Abstract
Mitochondrial division is mediated by the conserved dynamin-related GTPase DNM1L/DRP1. DNM1L assembles onto the surface of mitochondria and constricts this tubular organelle. Alterations in mitochondrial division are linked to many neurodegenerative diseases. However, the in vivo function of mitochondrial division is poorly understood. In our recent paper, we studied the physiological role of mitochondrial division in postmitotic neurons using the cre-loxP system. We found that the loss of DNM1L resulted in increased oxidative damage in mitochondria, impaired respiration and neurodegeneration in postmitotic neurons. Suggesting a decrease in mitochondrial turnover, mitophagy-related proteins such as LC3, SQSTM1/p62 and ubiqutin accumulated in division-defective mitochondria. These findings suggest that mitochondrial division functions as an important quality control mechanism that suppresses oxidative damage and neurodegeneration in vivo
Mitochondria continuously divide and fuse, and the balance between division and fusion determines organelle size, number and morphology. In many cell types, such as mouse embryonic fibroblasts (MEFs), the loss of mitochondrial division leads to interconnected, elongated mitochondria due to unopposed fusion, and the loss of mitochondrial fusion generates many small mitochondria due to ongoing division. Mitochondrial division is mediated by the dynamin-related GTPase DNM1L, which drives constriction by assembling into filaments around mitochondria through interactions with DNM1L receptor proteins. Studies on human diseases have suggested that neurons are highly dependent on mitochondrial division for their survival. For example, a mutation in DNM1L causes postneonatal death accompanied by neurodegeneration. In addition, alterations in mitochondrial division have been implicated in many aging-related neurodegenerative disorders such as Alzheimer, Huntington and Parkinson diseases. Therefore, it is important to understand the role of mitochondrial division in neurons and other cell types. However, it is largely unknown why mitochondria divide. In our recent paper, we investigated the physiological and cellular function of DNM1L in postmitotic neurons using animal and cell culture systems, with particular focus on cerebellar Purkinje cells since these neurons highly express DNM1L.
We deleted Dnm1l specifically in postmitotic Purkinje cells by crossing mice that carry a floxed allele of Dnm1l to a transgenic mouse line that expresses cre recombinase from the Purkinje cell-specific L7 promoter at 1 mo of age (L7-Drp1KO mice). Upon the loss of DNM1L, mitochondria, which are short tubules in the wild type, elongated and then became large spheres (). We found similar changes in mitochondrial morphology when Dnm1l was deleted in cultured Purkinje cells in vitro. This large, spherical morphology was in sharp contrast to the interconnected, long tubules of mitochondria observed in Dnm1l-null MEFs. The mitochondria in Dnm1l-null Purkinje cells accumulated oxidative damage as shown by immunofluorescence with anti-hydroxynonenal antibodies, which recognize peroxidation of proteins and lipids. This oxidative damage was responsible for the formation of large spherical mitochondria, since treating with antioxidants such as N-acetylcysteine, coenzyme Q10 and mito Q resulted in elongated mitochondria similar to those of Dnm1l-null MEFs. Supporting this notion, treating Dnm1l-null MEFs with hydrogen peroxide converted elongated mitochondrial tubules into large round structures.
Figure 1. Mitochondrial division is essential for the maintenance of mitochondrial function in Purkinje cells.
