, Spalax ehrenbergi. FASEB J 2009; 23:2327 - 35; http://dx.doi.org/10.1096/fj.08-122978; PMID: 19255257
- Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem 2008; 283:10892 - 903; http://dx.doi.org/10.1074/jbc.M800102200; PMID: 18281291
- Thomas RL, Kubli DA, Gustafsson AB. Bnip3-mediated defects in oxidative phosphorylation promote mitophagy. Autophagy 2011; 7:775 - 7; http://dx.doi.org/10.4161/auto.7.7.15536; PMID: 21460627
- Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 2010; 22:124 - 31; http://dx.doi.org/10.1016/j.ceb.2009.11.014; PMID: 20034776
- Mendl N, Occhipinti A, Muller M, Wild P, Dikic I, Reichert AS. Mitophagy in yeast is independent of mitochondrial fission and requires the stress response gene WHI2. J Cell Sci 2011; 124:1339 - 50; http://dx.doi.org/10.1242/jcs.076406; PMID: 21429936
- Pan T, Rawal P, Wu Y, Xie W, Jankovic J, Le W. Rapamycin protects against rotenone-induced apoptosis through autophagy induction. Neuroscience 2009; 164:541 - 51; http://dx.doi.org/10.1016/j.neuroscience.2009.08.014; PMID: 19682553
- Sardiello M, Palmieri M. di RA, Medina DL, Valenza M, Gennarino VA, Di MC, Donaudy F, Embrione V, Polishchuk RS, Banfi S, Parenti G, Cattaneo E, Ballabio A. A gene network regulating lysosomal biogenesis and function. Science 2009; 325:473 - 7; PMID: 19556463
- Settembre C, Di MC, Polito VA, Garcia AM, Vetrini F, Erdin S, et al. TFEB links autophagy to lysosomal biogenesis. Science 2011; 332:1429 - 33; http://dx.doi.org/10.1126/science.1204592; PMID: 21617040
- Khan S, Salloum F, Das A, Xi L, Vetrovec GW, Kukreja RC. Rapamycin confers preconditioning-like protection against ischemia-reperfusion injury in isolated mouse heart and cardiomyocytes. J Mol Cell Cardiol 2006; 41:256 - 64; http://dx.doi.org/10.1016/j.yjmcc.2006.04.014; PMID: 16769083
- Buss SJ, Muenz S, Riffel JH, Malekar P, Hagenmueller M, Weiss CS, et al. Beneficial effects of Mammalian target of rapamycin inhibition on left ventricular remodeling after myocardial infarction. J Am Coll Cardiol 2009; 54:2435 - 46; http://dx.doi.org/10.1016/j.jacc.2009.08.031; PMID: 20082935
- Bjørkøy G, Lamark T, Pankiv S, Overvatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol 2009; 452:181 - 97; http://dx.doi.org/10.1016/S0076-6879(08)03612-4; PMID: 19200883
- Kawai A, Uchiyama H, Takano S, Nakamura N, Ohkuma S. Autophagosome-lysosome fusion depends on the pH in acidic compartments in CHO cells. Autophagy 2007; 3:154 - 7; PMID: 17204842
- Patterson GH, Knobel SM, Sharif WD, Kain SR, Piston DW. Use of the green fluorescent protein and its mutants in quantitative fluorescence microscopy. Biophys J 1997; 73:2782 - 90; http://dx.doi.org/10.1016/S0006-3495(97)78307-3; PMID: 9370472
- Bocharov EV, Pustovalova YE, Pavlov KV, Volynsky PE, Goncharuk MV, Ermolyuk YS, et al. Unique dimeric structure of BNip3 transmembrane domain suggests membrane permeabilization as a cell death trigger. J Biol Chem 2007; 282:16256 - 66; http://dx.doi.org/10.1074/jbc.M701745200; PMID: 17412696
- Kubli DA, Quinsay MN, Huang C, Lee Y, Gustafsson AB. Bnip3 Functions as a Mitochondrial Sensor of Oxidative Stress during Myocardial Ischemia and Reperfusion. Am J Physiol Heart Circ Physiol 2008; http://dx.doi.org/10.1152/ajpheart.00552.2008; PMID: 18790835
- Zhang L, Li L, Liu H, Borowitz JL, Isom GE. BNIP3 mediates cell death by different pathways following localization to endoplasmic reticulum and mitochondrion. FASEB J 2009; 23:3405 - 14; http://dx.doi.org/10.1096/fj.08-124354; PMID: 19535684
- Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep 2010; 11:45 - 51; http://dx.doi.org/10.1038/embor.2009.256; PMID: 20010802
- Schwarten M, Mohrluder J, Ma P, Stoldt M, Thielmann Y, Stangler T, et al. Nix directly binds to GABARAP: a possible crosstalk between apoptosis and autophagy. Autophagy 2009; 5:690 - 8; http://dx.doi.org/10.4161/auto.5.5.8494; PMID: 19363302
- Gang H, Hai Y, Dhingra R, Gordon JW, Yurkova N, Aviv Y, et al. A novel hypoxia-inducible spliced variant of mitochondrial death gene Bnip3 promotes survival of ventricular myocytes. Circ Res 2011; 108:1084 - 92; http://dx.doi.org/10.1161/CIRCRESAHA.110.238709; PMID: 21415393
