Abstract
Suppression of autophagy has been increasingly recognized as a novel cancer therapeutic approach. Andrographolide (Andro), a diterpenoid lactone isolated from an herbal plant Andrographis paniculata, is known to possess anti-inflammatory and anticancer activity. In this study, we sought to examine the effect of Andro on autophagy, and to evaluate whether such effect is relevant to the sensitization effect of Andro on apoptosis induced by DNA damage agents in cancer cells. First, we found that Andro is able to significantly enhance autophagic markers in various cancer cell lines, including GFP-LC3 puncta and LC3-II level. Interestingly, Andro treatment also led to marked increase of p62 protein level and addition of chloroquine (CQ) failed to further enhance either LC3-II or p62 level, indicating that Andro is likely to suppress autophagic flux at the maturation and degradation stage. Next, we provided evidence that Andro inhibits autophagosome maturation not by affecting the lysosomal function, but by impairing autophagosome-lysosome fusion. Lastly, we demonstrated that treatment with cisplatin, a DNA damage agent, induces autophagy in cancer cells. Importantly, Andro is capable of sensitizing cisplatin-induced cell killing determined with both short-term apoptosis assays and long-term clonogenic test, via suppression of autophagy, a process independent of p53. In summary, these observations collectively suggest that Andro could be a promising anti-cancer agent in combination therapy via its potent inhibitory effect on autophagy by disrupting autophagosome-lysosome fusion.
Introduction
Autophagy is an evolutionarily conserved process by which cytoplasmic materials including damaged proteins and organelles are sequestered for lysosome-dependent degradation.Citation1,Citation2 In mammalian cells there are three types of autophagy: macroautophagy, microautophagy and chaperone-mediated autophagy. Macroautophagy (referred as autophagy hereafter) is characterized by the formation of a cytosolic double-membrane vesicle, the autophagosome. Autophagy is a dynamic process consisting of several consecutive stages: (1) induction or initiation, which involves formation of phagophore and is negatively regulated by mammalian target of rapamycin (mTOR) via the ULK1/Atg1 complex; (2) nucleation, which is mediated by the Beclin 1 and hVps34/class III PtdIns3K complex; (3) elongation, which is a critical step in forming the complete autophagosomes controlled by two ubiquitin-like conjugation systems (the Atg12–Atg5 and LC3/Atg8 system); and finally (4) maturation and degradation, which involves fusion with endosomes-lysosomes to form autolysosomes and degradation of the inner membrane together with its luminal contents.Citation3-Citation5
Recently, increasing evidence has suggested that autophagy is closely involved in the development of many important human diseases, including cancer, neurodegenerative diseases and metabolic disorders.Citation2,Citation6 The role of autophagy in cancer is rather complex and one important understanding is that autophagy is a double-edged sword in cancer. On the one hand, during the tumorigenesis and oncogenic transformation processes, autophagy is believed to function as a tumor suppressor mechanism;Citation7-Citation9 and on the contrary, once the tumor is formed, autophagy in the tumor cells will provide a survival advantage to resist cell death induced by cancer therapy.Citation10,Citation11 Although there are reports showing that promotion of autophagy enhances the therapeutic efficacy by inducing autophagy-dependent cell death or autophagic cell death in cancer cells,Citation12,Citation13 currently the overwhelming evidence tends to suggest otherwise: suppression of autophagy by either chemical inhibitors or by knockdown of Atg genes effectively sensitizes cancer cells to chemotherapeutic agents and radiotherapy.Citation10,Citation11,Citation14-Citation16 More importantly, many of the cancer therapeutic agents are capable of inducing autophagy via various pathways.Citation16,Citation17 Among various cancer therapeutic agents, cisplatin is one of the most important anticancer drugs used in the treatment of solid tumors, especially for ovarian, testicular, cervical and small cell lung carcinomas.Citation18 The main action of cisplatin is to cause DNA damage and ultimately triggers apoptosis.Citation18 Similar to many other cancer therapeutics, chemoresistance is one of the major obstacles in its clinical application.Citation19 Recent studies have revealed that cisplatin is capable of inducing autophagy in a number of human cancer cells and such autophagy acts as a prosurvival mechanism and is thus associated with reduced sensitivity to cisplatin-induced cell killing.Citation20-Citation26 It is thus possible that suppression of autophagy would serve as a logical strategy to overcome the chemoresistance of cisplatin.
Andrographolide (Andro) is one of the major active components in Andrographis paniculata, a traditional herbal medicine which has been widely used in treatment of various disorders including respiratory infection, bacterial dysentery, diarrhea and fever.Citation27 Andro has been shown to possess potent anti-inflammatory effect, which is executed by inhibiting the NFκB pathway and regulating expression of various cytokines.Citation28 Recently, there is emerging evidence showing the anticancer function of Andro, including those from our own laboratory, with the following main findings: (1) Andro is able to induce apoptosis in various human cancer cells;Citation29,Citation30 (2) Andro induces cell cycle arrest and growth inhibition;Citation31,Citation32 (3) Andro is capable of sensitizing human cancer cells to other cancer therapeutics such as TRAIL, doxorubicin and 5-fluorouracil;Citation33-Citation35 and (4) Andro suppresses cancer cell invasion and migration, thus may play a possible role in antimetastasis.Citation36 Taken together, it appears that Andro is a compound with great potential as a cancer therapeutic agent. At present, there is no report regarding the effect of Andro on autophagy. A very recent report has provided some clues that Andro is able to affect receptor trafficking by inhibiting receptor movement from the late endosomes to lysosomes in human cancer cells,Citation37 suggesting that Andro may interfere with the late stage of autophagy via its effect on endosomes and lysosomes.
In this study, we found that Andro is able to suppress autophagy by interfering with the fusion of autophagosome with the lysosome, through which it sensitizes human cancer cells to cisplatin-induced cell death in a p53-independent manner. At present autophagy suppression with small molecule inhibitors is increasingly recognized as a novel strategy in combination chemotherapy. Data from this study will thus open a new horizon of anticancer drug development by targeting endosomes/lysosomes and autophagy.
