Abstract
Tumorigenesis and the efficacy of cancer therapeutics are both defined by the balance between autophagy and apoptosis. High-mobility group box 1 (HMGB1) is a DNA chaperone and extracellular damage-associated molecular pattern molecule (DAMP) with pro-autophagic activity. TP53/p53 plays a transcription-dependent and -independent role in the regulation of apoptosis, autophagy, metabolism, cell cycle progression, and many other processes. Both HMGB1 and TP53 are tightly linked with the development of cancer, associated with many of the hallmarks defining the altered biology of cancer. We have demonstrated that TP53-HMGB1 complexes regulate the balance between apoptosis and autophagy through regulation of the cytosolic localization of the reciprocal binding partner, whereby increased cytosolic HMGB1 enhances autophagy and increased cytosolic TP53 enhances apoptosis in colon cancer cells. We found that HMGB1-mediated autophagy promotes cell survival in TP53-dependent processes, and that TP53 inhibits autophagy through negative regulation of HMGB1-BECN1 complexes. Nuclear localization of TP53 and HMGB1 in tumors from patients with colon adenocarcinoma had a positive trend with survival time from diagnosis. Thus, HMGB1 and TP53 are critical in the crossregulation of apoptosis and autophagy and central to colon cancer biology.
Keywords: :
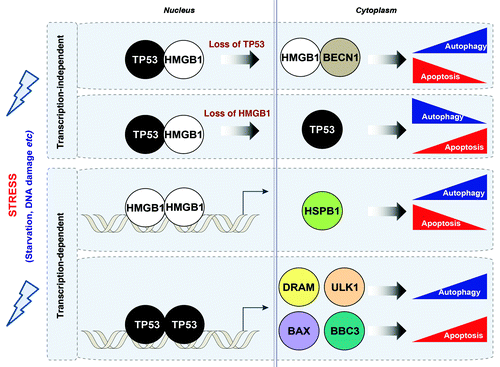
The balance between “programmed cell survival” (autophagy) and “programmed cell death” (apoptosis) is essential in respectively enhancing or suppressing tumorigenesis and regulating the response to therapy. Although various cell signals have been identified as crossregulators between these pathways, a more detailed understanding of the mechanisms by which the crosstalk between autophagy and apoptosis occurs is critical to our understanding of cancer biology and development of new therapeutic agents. HMGB1 and TP53 are both implicated in the regulation of autophagy and apoptosis. HMGB1 plays a nuclear role in promoting chromatin access to transcriptional complexes and extracellularly as a damage-associated molecular pattern protein important in inflammation, cell differentiation, cell migration and tumor progression. Endogenous HMGB1 enhances autophagy through transcription-dependent and independent mechanisms. Cytosolic HMGB1 enhances autophagy through sustaining BECN1- PtdIns3KC3 complex activation. Exogenous HMGB1 promotes autophagy via binding to the receptor for advanced glycation end products (RAGE). TP53, the most commonly mutated gene in human cancers, also promotes apoptosis through both transcription-dependent and -independent mechanisms. Interestingly, cytosolic TP53 inhibits autophagy through a transcription-independent mechanism and nuclear TP53 stimulates autophagy through a transcription-dependent mechanism. HMGB1 and TP53 form complexes that regulate the cytoplasmic localization of the reciprocal binding protein and subsequent levels of autophagy ().
Figure 1. Relationship between HMGB1 and TP53 in the crossregulation of apoptosis and autophagy. HMGB1 and TP53 play transcription-dependent and -independent roles in the regulation of autophagy and apoptosis. Stress signals including serum starvation and DNA damage promote interactions between TP53 and HMGB1 in the nucleus and cytoplasm. Loss of TP53 increases cytosolic HMGB1 leading to increased binding to BECN1, thereby promoting autophagy, and decreasing apoptosis. In contrast, loss of HMGB1 increases cytosolic TP53 and apoptosis, and decreases autophagy. In the nucleus, HMGB1 regulates expression of heat shock protein β-1 (HSPB1/HSP27). HSPB1 inhibits apoptosis through an ability to interact with key components of the apoptotic signaling pathway, as we have recently shown. HSPB1 and its phosphorylation promotes autophagy through regulation of the actin cytoskeletal response, which is required to promote the processes of autophagy and mitophagy. DRAM and ULK1 have recently been identified as TP53 transcriptionally-regulated genes through which TP53 promotes autophagy in contrast to its cytosolic role. In addition, BAX and the TP53 upregulated modulator of apoptosis (BBC3/PUMA) are TP53-targeted genes that induce the mitochondrial apoptotic pathway.
HMGB1 Binds TP53 within the Nucleus and Cytosol
We used biolayer interferometry to determine the dissociation constant for the binding of HMGB1 and TP53. The KD for HMGB1 and TP53 interactions is 1.15 × 10−9 ± 0.03 M and 1.83 × 10−9 ± 0.44 M when HMGB1 and TP53 are coupled to the amine reactive biosensors, respectively. We found that oxidation has minimal effect on the binding affinity, whereas reduction of TP53 with tris 2-carboxyethyl phosphine abrogates interactions with HMGB1, consistent with a role for the disulfide bond within HMGB1. When the A box and B box of HMGB1 are coupled to the biosensors, the respective KD’s are 6.38 × 10−9 M and 14.5 × 10−9, respectively. These findings were validated using known targets and nonspecific targets for HMGB1 and TP53, and biotinylated TP53 and HMGB1 coupled with streptavidin biosensors. Using a global fit over a dilution series, biotinylated HMGB1 and biotinylated TP53 demonstrate KD’s of 9.47 × 10−9 ± 1.47 M and KD of 7.35 × 10−8 ± 8.10 M for TP53 and, respectively.
