Abstract
Oxygen (O2), while essential for aerobic life, can also cause metabolic toxicity through the excess generation of reactive oxygen species (ROS). Pathological changes in ROS production can originate through the partial reduction of O2 during mitochondrial electron transport, as well as from enzymatic sources. This phenomenon, termed the oxygen paradox, has been implicated in aging and disease, and is especially evident in critical care medicine. Whereas high O2 concentrations are utilized as a life-sustaining therapeutic for respiratory insufficiency, they in turn can cause acute lung injury. Alveolar epithelial cells represent a primary target of hyperoxia-induced lung injury. Recent studies have indicated that epithelial cells exposed to high O2 concentrations die by apoptosis, or necrosis, and can also exhibit mixed-phenotypes of cell death (aponecrosis). Autophagy, a cellular homeostatic process responsible for the lysosomal turnover of organelles and proteins, has been implicated as a general response to oxidative stress in cells and tissues. This evolutionarily conserved process is finely regulated by a complex interplay of protein factors. During autophagy, senescent organelles and cellular proteins are sequestered in autophagic vacuoles (autophagosomes) and subsequently targeted to the lysosome, where they are degraded by lysosomal hydrolases, and the breakdown products released for reutilization in anabolic pathways. Autophagy has been implicated as a cell survival mechanism during nutrient-deficiency states, and more generally, as a determinant of cell fate. However, the mechanisms by which autophagy and/or autophagic proteins potentially interact with and/or regulate cell death pathways during high oxygen stress, remain only partially understood.
In our recent studies we have observed that exposure to hyperoxia (> 95% O2, 72 h), a model of acute lung injury in mice, activates morphological and biochemical markers of autophagy. Hyperoxia induces the time-dependent expression of the autophagic protein microtubule-associated protein 1 light chain 3B (LC3B) in mouse lung. Furthermore, hyperoxia promotes the enzymatic conversion of LC3B from its cytosolic form LC3B-I to its phosphatidylethanolamine-conjugated (active) form LC3B-II in lung tissue. On the basis of these observations, we hypothesized that autophagic proteins may determine cell fate in the context of hyperoxia. We therefore investigated the mechanisms by which LC3B can influence epithelial cell apoptosis during hyperoxia.
Using cultured epithelial (Beas-2B) and primary human bronchial epithelial (HBE) cells, we found that hyperoxia induces the time-dependent expression of LC3B, and conversion of LC3B-I to LC3B-II. These observed changes in hyperoxia-mediated gene expression are accompanied by morphological and biochemical indices of autophagosome formation in Beas-2B cells as determined by electron microscopy, and the formation of green fluorescent protein (GFP)-LC3 puncta in transfected cells. The increase in total LC3B was regulated at a transcriptional level, corresponding to increased LC3B mRNA synthesis, after hyperoxia.
We found that the transcriptional regulator c-Jun-NH2-terminal kinase (JNK), a mitogen-activated protein kinase commonly associated with the regulation of apoptosis, is essential for the oxygen-dependent regulation of LC3B. These experiments illustrate that autophagy and apoptosis share common elements of their respective regulatory pathways. It remains to be determined whether JNK activation represents a regulatory checkpoint for progression to either or both pathways (). Furthermore, the activation of LC3B by hyperoxia is clearly ROS-dependent, since it can be inhibited by general and mitochondria-targeted antioxidants.
Figure 1. Schematic of the proposed pathways. Hyperoxia increases cellular reactive oxygen species (ROS) production which promotes the expression and activation of LC3B, through a pathway dependent on c-Jun-NH2-terminal kinase (JNK). We describe dynamic interactions between LC3B and the apoptotic regulator Fas. The interactions of LC3B and Fas depend on phospho-caveolin-1. LC3B provides transient cytoprotection against hyperoxia by sequestering Fas. This association precludes formation of the death-inducing signaling complex (DISC), which consists of Fas, Fas-associated death domain protein (FADD) and caspase-8. The balance between LC3B-cav-1-Fas complex formation determines cell fate.