Results
Andro enhances the level of autophagy markers by suppressing autophagic flux in human cancer cells
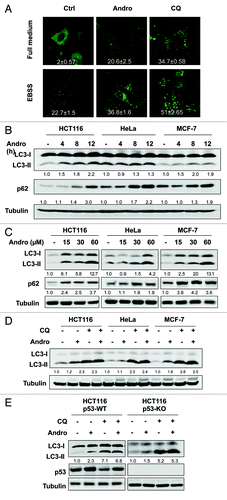
In order to examine the effect of Andro on autophagy, we first tested the changes of the GFP-LC3 distribution pattern in MEF cells with stable expression of the GFP-LC3, which is a well known marker for autophagosome formation.Citation38 As shown in , Andro enhanced the GFP-LC3 punctation in cells cultured in full-medium, as well as in cells under starvation (cultured in EBSS). Such an effect of Andro was found to be similar to that of CQ, although to a much lesser extent. Next, we checked the changes of the LC3 conversion by western blot. The protein level of LC3-II was markedly enhanced in a time- and dose-dependent manner in three human cancer cell lines treated with Andro (). Since the increase of GFP-LC3 puncta or LC3-II level may represent either the increased generation of autophagosomes (promotion of autophagic flux) or a blockage in autophagosomal maturation and degradation (suppression of autophagy at its late stage).Citation39,Citation40 Therefore, we examined the changes of autophagic flux by Andro by the following two approaches: (1) tracking the p62 protein level and (2) combined treatment with CQ. p62 is selectively incorporated into the autophagosome through direct binding to LC3 and is efficiently degraded by autophagy.Citation41 Andro was able to significantly enhance the p62 protein level in all three cancer cell lines tested (). Moreover, Andro failed to further enhance the LC3-II level in cells treated with CQ (). Notably, the increase of LC3-II by Andro alone was much lesser than that of CQ alone. Thus, these results suggest that the increased autophagic markers (GFP-LC3 puncta and LC3-II) in Andro-treated cells are unlikely due to enhanced autophagic flux, but rather by suppression of the late maturation and degradation stage, an effect similar to that of CQ, although to a lesser extent. Finally, we compared the effect of Andro in HCT116 cells with or without p53 and found that the effect of Andro on autophagy is p53-independent ().
Figure 1. Andro enhances the level of autophagy markers by suppressing autophagic flux in human cancer cells. (A) Effect of Andro on GFP-LC3 punctation. GFP-LC3-expressing stable MEF cells were treated with Andro (30 µM) or CQ (20 µM) in full-medium or EBSS for 6 h. The GFP-LC3 punctation was observed under a confocal microscope (× 600) and representative images are shown. The number of GFP-LC3 puncta per cell was quantified and presented as mean ± SD from 50 randomly selected cells based on one batch of three independent experiments. (B) and (C) Andro promotes LC3-II conversion and p62 accumulation in time-dependent and dose-dependent patterns. HCT116, HeLa and MCF7 cells were treated with Andro (30 µM) for indicated time courses, or treated with Andro at indicated doses for 12 h. LC3-II and p62 were quantified and the folds of increase were presented. (D) Andro does not promote autophagic flux. HCT116, HeLa and MCF7 cells were pretreated with chloroquine CQ (5 µM) for 30 min, followed by treatment with Andro (30 µM) for another 12 h. Cell lysates were collected and subject to western blot for detecting LC3 conversion. (E) Andro’s effect on autophagy is p53-independent. HCT116 p53-WT and p53-KO cells were treated as indicated in (D); and cell lysates were subject to western blot. LC3-II conversion was quantified accordingly.
Andro suppresses autophagy at its late stage without affecting lysosomal functions
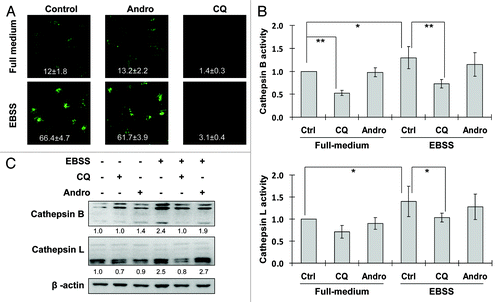
The data shown earlier suggest that Andro blocks the autophagic flux at the late stage of autophagy. Therefore, here we attempted to examine the effect of Andro on lysosomal functions. The intralysosomal pH is a critical factor in determining the cathepsin enzymatic activity and lysosomal functions.Citation42 We thus examined the effect of Andro on intralysosomal pH by using the Lysosensor which exhibits a pH-dependent increase in fluorescence intensity upon acidification.Citation43 As shown in , EBSS treatment dramatically enhanced the fluorescence intensity, indicating the decreased intralysosomal pH. Andro treatment did not affect intralysosomal pH, whereas treatment with CQ effectively abolished the fluorescence, indicating the neutralization of the intralysosomal pH (in full medium or EBSS). Moreover, we tested whether Andro can affect the enzymatic activity of cathepsins, the most important group of proteases inside lysosome,Citation44 via the following two methods: (1) detection of fluorescence intensity of cathepsin B and L substrates and (2) activated cathepsin B and L by western blot. As shown in , treatment with Andro failed to alter cathepsin B and L activities, while CQ was found to be highly effective in suppression of both cathepsin B and L activities. Taken together, these results collectively demonstrate that Andro has no adverse effect on lysosomal function per se.
Figure 2. Andro has no adverse effect on lysosomal function. (A) Effect of Andro on intralysosomal pH. HeLa cells were treated with Andro (30 µM) or CQ (50 µM) in full-medium or EBSS for 3 h. At the end of treatment, cells were stained with LysoSensor (5 µM) to evaluate lysosomal pH change. The fluorescence intensity was observed under a confocal microscope (× 600) and measured in 10 randomly selected fields and the relative fluorescence intensities were presented as mean ± SD. (B) Effect of Andro on cathepsin B/L enzyme activity. HeLa cells were treated as described in (A). Cell lysates were subject to cathepsin B/L activity assay. The changes of cathepsin activity are shown as relative peak height and are presented as means ± SD from three independent experiments (*p < 0.05; **p < 0.01). (C) Detection of cathepsin B/L activation by western blot. HeLa cells were treated as indicated in (B), and cell lysates were collected and subject to western blot. In (C), the densities of the respective mature form of cathepsin B and cathepsin L were quantified and the folds of increase were presented accordingly.
Andro inhibits autolysosome formation by interfering with the fusion of autophagosome with lysosome
In order to further understand the inhibitory effect of Andro on autophagy, here we transfected a tandem mRFP-GFP-LC3 construct (tfLC3) into HeLa cells and examined autophagosome maturation and autolysosome formation.Citation45 Essentially, this assay is based on the fact that the low pH inside the lysosome quenches the fluorescent signal of GFP, whereas mRFP exhibits more stable fluorescence in acidic compartments. Therefore, if the autolysosome maturation proceeds normally, it would give rise to more red-only puncta. Conversely, if the autophagosome does not fuse with lysosome or the lysosome function is impaired, most of the puncta should exhibit both red and green signals and appear to be yellow. As shown in , treatment with Andro (in full medium for 6 h) enhanced both GFP and mRFP puncta formation and colocalization, which is similar to that of CQ, although to a lesser extent. Such findings thus raise the possibility that the Andro may inhibit autolysosome formation by interfering fusion of autophagosome with lysosome.