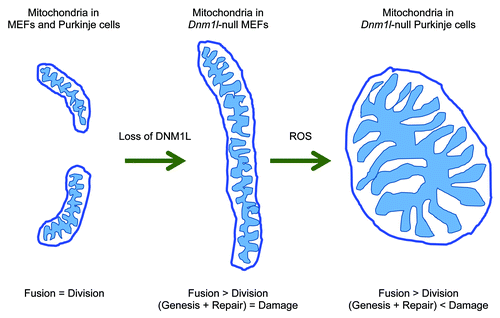
Dnm1l-null Purkinje neurons gradually decreased between the age of 3 mo and 6 mo in L7-Drp1KO mice. This neurodegeneration was correlated with decreased function of the cerebellum as measured by rotarod tests, which examine the ability of the mice to coordinate their body movements. As a likely cause of neurodegeneration, we found dramatic decreases in the activities of electron transport chain complexes (NADH dehydrogenase and cytochrome c oxidase) 2 mo prior to the onset of Dnm1l-null Purkinje cell death. Consistent with the in vivo results, loss of DNM1L also accelerated cell death in cultured Purkinje cells in vitro. Dnm1l-null Purkinje cells did not show TUNEL staining or the activation of CASP3, suggesting that Purkinje cells die via a nonapoptotic mechanism. Similar to the mitochondrial shape defects, the death of Dnm1l-null Purkinje cells was also rescued by antioxidant treatment both in vivo and in vitro. This suggests that oxidative damage builds up and leads to neurodegeneration when mitochondria fail to divide in Purkinje cells. The cell death induced by defects in mitochondrial division was specific to neurons. Unlike Dnm1l-null Purkinje cells, Dnm1l-null MEFs accumulated no oxidative damage, maintained normal respiration and proliferated without increased cell death.
What is the mechanism that increases oxidative damage in mitochondria in the absence of DNM1L? We speculate that oxidative damage may accumulate due to compromised turnover of mitochondria by autophagy. In the absence of mitochondrial turnover, oxidative damage would gradually accumulate and eventually make mitochondria dysfunctional in neurons. Previous studies have suggested that decreasing the size of mitochondria by division promote their efficient engulfment by autophagosomes during the process of mitochondria-selective autophagy, called mitophagy. We found an increase in the association of two autophagy components, LC3 and SQSTM1, with enlarged mitochondria in Dnm1l-null Purkinje cells, suggesting that engulfment of these organelles is impaired due to their increased size. Furthermore, it has been suggested that the E3 ubiquitin ligase PARK2/parkin, which is defective in a familial form of Parkinson disease, is recruited to dysfunctional mitochondria and ubiquitinates mitochondrial proteins to promote mitophagy. We found elevated levels of ubiquitinated proteins associated with mitochondria in an age-dependent manner in the Purkinje cells of L7-Drp1KO mice. However, surprisingly and interestingly, the ubiquitination of mitochondrial proteins was independent of PARK2, since L7-Drp1KO::parkinKO mice also had elevated levels of mitochondrial protein ubiquitination in Purkinje cells as compared with the wild type. It should be noted that once mitochondria become at least partially dysfunctional, their inefficiently coupled respiration would increase the production of reactive oxygen species (ROS) beyond the basal level, further recruiting autophagic components to mitochondria. Therefore, the accumulation of LC3, SQSTM1 and ubiquitinated proteins may reflect both incomplete mitophagy as well as its activation.
In summary, our recent study shows that mitochondrial division is essential for the suppression of oxidative damage and therefore the survival of postmitotic neurons in vivo and in vitro. Our findings also strongly support the emerging hypothesis that mitochondrial division maintains mitochondrial functions by promoting mitophagy in order to facilitate the remodel of oxidative damage. Postmitotic neurons contain high ROS levels and do not proliferate; therefore a mitochondrial quality control mechanism is essential. In contrast, actively proliferating cells such as MEFs do not require mitochondrial division and appear to respire normally with interconnected, elongated mitochondrial tubules when division is impaired. In MEFs, mitochondria are likely produced continuously during cell proliferation and therefore the effect of any oxidative damage may be diluted by the newly formed mitochondria. Also, basal ROS levels may be lower in MEFs than in neurons. However, more work is needed to further test this model. For example, the levels of oxidative stress in mitochondria and the rates of mitochondrial turnover in neurons and other cells in vivo are still unknown. Also, it would be important to determine which E3 ligases ubiquitinate mitochondrial proteins in the absence of mitochondrial division. Finally, we would like to identify the targets of oxidative damage; is there a small set of specific molecules that are affected, thereby causing changes in mitochondrial shape and function? Answering these questions would help us better understand the role of mitochondrial division and the human diseases associated with its alterations.
Acknowledgments
We thank Miho Iijima and Kristen F. Swaney for critically reading the manuscript. This work was supported by an NIH grant to H.S. (GM089853).