- Liu BF, Anbarasu K, Liang JJ. Confocal fluorescence resonance energy transfer microscopy study of protein-protein interactions of lens crystallins in living cells. Mol Vis 2007; 13:854 - 61; PMID: 17615546
- Ding WX, Ni HM, Li M, Liao Y, Chen X, Stolz DB, et al. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J Biol Chem 2010; 285:27879 - 90; http://dx.doi.org/10.1074/jbc.M110.119537; PMID: 20573959
- Feldstein AE, Werneburg NW, Li Z, Bronk SF, Gores GJ. Bax inhibition protects against free fatty acid-induced lysosomal permeabilization. Am J Physiol Gastrointest Liver Physiol 2006; 290:G1339 - 46; http://dx.doi.org/10.1152/ajpgi.00509.2005; PMID: 16484678
- Kågedal K, Johansson AC, Johansson U, Heimlich G, Roberg K, Wang NS, et al. Lysosomal membrane permeabilization during apoptosis–involvement of Bax?. Int J Exp Pathol 2005; 86:309 - 21; http://dx.doi.org/10.1111/j.0959-9673.2005.00442.x; PMID: 16191103
- Claycomb WC, Lanson NA Jr., Stallworth BS, Egeland DB, Delcarpio JB, Bahinski A, et al. HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci USA 1998; 95:2979 - 84; http://dx.doi.org/10.1073/pnas.95.6.2979; PMID: 9501201
- Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010; 465:942 - 6; http://dx.doi.org/10.1038/nature09076; PMID: 20526321
- Bruick RK. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc Natl Acad Sci USA 2000; 97:9082 - 7; http://dx.doi.org/10.1073/pnas.97.16.9082; PMID: 10922063
- Smiley ST, Reers M, Mottola-Hartshorn C, Lin M, Chen A, Smith TW, et al. Intracellular heterogeneity in mitochondrial membrane potentials revealed by a J-aggregate-forming lipophilic cation JC-1. Proc Natl Acad Sci USA 1991; 88:3671 - 5; http://dx.doi.org/10.1073/pnas.88.9.3671; PMID: 2023917
- Mancini M, Anderson BO, Caldwell E, Sedghinasab M, Paty PB, Hockenbery DM. Mitochondrial proliferation and paradoxical membrane depolarization during terminal differentiation and apoptosis in a human colon carcinoma cell line. J Cell Biol 1997; 138:449 - 69; http://dx.doi.org/10.1083/jcb.138.2.449; PMID: 9230085
- Kubli DA, Ycaza JE, Gustafsson AB. Bnip3 mediates mitochondrial dysfunction and cell death through Bax and Bak. Biochem J 2007; 405:407 - 15; http://dx.doi.org/10.1042/BJ20070319; PMID: 17447897
- Landes T, Emorine LJ, Courilleau D, Rojo M, Belenguer P, Arnaune-Pelloquin L. The BH3-only Bnip3 binds to the dynamin Opa1 to promote mitochondrial fragmentation and apoptosis by distinct mechanisms. EMBO Rep 2010; 11:459 - 65; http://dx.doi.org/10.1038/embor.2010.50; PMID: 20436456
- Boya P, Andreau K, Poncet D, Zamzami N, Perfettini JL, Metivier D, et al. Lysosomal membrane permeabilization induces cell death in a mitochondrion-dependent fashion. J Exp Med 2003; 197:1323 - 34; http://dx.doi.org/10.1084/jem.20021952; PMID: 12756268
- Diwan A, Matkovich SJ, Yuan Q, Zhao W, Yatani A, Brown JH, et al. Endoplasmic reticulum-mitochondria crosstalk in NIX-mediated murine cell death. J Clin Invest 2009; 119:203 - 12; PMID: 19065046
- Dagda RK, Zhu J, Kulich SM, Chu CT. Mitochondrially localized ERK2 regulates mitophagy and autophagic cell stress: implications for Parkinson's disease. Autophagy 2008; 4:770 - 82; PMID: 18594198
- Boya P, Kroemer G. Lysosomal membrane permeabilization in cell death. Oncogene 2008; 27:6434 - 51; http://dx.doi.org/10.1038/onc.2008.310; PMID: 18955971
- Olsson GM, Brunmark A, Brunk UT. Acridine orange-mediated photodamage of microsomal- and lysosomal fractions. Virchows Arch B Cell Pathol Incl Mol Pathol 1989; 56:247 - 57; http://dx.doi.org/10.1007/BF02890023; PMID: 2565619
- Wattiaux R, Wattiaux-De CS, Ronveaux-Dupal MF, Dubois F. Isolation of rat liver lysosomes by isopycnic centrifugation in a metrizamide gradient. J Cell Biol 1978; 78:349 - 68; http://dx.doi.org/10.1083/jcb.78.2.349; PMID: 211139
- Oberle C, Huai J, Reinheckel T, Tacke M, Rassner M, Ekert PG, et al. Lysosomal membrane permeabilization and cathepsin release is a Bax/Bak-dependent, amplifying event of apoptosis in fibroblasts and monocytes. Cell Death Differ 2010; 17:1167 - 78; http://dx.doi.org/10.1038/cdd.2009.214; PMID: 20094062