Figure 3. Andro inhibits autolysosome formation by interfering fusion of autophagosome with lysosome. (A) Effects of Andro and CQ on autophagosome maturation. HeLa cells were first transiently transfected with the tfLC3 vector for 48 h, then treated with Andro (30 µM) or CQ (5 µM) for 6 h in full medium. At the end of treatment, cells were observed for the change of both green and red fluorescence using a confocal microscope (× 600). (B) HeLa cells transfected with GFP-mRFP-LC3 were treated as in (A) and the number of GFP- LC3 and mRFP-LC3 puncta per cell in different group was quantified. 50 cells were randomly selected and counted, and data (mean ± SD) are representative of three independent experiments. (C) Andro interferes with the fusion of the autophagosome with the lysosome. GFP-LC3-expressing stable MEF cells were treated with Andro (30 µM), bafilomycin A1 (100 nM) or CQ (20 µM), respectively, in full-medium or EBSS for 6 h, then processed for LAMP-1 immunostaining and observed under a confocal microscope (× 600). The enlarged areas were presented to show the colocalization of GFP-LC3 (in green) with LAMP-1 (in red). (D) GFP-LC3 MEF cells were treated as in (C) and the GFP-LC3/LAMP1 colocalization was quantified with a colocalization tool from Olympus Fluoview software (Olympus). Data (mean ± SD) are representative of three independent experiments.
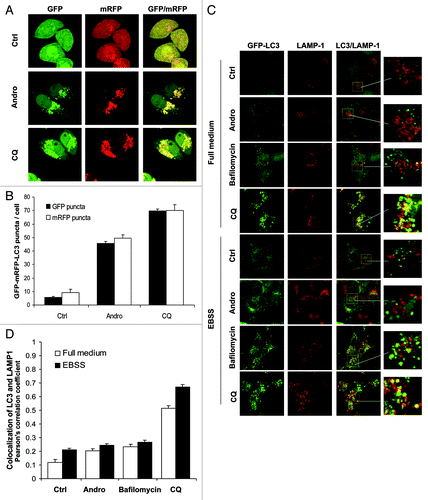
To confirm the above hypothesis, we next examined the autophagosome-lysosome fusion process by tracking the late endosome/lysosome marker LAMP-1 to detect its colocalization with the autophagosomal marker GFP-LC3 in GFP-LC3-expressing stable MEF cells.Citation46 In this study, we used bafilomycin A1 as a positive control, which was known as a vacuolar H+-ATPase inhibitor and can inhibit the fusion between autophagosome and lysosome.Citation47 As shown in , Andro treatment induced the GFP-LC3 punctation in cells cultured in either full-medium or EBSS, and in most of GFP-LC3 puncta, GFP-LC3 were not colocalized with LAMP-1. Such changes were in fact rather similar to that in cells treated with bafilomycin A1. In contrast, in cells treated with CQ, there was extensive colocalization GFP-LC3 and LAMP-1 in cells cultured in full-medium (basal autophagy) or EBSS (induced autophagy) (). Such observations were confirmed by a quantification analysis for the colocalization coefficiency as presented in . Thus, data from this part of our study present clear evidence that Andro inhibits autophagosome maturation not by affecting the lysosomal function, but by impairing autophagosome-lysosome fusion, thus further strengthening our earlier observations that the enhanced autophagic marker in cells treated with Andro is due to the suppression of autophagy at the late stage.
Cisplatin induces autophagic flux in human cancer cells in a p53-independent manner
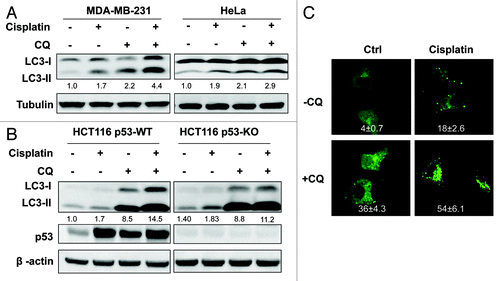
In this part of our study, we studied whether cisplatin is able to induce autophagy and if so, what is the function of autophagy in cisplatin-induced cell death. As shown in , cisplatin enhanced LC3-II conversion in two human cancer cell lines. Importantly, the addition of CQ further increased the LC3-II level, suggesting the fact that cisplatin is capable of increasing autophagic flux in cancer cells. Moreover, we observed similar effects by cisplatin on LC3-II in HCT116 p53-WT and p53-KO cells (), indicating the possibility that induction of autophagy by cisplatin is p53-independent. Such a finding was further supported by the data from , in which cisplatin was found to enhance the GFP-LC3 puncta formation which was further increased by CQ. Our data are generally consistent with an earlier report for cisplatin-induced autophagy.Citation48
Figure 4. Cisplatin induces autophagy in human cancer cells. (A) Cisplatin promotes autophagy flux in human cancer cells. MDA-MB-231 and HeLa cells were pretreated with CQ (5 µM) for 30 min, followed by cisplatin (10 µg/ml) for another 12 h, cell lysates were collected and subject to western blot. (B) Cisplatin’s effect on autophagy is p53-independent. HCT116 p53-WT and p53-KO cells were treated as described in (A), and cell lysates were collected and subject to western blot. In (A and B) LC3-II was quantified and the folds of increase were presented accordingly. (C) Induction of GFP-LC3 punctation by cisplatin. Same treatment as (A) was performed on MEFs with stable expression of GFP-LC3. The GFP-LC3 distribution pattern was observed under a confocal microscope (× 600) and the number of GFP-LC3 dots per cell was quantified. Data (mean ± SD) are representative of three independent experiments.
