Abstract
While anti-angiogenic therapy was initially greeted enthusiastically by the cancer community, initial successes with this therapeutic modality were tempered by the failure of angiogenesis inhibitors to produce sustained clinical responses in most patients, with resistance to the inhibitors frequently developing. We recently reported that hypoxia increases after the devascularization caused by anti-angiogenic therapy, consistent with the goals of these therapies, but that some tumor cells become resistant and survive the hypoxic insult elicited by anti-angiogenic therapy through autophagy by activating both AMPK and HIF1A pathways. These findings suggest that modulating the autophagy pathway may someday allow anti-angiogenic therapy to fulfill its therapeutic potential. However, further work will clearly be needed to develop more potent and specific autophagy inhibitors and to better understand the regulators of autophagy in malignant cells.
Keywords: :
The hypothesis that tumor progression can be curbed by anti-angiogenic agents targeting abnormal tumor blood vessels has been confirmed by preclinical evidence and evidence from clinical trials over the past three decades. However, these initial successes were tempered by the failure of angiogenesis inhibitors to produce enduring clinical responses in most patients. For example, in clinical trials of the vascular endothelial growth factor (VEGF) neutralizing antibody bevacizumab in glioblastoma treatment, there were 40–60% rates of radiographic progression after initially successful treatment, consistent with the development of acquired resistance to anti-angiogenic therapy, a state that we have found exhibits a poor prognosis and poor response to available treatments. The molecular basis of the adaptive responses of tumors to anti-angiogenic treatments causing the lack of sustained responses seen to date remains undefined. We hypothesized that the regression of blood vessels caused by anti-angiogenic therapy increases tumor hypoxia, and that this hypoxia mediates the adaptive response to anti-angiogenic therapy.
Cellular stressors have long been known to activate autophagy, a pathway in which double-membrane vesicles form and engulf protein aggregates, cytoplasm and organelles that are then delivered to lysosomes for degradation. Several cancer therapies, including DNA-damaging chemotherapeutics such as temozolomide, and radiation induce autophagy in culture and animal models, and the autophagic response to many of these treatments is cytoprotective. Radiation therapy promotes autophagy by upregulating transcription of several autophagy essential genes such as BECN1, ATG3, ATG4, ATG5 and ATG12, with a survival-promoting effect confirmed by autophagy inhibition. Other studies have shown that some chemotherapy agents like histone deacetylase (HDAC) inhibitors and cisplatin induce autophagy by increasing production of reactive oxygen species (ROS) in mitochondria.
These observations reflecting autophagy as an adaptive response to radiation therapy and conventional DNA damaging chemotherapy have been augmented by our recent finding that autophagy is an adaptive response to anti-angiogenic treatment. We found that the devascularization caused by anti-angiogenic therapy increases tumor hypoxia, consistent with the goals of these therapies, but that some tumor cells survive the hypoxic insult through autophagy by activating both AMPK and HIF1A pathways ().
Figure 1. Simplified scheme of nonselective vs. selective autophagy, and how they might be affected in cancer cells by oncogenic pathways and stressors in the microenvironment such as the hypoxia triggered by anti-angiogenic therapy. Shown are regulators of nonselective vs. selective autophagy in tumor cells. Hypoxia, as caused by anti-angiogenic therapy, influences both nonselective and selective autophagy, with mechanisms more clearly identified for the former. Abbreviations used: ROS, reactive oxygen species; HIF1A, hypoxia-inducible factor-1α; AMPK, AMP-activated protein kinase; and EGLN1/PHD2, prolyl hydroxylase domain-containing protein 2.