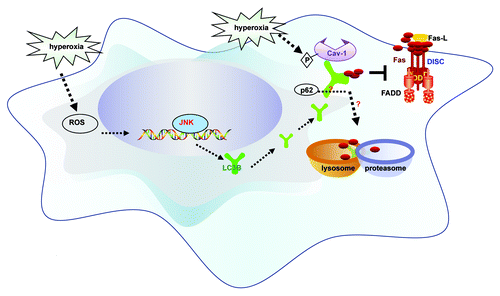
We then hypothesized that LC3B could play a functional role in hyperoxia-induced epithelial cell apoptosis. Genetic interference of LC3B promotes DISC formation and hyperoxia-induced cell death in epithelial cells. Conversely, overexpression of LC3B confers cytoprotection after hyperoxia, by inhibiting DISC formation, the cleavage of caspase-3 and initiation of apoptosis. Based on these experiments we concluded that LC3B acts as a critical prosurvival factor in oxygen-dependent cytotoxicity.
The Fas-dependent apoptotic pathway plays a major signaling role in hyperoxia-induced epithelial cell apoptosis. Fas associates with Fas-associated death domain protein and caspase-8, to form the death-inducing signaling complex (DISC). The activation of caspase-8 by DISC subsequently initiates apoptosis. In our recent study, we demonstrated that hyperoxia-induced LC3B physically regulates the Fas-dependent apoptotic pathway, further providing evidence of a regulatory overlap between apoptosis and autophagy. We show that LC3B cross-regulates the Fas apoptotic pathway during oxygen-dependent cell death by physically interacting with Fas-DISC. Short-term hyperoxia promotes this interaction, whereas the complex dissociates with prolonged hyperoxia. The interaction of LC3B and Fas proteins requires that the caveolae scaffolding protein caveolin-1 (cav-1) participates as an intermediate. Caveolin-1 maintains the integrity of plasma membrane caveolae, and modulates the activity of a number of membrane associated signaling proteins (i.e., endothelial nitric oxide synthase) through direct interaction. Substitution mutagenesis revealed that the interaction of LC3B and Fas is mediated by caveolin-1 (cav-1) tyrosine 14, a critical site for regulatory phosphorylation. Taken together, hyperoxia-induced LC3B activation inhibits the Fas apoptotic pathway through complex formation and thus confers cytoprotection in lung epithelial cells. The precise role of the caveolin-1 phosphorylation state in regulating association and dissociation of Fas-LC3B complexes remains incompletely elucidated.
Based on our observations with LC3B, we speculate that other autophagic proteins may also have dynamic interactions with the Fas-dependent pathway. Specifically, the selective autophagic substrate p62/SQSTM1 interacts with LC3B, and this interaction facilitates the selective autophagic degradation of ubiquitinated proteins. Thus, the interaction of p62/SQSTM1 and LC3B may provide further complexity in the mechanism by which LC3B regulates Fas. These potential associations warrant further investigation.
We conclude that LC3B exerts a protective function during oxygen toxicity by interfering with DISC processing. These studies lend further support to the concept that autophagic proteins, such as LC3B, participate in signaling networks that maintain a dynamic equilibrium, which determines the fate of cells under stress. Thus, autophagic proteins may be selected as potential therapeutic targets to modulate outcome in diseases where cell death is implicated in the pathogenic process (i.e., acute lung injury). It should be noted that the functional roles of autophagic proteins in cell death appear to vary with model system, duration and strength of toxin exposure, and also with cell/tissue type. Thus, a further understanding of these processes must be obtained before targeting the autophagic pathway in human disease.
| Abbreviations: | ||
| cav-1 | = | caveolin-1 |
| DISC | = | death-inducing signaling complex |
| GFP | = | green fluorescent protein |
| JNK | = | Jun-NH2-terminal kinase |
| LC3B | = | microtubule-associated protein 1 light chain 3B |
| p62/SQSTM1 | = | 62 kiloDalton protein, selective autophagic substrate |
| ROS | = | reactive oxygen species |