Prosurvival function of autophagy in cell death induced by cisplatin
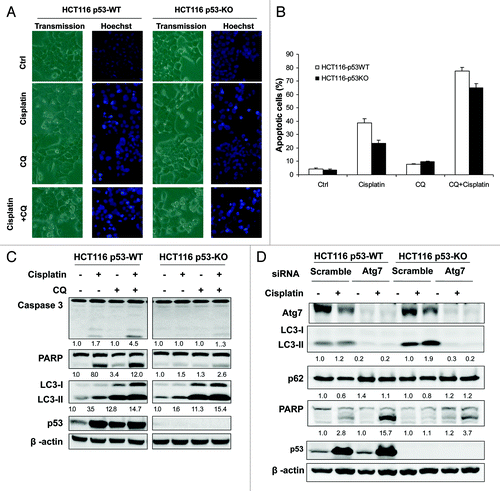
Here we attempted to determine whether the autophagy induced by cisplatin serves as a prosurvival or prodeath mechanism. We first used CQ to inhibit autophagy and tested its effect on cisplatin-induced apoptotic cell death. Cells treated with CQ markedly enhanced cisplatin-induced apoptotic cell death in both HCT116 p53-WT and p53-KO cells as determined by cell morphology and Hoechst staining (), as well as PARP and caspase 3 cleavage detected by western blot (). The blockage of autophagy by CQ treatment was confirmed by the increase of LC3-I to II conversion (). Importantly, although cisplatin-induced cell death is largely p53-depdendent, we found that CQ was able to increase cisplatin-induced cell death in both p53-WT and KO cells (). Similar effects were also found when cells were treated with bafilomycin A1 (data not shown). In order to further elucidate the role of autophagy in cisplatin-induced cell death, we manipulated the autophagy level by knockdown of Atg7 and examined the effect on cisplatin-induced cell death. As shown in , suppression of autophagy by knockdown of Atg7 significantly sensitized cells to cisplatin-induced cell death, as determined by cell morphology and PARP and caspase 3 cleavage (). The effect of such knockdown on autophagy was confirmed by reduced LC3-I to II conversion and Atg 7 protein level (). These results thus collectively demonstrate that autophagy plays a prosurvival role in cisplatin-induced apoptotic cell death.
Figure 5. Prosurvival function of autophagy in cell death induced by cisplatin. (A-C) CQ sensitizes cells to cisplatin-induced cell death. HCT116 p53-WT and p53-KO cells were pretreated with CQ (50 µM) for 30 min followed by cisplatin (10 µg/ml) for another 24 h. At the end of treatment, cell death was evaluated by Hoechst staining (A and B) and western blot (C). (B) The Hoechst staining data was presented as the percentage of cells with evident nuclear condensation in 200 randomly selected cells. Data (mean ± SD) are representative of three independent experiments. In (C), the cleavage of caspase 3 and PARP, and LC3-II were quantified and the folds of increase were presented accordingly. (D) Knockdown of Atg7 enhanced cisplatin-induced cell death. After transfection with scramble or Atg7 siRNA oligos for 48 h, HCT116 p53-WT and p53-KO cells were treated with cisplatin (10 µg/ml) for 24 h, and cell death was determined by western blot.
Andro sensitizes cisplatin-induced cell death via suppression of autophagy
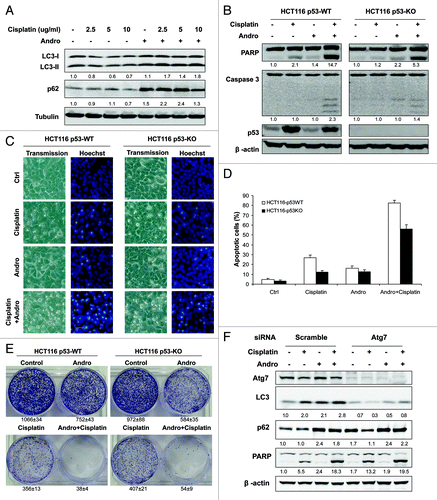
Since our data suggest that autophagy plays a prosurvival role in cisplatin-induced cell death, whereas Andro is able to suppress autophagy by interfering the fusion of autophagosome with lysosome, here we tested the effect of Andro on cisplatin-induced cell death. As shown in , Andro increased cisplatin-induced LC3-II conversion and p62 protein level, thus confirming that Andro is able to suppress cisplatin-induced autophagy. More importantly, in both HCT116 p53-WT and p53-KO cells, Andro significantly augmented cisplatin-induced apoptotic cell death, as evidenced by increased cleavage of caspase-3 and PARP (). Such findings are consistent with the cell morphology and Hoechst staining results (). Notably, the sensitization effect of Andro on cisplatin-induced cell death was believed to be p53-independent, based on the results that Andro was also effective in enhancing cisplatin-induced apoptosis in p53-KO HCT116 cells, although there was still more severe cell death in p53-WT cells with the combined treatment of Andro and cisplatin (). In addition, to test the long-term effect of Andro on cisplatin-induced cell killing, we performed the colony formation assay in both HCT116 p53-WT and p53-KO cells. As showed in , although Andro and cisplatin alone could reduce the number colony formation to a certain extent, combined treatment with Andro and cisplatin almost completely suppressed colony formation. Finally, in order to provide more evidence to support the notion that Andro sensitizes cisplatin-induced apoptotic cell death via suppression of autophagy, we compared the effect of Andro in cells with or without Atg7 knockdown. As shown in , Atg7 knockdown significantly enhanced cisplatin-induced apoptosis (measured by PARP cleavage), being consistent with the data in . Importantly, Andro’s sensitization effect on cisplatin-induced cell is rather marginal in cells with Atg7-knockdown, comparing to that in the control-knockdown cells (). Such findings thus support our conclusion that Andro-induced sensitization effect is due to its inhibition on autophagy.
Figure 6. Andro blocks cisplatin-induced autophagy and sensitizes cell death in a p53-independent manner. (A) Andro increases cisplatin-induced LC3-II and p62 protein level. HeLa cells were pretreated with Andro (30 µM) for 30 min before exposed to various doses of cisplatin for 12 h. (B) Andro promotes cisplatin-induced apoptosis in HCT116 p53-WT and p53-KO cells. Cells were pretreated with Andro (30 µM) for 30 min followed by treatment of cisplatin (10 µg/ml) for 24 h and then cell lysates were collected for detection of cleavages of PARP and caspase 3. (C and D) Evaluation of cell death using Hoechst staining. HCT116 p53-WT and p53-KO cells were treated as described in (B), respectively. At the end of treatment, cell death was evaluated by morphological change which was visualized by Hoechst staining. Representative images were taken by a phase-contrast microscope (x200). (D) The numeric data were presented as the percentage of cells with evident nuclear condensation in 200 randomly selected cells. Data (mean ± SD) are representative of three independent experiments. (E) HCT116 p53-WT and p53-KO cells were treated as indicated for 12 h before reseeded in 6-well plates (5,000 cells/well) respectively. After two weeks, the survival colonies were stained by 0.5% crystal violet for 1 h and Representative images of HCT116 cells were photographed using digital camera. Data (mean ± SD) are representative of three independent experiments. (F) Andro failed to significantly sensitize cell death in Atg7-knockdown cells. After transfection with scramble or Atg7 siRNA oligos for 48 h, HCT116 p53-WT cells were treated as indicated for 24 h, and cell death was determined by western blot. In (A, B and F), the densities of LC3-II, p62 and the respective cleaved band of caspase 3 and PARP were quantified and the folds of increase were presented accordingly.