To validate these findings in vitro, we serum-starved HCT116 cells to enhance autophagy, then performed immunprecipitation of whole cell, nuclear and cytosolic lystates with HMGB1 antibody with sequential western blot for TP53. We demonstrated increased HMGB1-TP53 complex formation in whole cell lysates following starvation. Complex formation was also identified in the nuclear and cytosolic extracts, specifically in the nucleus following starvation. Confocal microscopy also demonstrated increased HMGB1-TP53 colocalization in the nucleus and cytosol following starvation.
TP53-HMGB1 Complexes Regulate Cytosolic Translocation of the Reciprocal Protein and Levels of Autophagy
To determine the role of HMGB1-TP53 complexes in regulating autophagy we serum-starved HCT116 TP53 knockout (TP53−/−) and HMGB1 knockdown (HMGB1 KD) cells to enhance autophagy and performed western blot analysis of SQSTM1/p62 and microtubule-associated protein 1 light chain 3 (LC3) and quantified LC3 puncta by confocal microscopy. LC3 is cleaved (LC3-I) and then conjugated to phosphatidylethanolamine (LC3-II) when autophagy is induced. SQSTM1, a scaffolding protein that binds to and delivers ubiquitinated proteins to the autophagosome, is degraded during lysosomal fusion. TP53−/− cells have increased autophagy, as demonstrated by increased levels of LC3-II, accumulation of LC3 puncta, and decreased levels of SQSTM1 under basal conditions relative to TP53+/+ cells, with a further increase in autophagy in response to starvation. TP53−/− cells and cancer cell lines with endogenous mutant TP53 (DLD-1 and HT-29) have increased cytosolic HMGB1. Knockdown or pharmacological inhibition of HMGB1 with ethyl pyruvate in TP53−/− cells attenuates TP53 knockout-induced autophagy. We found that cytosolic HMGB1 promotes autophagy through increased complex formation with BECN1.
Conversely, starvation of HCT116 HMGB1 KD cells decreases levels of autophagy as demonstrated by decreased LC3-II expression and LC3 puncta and increased SQSTM1 expression. HMGB1−/− cells have increased levels of cytosolic TP53 and decreased levels of TP53 in the nucleus relative to HMGB1+/+ cells, with a further accentuation in these differences following starvation. We found that TP53 is not required for HMGB1-sustained autophagy as KD of TP53 in HMGB1−/− cells does not restore LC3 puncta formation. In response to DNA damaging agents, nuclear TP53 promotes autophagy through upregulation of damage-regulated autophagy modulator (DRAM) and Unc-51-like kinase 1 (ULK1; Atg1 in yeast). We found that knockout of TP53, but not knockdown of HMGB1, impairs upregulation of DRAM and ULK1 in response to DNA damaging agents including adriamycin and etoposide, suggesting that TP53-mediated expression of DRAM and ULK1 during DNA damage is HMGB1-independent. These findings demonstrate that HMGB-TP53 nuclear complexes limit the level of cytosolic TP53. In the absence of HMGB1, there is enhanced TP53 translocation to the cytosol which limits levels of autophagy.
During TP53-Dependent Apoptosis HMGB1-Mediated Autophagy Promotes Cell Survival
Autophagy has been identified as a double-edged sword, which both promotes and inhibits cell death during TP53-dependent apoptosis. To determine the role of HMGB1 in TP53-dependent apoptosis we knocked down HMGB1 in HCT116 cells, which restores and increases sensitivity of TP53−/− and TP53+/+ cells, respectively, to adriamycin- and etoposide-induced apoptosis as evaluated by flow cytometry and a clonogenic survival assay. We demonstrated that this increased sensitivity to these DNA damaging agents is mediated by decreased levels of autophagy as evaluated by LC3 puncta with HMGB1 knockdown. Autophagy inhibitors 3-methyladenine and wortmannin also increase adriamycin- and etoposide-induced apoptosis in TP53−/− cells. Knockdown of HMGB1 in TP53−/− cells also promotes BAX translocation, downstream in the TP53 apoptosis pathway, thereby promoting cytochrome c release and CASP9 activation. Thus, HMGB1-mediated autophagy promotes cell survival during TP53-dependent apoptosis.
Subcellular Localization of TP53 and HMGB1 is Important in Human Colon Cancer
To determine the clinical significance of complex formation and subcellular localization of HMGB1 and TP53, we analyzed a tissue microarray from patients with normal colons, normal tissues adjacent to tumor, adenomas and adenocarcinomas of the colon using two independent scientists who were blinded to the histological diagnosis and grade as well as applying imaging cytometric analysis. Tumors from patients with adenocarcinoma had significantly greater overall total HMGB1 (n = 8, paired t-test, p = 0.00031) and nuclear HMGB1 (n = 8, paired t-test, p = 0.023) than normal colon. Linear regression was used to determine whether HMGB1 or TP53 expression was associated with survival. There was a statistically significant association with TP53 expression and time of survival following the first recurrence (p < 0.00761) by automated and manual scoring. Nuclear TP53 expression (p = 0.059, manual scoring) and nuclear HMGB1 expression (p = 0.068, automated scoring) demonstrate positive trends with survival time from diagnosis that are not statistically significant. Thus, the subcellular localization of HMGB1 and TP53 are likely related to their roles in regulating survival and further studies are necessary to characterize this relationship.
Acknowledgments
This project was funded by a grant from the NIH 1 P01 CA 101944-04 (M.T.L.) and funding provided by the Department of Surgery, University of Pittsburgh (D.T. and M.T.L.).