Abstract
Pancreatic ductal adenocarcinoma (PDA), the fourth leading cause of cancer death in the United States, is a complex disease that arises in the setting of genetic alterations (KRAS, BRCA1, SMAD4, CDKN2A/p16INK4a and TP53), epigenetic perturbations (MIR155, acetylation and methylation) and epicellular events (diabetes and inflammation). We have demonstrated that the advanced glycation end product-specific receptor (AGER, also called RAGE) contributes to pancreatic tumorigenesis. Targeted ablation of AGER diminishes the amount of autophagic flux and attenuates the development of early pancreatic intraepithelial neoplasia (PanIN) lesions in a murine model of KRAS-drivien carcinogenesis. Autophagy (programmed cell survival), a metabolic process of lysosome-mediated self-digestion, promotes pancreatic cancer growth. In pancreatic tumor cell lines, AGER-mediated autophagy promotes interleukin-6 (IL6)-induced phosphorylation of signal transducer and activator of transcription 3 (pSTAT3) and mitochondrial localization of pSTAT3. Enhanced mitochondrial pSTAT3 increases the pool of available ATP and increases cellular proliferation. Moreover, we observed a positive feedback loop between activation of autophagy and the IL6-pSTAT3 pathway, perhaps different from the role of cytosolic nonphosphorylated STAT3, which has been reported to inhibit autophagy. These AGER-dependent changes were found during the earliest stages of pancreatic cancer development. These observations of inflammation and altered metabolism in PDA provide a pathological link to early precursor lesion development. Thus, AGER is an important inflammatory mediator that modulates crosstalk between prosurvival pathways, IL6-pSTAT3 and autophagy, in PDA tumor cells, and contributes to early PanIN formation.
Upregulation of AGER during Pancreatic Cancer Development
AGER is a transmembrane receptor of the immunoglobulin gene superfamily encoded within the class III region of the major histocompatibility locus. It was first characterized in bovine lung in 1992 and for the past two decades in the setting of diabetes, sepsis, inflammation, cancer and Alzheimer disease. The interaction between AGER and its ligands, including high mobility group box 1 (HMGB1), promotes pro-inflammatory signal pathway activation. Our previous studies found that AGER is expressed in most mouse and human pancreatic cancer cell lines. Overexpression of AGER inhibits cell death and increases drug resistance following exposure to chemotherapeutic agents in pancreatic cancer cells. Interestingly, AGER is overexpressed in human PDA specimens but not in the adjacent normal ducts. Furthermore, we observed increasing AGER expression concurrent with progression of pancreatic PanIN lesions in Pdx1-Cre:KrasG12D/+ (KC mice), a spontaneous transgenic mouse model of pancreatic cancer. These findings suggest that AGER might play a role in pancreatic cancer development.
AGER Ablation Attenuates Development of Pancreatic Cancer Precursors (PanIN) in the KC Spontaneous Mouse Model of Pancreatic Cancer
Ager null mice have been utilized in biochemical and physiological experiments to address the roles of AGER in progression of various pathological conditions including sepsis and diabetes. In this study, we generated a novel transgenic murine strain (Ager−/−:Pdx1-Cre:KrasG12D/+, KCR mice), backcrossing Ager null mice to the KC strain. Knockout of Ager delayed mutant Kras-driven PanIN lesion progression. Moreover, knockout of Ager prolongs murine survival, and decreases the incidence of nonpancreatic tumors of the head, neck and vulva, suggesting that AGER is important for mutant Kras-driven carcinogenesis. Both apoptosis (“programmed cell death”) and autophagy (“programmed cell survival”) are important regulators of pancreatic tumorigenesis. Consistent with our previous observations that AGER promotes autophagy and decreases apoptosis in murine and human pancreatic tumor cell lines; we found that targeted ablation of AGER in KC mice leads to increased apoptosis and decreased autophagy/proliferation in the emerging pancreatic tumor microenvironment, supporting our notion that AGER plays an important role in regulation of tumor cell death and survival.
AGER Promotes Secretion of IL6 and pSTAT3 Targeting to Mitochondria
Cytokines play an important role in cancer pathogenesis and have potential as tumor markers, therapeutic targets, or as treatments themselves. The production and release of various epithelial survival factors such as the pleiotropic cytokine IL6, a major mediator of inflammation and activator of STAT3, serves to block apoptosis in the tumor microenvironment. Recent studies have shown that oncogenic RAS-induced secretion of IL6 is required for tumorigenesis. Serum IL6 is elevated in patients with pancreatic cancer. We observed decreased levels of serum IL6 in KCH mice when compared with KC mice. Consistent with this, we observed that knockdown of AGER in pancreatic tumor cells lines not only decreases IL6 autocrine/paracrine secretion, but also inhibits IL6-induced cell proliferation. pSTAT3 mediates IL6-dependent enhancement of cell survival and proliferation. Recent studies indicate that mitochondrial pSTAT3 regulates cellular respiration and supports RAS-dependent oncogenic transformation. Phosphorylated Ser727 is important for the mitochondrial localization and function of STAT3. We demonstrated that targeted ablation of AGER in vivo and in vitro significantly inhibits IL6-induced pSTAT3 at Ser727 as well as limiting the subsequent mitochondrial localization of pSTAT3 ().