Discussion
Autophagy generally serves as a prosurvival mechanism under stress conditions and suppression of autophagy has been increasingly recognized as a novel therapeutic strategy in combination with cancer therapy.Citation11,Citation13,Citation17 At present, there are only a limited number of small molecular chemical inhibitors used in autophagy research. The phosphoinositide 3-kinase (PtdIns3K) inhibitors, such as 3-methyadenine (3-MA) and wortmannin, are not class III PtdIns3K specific as they also block the class I PtdIns3K which is a negative regulator of autophagy via mTOR.Citation40,Citation49,Citation50 Moreover, our recent work has shown that 3-MA can even promote autophagy when treated with prolonged period time in no-starvation conditions.Citation51 Hence, the potential clinical usage of this group of autophagy inhibitors is rather limited. The second group of autophagy chemical inhibitors are those inhibiting the late stage of autophagy, such as CQ and bafilomycin A1, via their inhibitory effects on lysosomal function and autophagosome-lysosome fusion, respectively.Citation40 CQ is probably the only autophagy inhibitor currently in clinical trial (phase I and II) in combination with various cancer therapeutic agents in different tumors.Citation11 Based on the above, there is an urgent need to develop specific autophagy inhibitors that are clinically viable.
In the first part of our study we present solid evidence that Andro is a potent autophagy inhibitor via disrupting the fusion of autophagosome with lysosome, based on the following observations: (1) Andro increases autophagic markers by inhibiting autophagic flux; (2) Andro has no adverse effect on either lysosomal pH or cathepsin enzyme activity; and (3) Andro prevents the co-localization of GFP-LC3 (marker for autophagosome) with LAMP-1 (marker for lysosome). Indeed such effects by Andro are similar to that of bafilomycin A1, which is known to block the autophagosome-lysosme fusion process.Citation47 Data from this study thus expand our current knowledge about the bioactivity of Andro. At present, the underlying molecular mechanisms for Andro’s effects on the fusion process remain to be further investigated.
Another important finding from our study is that cisplatin induces autophagy and such autophagy serves as a prosurvival mechanism against cisplatin-induced apoptosis. In fact our results are generally consistent with some of the earlier reports.Citation20-Citation26 For instance, in esophageal squamous cell carcinoma cells, suppression of autophagy by 3-methyladenine greatly enhanced cisplatin-induced apoptosis.Citation23 Although the role of autophagy in cell death and cell survival is still controversial, there is increasing understanding that autophagy plays a prosurvival function in cells under stress conditions, such as starvation, oxidative stress, metabolic stress and DNA damage.Citation6,Citation7 Moreover, there is evidence showing that autophagy is involved in the resistance of cancer cells to chemotherapy.Citation11,Citation16 Therefore, suppression of autophagy becomes a logical approach in combination therapy to overcome the resistance and to enhance the therapeutic efficacy. In this study, due to the unique inhibitory function of Andro on autophagy, Andro is able to markedly sensitize cisplatin-induced cell killing measured with both short-term apoptosis assays and long-term clonogenic test. Such observations thus suggest that Andro or similar compounds could be further developed as cancer therapeutic agents by targeting autophagy.
p53 is the most important tumor suppressor in human cancer. Right now, the role of p53 in autophagy is still controversial. The nuclear p53 is known to promote autophagy, whereas the cytosolic p53 has been reported to inhibit autophagy.Citation52,Citation53 In this study, we evaluated the involvement of p53 in the function of Andro and cisplatin. Although Andro is known to activate p53 via a defined pathway involving ROS and JNK,Citation34 the inhibitory effect of Andro on autophagy is found to be p53-independent. Moreover, although p53 is an important mediator in cisplatin-induced apoptosis,Citation19 sensitization to cisplatin-induced cell death via autophagy inhibition is also p53-independent. Based on the well-known facts that cancer cells without a functional p53 are more resistant to cancer therapeutic agent, it is thus believed that Andro offers an ideal model compound in cancer combination therapy, especially in those cancers with p53 mutation or deletion.
In summary, we show for the first time that Andro is able to suppress autophagy by interfering the fusion of autophagosome with lysosome and via which Andro sensitizes cisplatin-induced cell death in human cancer cells. At present, suppression of autophagy has been increasingly recognized as a novel cancer therapeutic approach and there are only a very limited number of small molecule inhibitors are available for animal models and clinical trials. Therefore, data from this study will help to develop a novel autophagy inhibitor with great potential in clinical application for combination cancer chemotherapy, especially in cancers with p53 mutation or deletion.
Materials and Methods
Chemicals, reagents and antibodies
The chemicals used in our experiments were: Andro (Sigma, 365645), cisplatin (Sigma, P4394), chloroquine (CQ, Sigma, C6628), bafilomycin A1 (Sigma, B1793), cathepsin B substrate (Z-RR-AMC, Calbiochem, 219392), cathepsin L (Ac-HRYR-ACC, Calbiochem, 219497). LysoSensor Yellow/Blue DND-160 (Invitrogen, L7545). The antibodies used in our experiments were: anti-microtubule-associated protein 1 light chain 3 (LC3) antibody (Sigma, L7543), anti-poly (ADP-ribose) polymerase (PARP) antibody (Cell Signaling Technology, 9542), anti-caspase-3 antibody (Cell Signaling Technology, 9662), cathepsin B antibody (Santa Cruz, SC-13985), cathepsin L antibody (Santa Cruz, SC-6498), anti-p53 antibody (Santa Cruz, SC126), anti-Atg7 antibody (ProScience 3617), anti-p62 antibody (Abnova, H00008878-2C11), anti-lysosome-associated membrane protein (LAMP-1) antibody (Developmental Studies Hybridoma Bank, 1D4B).
Cell culture and treatments
HeLa cells, MCF-7 cells and MDA-MB-231 cells were all obtained from the American Type Culture Collection. An isogenic pair of HCT116 colon cancer cell lines (p53 wild-type and knockout) was kindly provided by Dr. Bert Vogelstein (Johns Hopkins University).Citation54 The GFP-LC3-expressing stable MEF cells line was provided by Dr. N. Mizushima (Tokyo Medical and Dental University).Citation46 The mRFP-GFP tandem fluorescence-tagged LC3 construct (tfLC3) was provided by Dr. T. Yoshimori (Osaka University).Citation45 All cell lines were maintained in DMEM (Sigma, D1152) containing 10% fetal bovine serum (HyClone, SV30160.03) in a 5% CO2 atmosphere at 37°C. Andro and cisplatin were dissolved in DMSO and DMF at 100 mmol/L and 50 mg/mL as stock solution respectively.