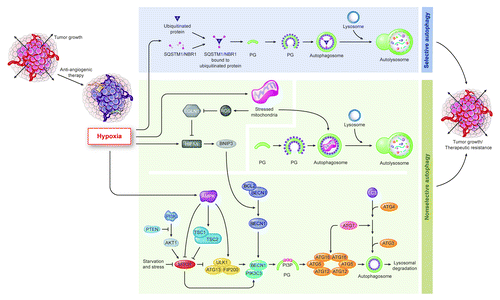
Our finding of hypoxia-induced autophagy in tumor cells as an adaptive response to the hypoxia caused by anti-angiogenic therapy can be expanded to determine the effect of hypoxia on cells in the tumor microenvironment. For example, we have found hypoxia does not induce autophagy in endothelial cells isolated from glioblastoma multiforme (GBM; unpublished data), consistent with our finding that the vessel density in GBMs resistant to anti-angiogenic therapy was suppressed and suggesting that tumors grow during anti-angiogenic therapy without increased endothelial survival. Furthermore, because hypoxia increases the cancer stem cell (CSC) population, one could hypothesize that hypoxia promotes autophagy in CSCs. Confirming this hypothesis would provide additional rationale for autophagy inhibition to prevent resistance to anti-angiogenic treatment.
The adaptive response of tumors to anti-angiogenic therapy may involve increased tumor cell invasiveness. Further studies will be needed to determine whether cells surviving anti-angiogenic therapy through autophagy exhibit increased invasiveness, as occurs in cells treated with a chemical that induces autophagy. Demonstration that tumor cells surviving anti-angiogenic therapy through autophagy exhibit increased invasiveness would suggest that autophagy inhibition could inhibit the invasion occurring after anti-angiogenic therapy by disrupting it at an earlier stage, which may be more effective than targeting invasion directly, as the numerous mediators of invasion make invasion difficult to pharmacologically disrupt.
Based on the preclinical evidence above, autophagy inhibition is currently being investigated as a way of modulating the response to cancer therapies in patients. Currently, the only FDA-approved agents able to inhibit autophagy are chloroquine, an antimalaria drug, and its derivative hydroxychloroquine, which block autophagy by disrupting lysosome/autolysosome acidification. One notable completed study was a randomized trial combining chloroquine with conventional treatment for glioblastoma with a benefit not quite significant. In addition, there are currently 22 phase I/II cancer clinical trials involving chloroquine or hydroxychloroquine open nationwide (www.clinicaltrials.gov), including two combining hydroxychloroquine with bevacizumab and conventional DNA-damaging chemotherapy, results of which could support the preclinical data we obtained showing a role for autophagy in resistance to anti-angiogenic therapy.
Despite these ongoing clinical efforts, the use of autophagy inhibition as a therapeutic strategy in cancer may need further preclinical evaluation to optimize the chances of success. Challenges in using autophagy inhibition as a therapeutic strategy include: (1) recognizing the dual roles for autophagy in tumors—cytoprotective or cytocidal depending on whether the tumor is in early or late stages of oncogenesis and the type of tumor; and (2) recognizing functional autophagy status in tumors, as some tumors may possess autophagy pathway defects, while others will have preserved autophagy capacity. Furthermore, based on the hypothesis that tumor cells exhibit minimal basal survival-promoting autophagy, and that autophagy may be most significant as an adaptive response to anticancer therapies, autophagy inhibition will likely be of minimal utility as a monotherapy. Therefore, the clinical trials of chloroquine and hydroxychloroquine to date have all combined these agents with treatments, which induce autophagy as an adaptive responsive.
Additional preclinical work will also be needed to develop autophagy inhibitors other than chloroquine or hydroxychloroquine. While preclinical studies like ours have suggested that these agents disrupt autophagy in animal models, other studies have shown that the ability of chloroquine to potentiate the effects of autophagy-inducing chemotherapies may occur independent of autophagy disruption. Furthermore, it has yet to be proven that chloroquine or hydroxychloroquine effectively block autophagy in human tumors, or how the genetic makeup of these tumors influences their susceptibility to these agents. Should chloroquine or hydroxychloroquine ultimately prove to be too nonspecific for clinical use as autophagy inhibitors, the development of more specific autophagy inhibitors will require focusing on kinases like ULK1/ATG1 and PIK3C3/VPS34, or proteases like ATG4 that specifically regulate the activation of autophagy and autophagosome formation, with minimal intracellular roles outside of autophagy.
Acknowledgments
Work was supported by funding to M.K.A. from the American Brain Tumor Association, the American Cancer Society, the James S. McDonnell Foundation, the NIH (5K02NS64167-2) and the UCSF Brain Tumor SPORE. A.J. is a Howard Hughes Medical Institute Research Fellow.