Figure 1. AGER, pSTAT3 and autophagy sustain bioenergetics during pancreatic cancer growth. Mutant KRAS is a master regulator of pancreatic cancer initiation and progression. AGER is expressed concurrently with progression of KRAS-promoted progression of pancreatic PanIN lesions. Autophagy is a process of degradation in which double-membrane autophagosomes sequester cytoplasmic material and fuse with lysosomes, where they are degraded and recycled to feed the tricarboxylic acid (TCA) cycle and support ATP production in mitochondria. Overexpression of AGER promotes autophagy by increasing ATG12–ATG5 conjugation and BECN1-PIK3C3 complex formation. In addition, AGER inhibits IL6 autocrine/paracrine secretion and IL6-induced phosphorylation of STAT3 at Ser727. Phosphorylation of STAT3 at this residue is important for its mitochondrial localization and enhancement of oxidative phosphorylation. There is also a positive feedback loop operative between autophagy and activation of mitochondrial pSTAT3. Thus the AGER-IL6-pSTAT3 autophagic pathway enhances bioenergetics and promotes pancreatic tumor growth.
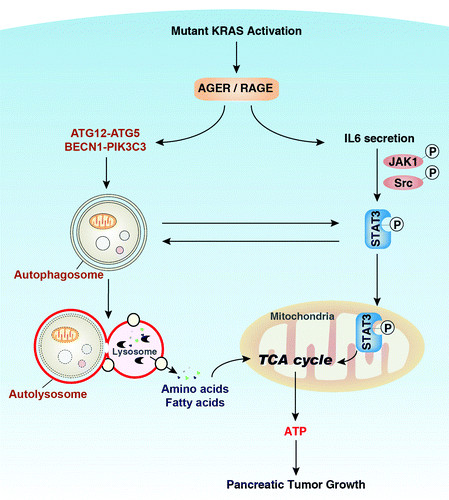
Autophagy is Required for Activation of the AGER-IL6-pSTAT3 Pathway
Autophagy is a metabolic stress response in which double-membrane autophagosomes sequester cytoplasmic material and fuse with lysosomes, where they are degraded and recycled. Autophagy promotes cell survival under a number of metabolic and inflammatory stress factors. The molecular mechanism of autophagy is highly conserved and is counter-regulated with apoptosis. Our previous studies have demonstrated that AGER promotes autophagy by regulating both MTOR and the BECN1-PIK3C3 complex, each of which are critical regulators expressed during autophagosome activation. Overexpression of AGER also increases IL6-induced ATG12–ATG5 conjugation, a critical step for the elongation of the initiating autophagosomal membrane. Other studies suggest that the autophagic process in cancer cells can contribute to their survival by STAT3 pathway activation and subsequent secretion of growth factors. We observed that IL6 and overexpression of STAT3 promotes autophagic flux in pancreatic tumor cells by enhancing LC3 turnover, suggesting that the IL6-pSTAT3 pathway is a positive regulator of autophagy in pancreatic cancer. Other studies have shown that IL6 inhibits autophagy in macrophages and that activated STAT3 is inversely correlated with BECN1 expression in tumors (malignant gliomas). Notably, knockout of Ager and knockdown of Atg5 diminish IL6-pSTAT3-mediated autophagy. These findings suggest that AGER is an important intermediary between autophagy and the IL6-pSTAT3 pathway that is operative during pancreatic cancer development ().
AGER Promotes ATP Production in Pancreatic Cancer Cells
Cancer cells use ATP as an energy source by increasing either oxidative phosphorylation, or, if the need is urgent, glycolysis. The degradation of cytosolic contents in the lysosome by autophagic processes supplies both amino acid and fatty acids for reutilization in anabolism and as substrates for metabolism to provide additional ATP. Since AGER-IL6-induced autophagic flux promotes mitochondrial localization of pSTAT3, we evaluated levels of ATP. Activation of the IL6-pSTAT3 pathway dose-dependently enhances mitochondrial respiratory chain complex I activity and ATP production in pancreatic tumor cell lines. Respiratory chain inhibitors (e.g., rotenone and 2-thenoyltrifluoroacetone) decrease IL6-induced ATP production and cell proliferation. Notably, knockdown of Ager/AGER by shRNA in murine and human pancreatic tumor cell lines significantly decreases IL6-STAT3-mediated mitochondrial respiratory chain complex I activity and ATP production. Consistent with these in vitro findings, KCR mice have significantly less absolute ATP amounts in the pancreas when compared with KC mice. These findings suggest that AGER expression has an impact upon autophagy-mediated bioenergetics by enhancing mitochondrial STAT3-mediated cellular respiration.
Conclusion
In this study we were able to link inflammation and bioenergetics, observations made in Ager knockdown tumor cell lines, to in vivo studies of early pancreatic carcinogenesis. We found that global knockout of Ager attenuates KRAS-driven development of PDA precursor lesions by decreasing signaling through the IL6-pSTAT3 autophagic pathway. Additional work is necessary to explore how AGER affects the transition from PanIN to PDA, and the differential role of nuclear or cytosolic STAT3/pSTAT3, and to elucidate in which cell types including inflammatory, stromal and endothelial cells within the tumor microenvironment AGER expression is most important.