Western blotting
At the end of the designated treatments, cells were lysed in M2 lysis buffer (20 mM Tris at pH 7, 0.5% NP-40, 250 mM NaCl, 3 mM EDTA, 3 mM EGTA, 2 mM dithiothreitol, 0.5 mM phenylmethylsulfonyl fluoride, 20 mM glycerol phosphate, 1 mM sodium vanadate and proteinase inhibitor cocktail). An equal amount of protein was resolved by SDS-PAGE and transferred onto PVDF membrane (Bio-Rad, 162-0177). After blocking with 5% nonfat milk, the membrane was probed with designated primary and secondary antibodies, developed with the enhanced chemiluminescence method (Thermo Scientific, 34076) and visualized with the Kodak Image Station 4000R (Kodak). The density of the various bands in western blot was quantified using the Kodak Image Station 4000R software (Kodak).
LAMP-1 immunofluorescence staining and confocal microscopy
Cells were seeded to a coverglass slide chamber (Lab-Tek, NUNC, 155411), and after the designated treatments, cells were washed with PBS, then fixed with 4% paraformaldehyde in PBS for 15 min at room temperature, and permeabilized with 50 mg/ml digitonin in PBS for 30 min. Cells were washed with PBS and blocked with 1% BSA in PBS for 30 min, then incubated with rat anti-mouse LAMP-1 primary antibody in a 1:100 dilution overnight at 4°C, followed by Alexa Fluor 633 goat anti-rat secondary antibody (Invitrogen, A21094). The cells were examined and recorded using a confocal microscope (Olympus Fluoview FV1000) and representative cells were selected and photographed. For HeLa cells transfected with tfLC3 or MEFs with stable expression of GFP-LC3-, cells were observed under a confocal microscope (Olympus Fluoview 2000) and representative cells were selected and photographed. The GFP-LC3 and mRFP-LC3 puncta were manually counted under a confocal microscope. For each group, 50 cells were randomly selected. The data presented were from one representative experiment of three independent repeats. The colocalization of LC3 and LAMP1 is analyzed by Olympus FluoView colocalization tool (Olympus).
Measurement of cathepsin B/L activity
Detection of cathepsin B/L activity in total cell extracts was conducted as described previously.Citation55 Briefly, cells were lysed in M2 buffer and the lysate (25 μg cellular protein) was then incubated with 50 μM of the fluorogenic cathepsin B/L substrate (Z-RR-AMC or Ac-HRYR-ACC) in 100 μl cell-free system buffer (10 mM HEPES-NaOH pH 7.4, 220 mM mannitol, 68 mM sucrose, 2 mM NaCl, 2.5 mM KH2PO4, 0.5 mM EGTA, 2 mM MgCl2, 5 mM pyruvate, 0.1 mM PMSF and 1 mM dithiothreitol) in a 96-well plate for 1 h at 37°C. The release of free 7-amino-4-methylcoumarin (AMC) or 7-mino-4-carbamoylmethylcoumarin (ACC) was monitored by a fluorometer (Tecan SpectraFluor Plus) at an excitation wavelength of 380 nm and an emission wavelength of 460 nm. Data are expressed as percentage of fluorescence intensity compared with the control group.
Measurement of intralysosomal pH
The LysoSensor Yellow/Blue DND-160 reagents exhibit a pH-dependent increase in fluorescence intensity upon acidification. HeLa cells were cultured on coverglass slide chamber, and after the designated treatments, cells were incubated with 5 μM LysoSensor reagent to evaluate lysosomal pH change. The fluorescence intensity was observed under a confocal microscope (Olympus Fluoview FV1000) and measured in 10 ramdonly selected fields with ImageJ software, and the relative fluorescence intensities (arbitrary unit) were presented as means ± SD.
Detection of cell death
In this study, several approaches were used to detect cell death quantitatively and qualitatively, including (1) morphological changes under phase-contrast microscopy and (2) Hoechst staining as described previouslyCitation55 Briefly, treated cells were stained with Hoechst (10 mg/ml) mixture solution (in PBS) at room temperature for 30 min. The staining of nuclei was visualized and captured under an inverted fluorescent microscope (Nikon ECLIPSE TE2000-S).
Colony formation assay
HCT116 cells were treated as designated for 12 h before reseeded in six-well plates (5,000 cells/well) respectively. After two weeks, the survival clones were stained by 0.5% crystal violet for 1h and photos were taken using digital camera and the colony was counted with the ImageJ software.
Small interfering RNA (siRNA) and transient transfection
The scrambled RNAi oligonucleotides and siRNAs targeting homo sapiens Atg7 (Dharmacon, L-020112-00) were transfected into HCT116 p53 wild-type (WT) and knockout (KO) cells using the DharmaFECT 4 Transfection Reagent (Dharmacon, T-2001-02) according to the manufacturer’s protocol. The tfLC3 vector, which has been described previously, was provided by Dr. Tamotsu Yoshimori.Citation45 The transfection was performed using Lipofectamine 2000 reagent according to the manufacturer’s protocol (Invitrogen, 11668-019).
Statistical analysis
All data presented are representatives from at least three independent experiments. The numeric data from the cathepsin B/L activity analysis are presented as means ± SD from three independent experiments and analyzed using Student's t-test. p value less than 0.05 was considered as statistically significant.
| Abbreviations: | ||
| 3-MA | = | 3-methyladenine |
| ACC | = | 7-amino-4-carbamoylmethylcoumarin |
| AMC | = | 7-amino-4-methylcoumarin |
| Andro | = | Andrographolide |
| CQ | = | chloroquine |
| DMEM | = | Dulbecco’s modified Eagle’s medium |
| EBSS | = | Earle’s Balanced Salt Solution |
| GFP | = | green fluorescence protein |
| KO | = | knockout |
| LC3 | = | microtubule-associated protein 1 light chain 3 |
| LAMP-1 | = | lysosome-associated membrane protein 1 |
| MEF | = | mouse embryonic fibroblast |
| mTOR | = | mammalian target of rapamycin |
| PARP | = | poly (ADP-ribose) polymerase |
| PtdIns3K | = | phosphatidylinositol-3-kinase |
| RFP | = | red fluorescence protein |
| ROS | = | reactive oxygen species |
| siRNA | = | small interfering RNA |
| tfLC3 | = | tandem fluorescence-tagged LC3 construct |
| WT | = | wild type |
Acknowledgments
We thank Dr. B Vogelstein for providing the p53-WT and p53-KO HCT116 cells, Dr. N Mizushima for the GFP-LC3-expressing stable MEF cells line, and Dr. T. Yoshimori for the mRFP-GFP tandem fluorescence-tagged LC3 construct (tfLC3). S Hu was in part supported by the Centre for Environmental and Occupational Health Research (CEOHR), Department of EPH, NUS. R.C. and Y.C. were supported by the Science Research Programme (SRP) in NUS for junior college students in Singapore. This study is supported in part by research grants from Singapore National Medical Research Council (NMRC/1118/2007) and Singapore Biomedical Research Council (BMRC /08/1/21/19/554) to H.M.S., China Natural Science Foundation grants 81028014 and 81172659 to H.M.S. and X.Z., as well as the NUS Toxicology Program to H.M.S. and C.N.O.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science 2000; 290:1717 - 21; http://dx.doi.org/10.1126/science.290.5497.1717; PMID: 11099404
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069 - 75; http://dx.doi.org/10.1038/nature06639; PMID: 18305538
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009; 43:67 - 93; http://dx.doi.org/10.1146/annurev-genet-102808-114910; PMID: 19653858
- Ohsumi Y, Mizushima N. Two ubiquitin-like conjugation systems essential for autophagy. Semin Cell Dev Biol 2004; 15:231 - 6; http://dx.doi.org/10.1016/j.semcdb.2003.12.004; PMID: 15209383
- Mizushima N, Ohsumi Y, Yoshimori T. Autophagosome formation in mammalian cells. Cell Struct Funct 2002; 27:421 - 9; http://dx.doi.org/10.1247/csf.27.421; PMID: 12576635
- Meijer AJ, Codogno P. Autophagy: regulation and role in disease. Crit Rev Clin Lab Sci 2009; 46:210 - 40; http://dx.doi.org/10.1080/10408360903044068; PMID: 19552522
- Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006; 10:51 - 64; http://dx.doi.org/10.1016/j.ccr.2006.06.001; PMID: 16843265
- Karantza-Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, Jin S, et al. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev 2007; 21:1621 - 35; http://dx.doi.org/10.1101/gad.1565707; PMID: 17606641
- Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer 2007; 7:961 - 7; http://dx.doi.org/10.1038/nrc2254; PMID: 17972889
- Chen N, Debnath J. Autophagy and tumorigenesis. FEBS Lett 2010; 584:1427 - 35; http://dx.doi.org/10.1016/j.febslet.2009.12.034; PMID: 20035753
- Chen N, Karantza V. Autophagy as a therapeutic target in cancer. Cancer Biol Ther 2011; 11:157 - 68; http://dx.doi.org/10.4161/cbt.11.2.14622; PMID: 21228626
- Kondo Y, Kondo S. Autophagy and cancer therapy. Autophagy 2006; 2:85 - 90; PMID: 16874083
- Moretti L, Yang ES, Kim KW, Lu B. Autophagy signaling in cancer and its potential as novel target to improve anticancer therapy. Drug Resist Updat 2007; 10:135 - 43; http://dx.doi.org/10.1016/j.drup.2007.05.001; PMID: 17627865
- Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, et al. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest 2007; 117:326 - 36; http://dx.doi.org/10.1172/JCI28833; PMID: 17235397
- White E, Karp C, Strohecker AM, Guo Y, Mathew R. Role of autophagy in suppression of inflammation and cancer. Curr Opin Cell Biol 2010; 22:212 - 7; http://dx.doi.org/10.1016/j.ceb.2009.12.008; PMID: 20056400
- Livesey KM, Tang D, Zeh HJ, Lotze MT. Autophagy inhibition in combination cancer treatment. Curr Opin Investig Drugs 2009; 10:1269 - 79; PMID: 19943199
- Maycotte P, Thorburn A. Autophagy and cancer therapy. Cancer Biol Ther 2011; 11:127 - 37; http://dx.doi.org/10.4161/cbt.11.2.14627; PMID: 21178393
- Barabas K, Milner R, Lurie D, Adin C. Cisplatin: a review of toxicities and therapeutic applications. Vet Comp Oncol 2008; 6:1 - 18; http://dx.doi.org/10.1111/j.1476-5829.2007.00142.x; PMID: 19178659
- Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene 2003; 22:7265 - 79; http://dx.doi.org/10.1038/sj.onc.1206933; PMID: 14576837
- Ren JH, He WS, Nong L, Zhu QY, Hu K, Zhang RG, et al. Acquired cisplatin resistance in human lung adenocarcinoma cells is associated with enhanced autophagy. Cancer Biother Radiopharm 2010; 25:75 - 80; http://dx.doi.org/10.1089/cbr.2009.0701; PMID: 20187799
- Periyasamy-Thandavan S, Jiang M, Wei Q, Smith R, Yin XM, Dong Z. Autophagy is cytoprotective during cisplatin injury of renal proximal tubular cells. Kidney Int 2008; 74:631 - 40; http://dx.doi.org/10.1038/ki.2008.214; PMID: 18509315
- O'Donovan TR, O'Sullivan GC, McKenna SL. Induction of autophagy by drug-resistant esophageal cancer cells promotes their survival and recovery following treatment with chemotherapeutics. Autophagy 2011; 7:509 - 24; http://dx.doi.org/10.4161/auto.7.5.15066; PMID: 21325880
- Liu D, Yang Y, Liu Q, Wang J. Inhibition of autophagy by 3-MA potentiates cisplatin-induced apoptosis in esophageal squamous cell carcinoma cells. Med Oncol 2011; 28:105 - 11; http://dx.doi.org/10.1007/s12032-009-9397-3; PMID: 20041317
- Kim HS, Hwang JT, Yun H, Chi SG, Lee SJ, Kang I, et al. Inhibition of AMP-activated protein kinase sensitizes cancer cells to cisplatin-induced apoptosis via hyper-induction of p53. J Biol Chem 2008; 283:3731 - 42; http://dx.doi.org/10.1074/jbc.M704432200; PMID: 18079115
- Harhaji-Trajkovic L, Vilimanovich U, Kravic-Stevovic T, Bumbasirevic V, Trajkovic V. AMPK-mediated autophagy inhibits apoptosis in cisplatin-treated tumour cells. J Cell Mol Med 2009; 13:3644 - 54; http://dx.doi.org/10.1111/j.1582-4934.2009.00663.x; PMID: 20196784
- Claerhout S, Verschooten L, Van Kelst S, De Vos R, Proby C, Agostinis P, et al. Concomitant inhibition of AKT and autophagy is required for efficient cisplatin-induced apoptosis of metastatic skin carcinoma. Int J Cancer 2010; http://dx.doi.org/10.1002/ijc.25300; PMID: 20196784
- Poolsup N, Suthisisang C, Prathanturarug S, Asawamekin A, Chanchareon U. Andrographis paniculata in the symptomatic treatment of uncomplicated upper respiratory tract infection: systematic review of randomized controlled trials. J Clin Pharm Ther 2004; 29:37 - 45; http://dx.doi.org/10.1046/j.1365-2710.2003.00534.x; PMID: 14748896
- Xia YF, Ye BQ, Li YD, Wang JG, He XJ, Lin X, et al. Andrographolide attenuates inflammation by inhibition of NF-kappa B activation through covalent modification of reduced cysteine 62 of p50. J Immunol 2004; 173:4207 - 17; PMID: 15356172
- Cheung HY, Cheung SH, Li J, Cheung CS, Lai WP, Fong WF, et al. Andrographolide isolated from Andrographis paniculata induces cell cycle arrest and mitochondrial-mediated apoptosis in human leukemic HL-60 cells. Planta Med 2005; 71:1106 - 11; http://dx.doi.org/10.1055/s-2005-873128; PMID: 16395645
- Zhou J, Zhang S, Ong CN, Shen HM. Critical role of pro-apoptotic Bcl-2 family members in andrographolide-induced apoptosis in human cancer cells. Biochem Pharmacol 2006; 72:132 - 44; http://dx.doi.org/10.1016/j.bcp.2006.04.019; PMID: 16740251
- Jada SR, Matthews C, Saad MS, Hamzah AS, Lajis NH, Stevens MF, et al. Benzylidene derivatives of andrographolide inhibit growth of breast and colon cancer cells in vitro by inducing G(1) arrest and apoptosis. Br J Pharmacol 2008; 155:641 - 54; http://dx.doi.org/10.1038/bjp.2008.368; PMID: 18806812
- Shi MD, Lin HH, Lee YC, Chao JK, Lin RA, Chen JH. Inhibition of cell-cycle progression in human colorectal carcinoma Lovo cells by andrographolide. Chem Biol Interact 2008; 174:201 - 10; http://dx.doi.org/10.1016/j.cbi.2008.06.006; PMID: 18619950
- Yang L, Wu D, Luo K, Wu S, Wu P. Andrographolide enhances 5-fluorouracil-induced apoptosis via caspase-8-dependent mitochondrial pathway involving p53 participation in hepatocellular carcinoma (SMMC-7721) cells. Cancer Lett 2009; 276:180 - 8; http://dx.doi.org/10.1016/j.canlet.2008.11.015; PMID: 19097688
- Zhou J, Lu GD, Ong CS, Ong CN, Shen HM. Andrographolide sensitizes cancer cells to TRAIL-induced apoptosis via p53-mediated death receptor 4 up-regulation. Mol Cancer Ther 2008; 7:2170 - 80; http://dx.doi.org/10.1158/1535-7163.MCT-08-0071; PMID: 18645026
- Zhou J, Ong CN, Hur GM, Shen HM. Inhibition of the JAK-STAT3 pathway by andrographolide enhances chemosensitivity of cancer cells to doxorubicin. Biochem Pharmacol 2010; 79:1242 - 50; http://dx.doi.org/10.1016/j.bcp.2009.12.014; PMID: 20026083
- Shi MD, Lin HH, Chiang TA, Tsai LY, Tsai SM, Lee YC, et al. Andrographolide could inhibit human colorectal carcinoma Lovo cells migration and invasion via down-regulation of MMP-7 expression. Chem Biol Interact 2009; 180:344 - 52; http://dx.doi.org/10.1016/j.cbi.2009.04.011; PMID: 19426720
- Tan Y, Chiow KH, Huang D, Wong SH. Andrographolide regulates epidermal growth factor receptor and transferrin receptor trafficking in epidermoid carcinoma (A-431) cells. Br J Pharmacol 2010; 159:1497 - 510; http://dx.doi.org/10.1111/j.1476-5381.2009.00627.x; PMID: 20233216
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19:5720 - 8; http://dx.doi.org/10.1093/emboj/19.21.5720; PMID: 11060023
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 2004; 15:1101 - 11; http://dx.doi.org/10.1091/mbc.E03-09-0704; PMID: 14699058
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313 - 26; http://dx.doi.org/10.1016/j.cell.2010.01.028; PMID: 20144757
- Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005; 171:603 - 14; http://dx.doi.org/10.1083/jcb.200507002; PMID: 16286508
- McGrath ME. The lysosomal cysteine proteases. Annu Rev Biophys Biomol Struct 1999; 28:181 - 204; http://dx.doi.org/10.1146/annurev.biophys.28.1.181; PMID: 10410800
- You JO, Auguste DT. The effect of swelling and cationic character on gene transfection by pH-sensitive nanocarriers. Biomaterials 2010; 31:6859 - 66; http://dx.doi.org/10.1016/j.biomaterials.2010.04.048; PMID: 20493524
- Guha S, Padh H. Cathepsins: fundamental effectors of endolysosomal proteolysis. Indian J Biochem Biophys 2008; 45:75 - 90; PMID: 21086720
- Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007; 3:452 - 60; PMID: 17534139
- Hosokawa N, Hara Y, Mizushima N. Generation of cell lines with tetracycline-regulated autophagy and a role for autophagy in controlling cell size. FEBS Lett 2007; 581:2623 - 9; PMID: 17553497
- Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct 1998; 23:33 - 42; http://dx.doi.org/10.1247/csf.23.33; PMID: 9639028
- Inoue K, Kuwana H, Shimamura Y, Ogata K, Taniguchi Y, Kagawa T, et al. Cisplatin-induced macroautophagy occurs prior to apoptosis in proximal tubules in vivo. Clin Exp Nephrol 2010; 14:112 - 22; http://dx.doi.org/10.1007/s10157-009-0254-7; PMID: 20013139
- Knight ZA, Shokat KM. Chemically targeting the PI3K family. Biochem Soc Trans 2007; 35:245 - 9; http://dx.doi.org/10.1042/BST0350245; PMID: 17371250
- Kong D, Yamori T. Phosphatidylinositol 3-kinase inhibitors: promising drug candidates for cancer therapy. Cancer Sci 2008; 99:1734 - 40; PMID: 18616528
- Wu YT, Tan HL, Shui G, Bauvy C, Huang Q, Wenk MR, et al. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem 2010; 285:10850 - 61; http://dx.doi.org/10.1074/jbc.M109.080796; PMID: 20123989
- Maiuri MC, Galluzzi L, Morselli E, Kepp O, Malik SA, Kroemer G. Autophagy regulation by p53. Curr Opin Cell Biol 2010; 22:181 - 5; http://dx.doi.org/10.1016/j.ceb.2009.12.001; PMID: 20044243
- Ryan KM. p53 and autophagy in cancer: guardian of the genome meets guardian of the proteome. Eur J Cancer 2011; 47:44 - 50; http://dx.doi.org/10.1016/j.ejca.2010.10.020; PMID: 21112207
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998; 282:1497 - 501; http://dx.doi.org/10.1126/science.282.5393.1497; PMID: 9822382
- Wu YT, Tan HL, Huang Q, Kim YS, Pan N, Ong WY, et al. Autophagy plays a protective role during zVAD-induced necrotic cell death. Autophagy 2008; 4:457 - 66; PMID: 18253089