Abstract
Autophagic responses to chemotherapeutic agents may vary greatly among different prostate cancer cells and have not been well characterized. In this study, we showed that valproic acid (VPA) induced conversion of LC3-I to LC3-II and formation of LC3 puncta, the typical markers of autophagy, in LNCaP and PC-3 cells. However, these markers were undetectable in DU145 cells upon autophagic stimulation, indicating a defect of autophagy in this cell line. Among several critical autophagy-related proteins, ATG5 and ATG12–ATG5 conjugates, which are essential for autophagy induction, were absent in DU145 cells. No canonical transcripts for full-length ATG5 but only two alternatively spliced ATG5 transcripts were identified in DU145 cells. These alternative transcripts lack one or two exons, leading to premature termination of ATG5 translation. Transfection of the wild-type ATG5 gene into DU145 cells rescued the production of ATG5 and ATG12–ATG5 conjugates, resulting in formation of LC3-II conjugates and LC3 puncta. Moreover, the levels of the SQSTM1 protein, which should be degradable as an autophagy adaptor, were much higher in DU145 than in LNCaP and PC-3 cells, but were significantly decreased after ATG5 restoration in DU145 cells. However, expression of wild-type ATG5 in DU145 or knockdown of ATG5 in LNCaP and PC-3 cells did not change the inhibitory effects of VPA on these cells. Collectively, these results indicated that VPA-induced autophagy in prostate cancer cells depended on ATG5 and more importantly, that the autophagy pathway was genetically impaired in DU145 cells, suggesting caution in interpreting autophagic responses in this cell line.
Introduction
Prostate cancer remains a significant health threat to men (especially among elderly men) and there is no cure therapy yet for advanced (hormone-refractory) prostate cancer. Recently, histone deacetylase (HDAC) inhibitors have been extensively investigated for their anti-prostate cancer activities.Citation1 Valproic acid (VPA), an HDAC inhibitor that is currently used as an anticonvulsant and antidepressant, has attracted much attention due to its anticancer properties. Through specific inhibition of class I and class II HDACs, VPA arrests the cell cycle at the G1 phase, and inhibits tumor cell growth and replication.Citation2,Citation3 Consistent with the strong expression of HDAC1, 2 and 3 (all belong to class I HDACs) in prostate cancer cells,Citation4 administration of VPA has shown promising antiprostate cancer activities through inhibition of cancer cell growth,Citation5,Citation6 induction of cell differentiationCitation7 and prostate-specific antigen (PSA) response in clinical trials.Citation8 More recent studies have indicated that HDAC inhibitors may induce autophagy in prostate cancer cells.Citation9 Despite these mechanistic investigations for VPA and its analogs, different prostate cancer types may show differential characteristics of autophagic responses to VPA treatment,Citation6,Citation10 and the underlying mechanism has not been well understood.
Autophagy is a highly conserved, homeostatic process by which intracellular constituents are delivered to lysosomes for degradation. Depending on the mode of delivery, there are three different types of autophagy, including macroautophagy, chaperone-mediated autophagy and microautophagy. Macroautophagy, simply referred to hereafter as autophagy, has been extensively investigated. By autophagy, redundant long-lived proteins and damaged organelles are degraded in autolysosomes, providing energy for cells to survive nutrient deprivation and to repair DNA damage. This is a multistep process in which the formation of the autophagosome necessitates the sequential modification of microtubule-associated protein 1 light chain 3 (LC3). After synthesis, the C-terminal of LC3 is first cleaved by a cysteine protease ATG4, to produce LC3-I with a molecular weight (MW) of 18 kDa. The product is localized diffusely throughout the entire cytoplasm. Upon induction of autophagy, a fraction of LC3-I is transferred to phosphotidylethanolamine (PE) to produce a new molecule LC3-PE conjugate (also known as LC3-II) with an apparent MW of 16 kDa.Citation11 LC3-II is associated with autophagosome and thus the amount of LC3-II and the formation of LC3 puncta are thought to be a faithful marker of the autophagosome.Citation12
Recent studies indicate that autophagy has been involved in tumor resistance to radio- and chemotherapy.Citation13,Citation14 Targeting autophagy is thought to be a potential approach to overcome the therapeutic resistance of cancer.Citation15 Indeed, inhibition of autophagy potentiates the anticancer activities of HDAC inhibitors.Citation16-Citation18 Although VPA has been shown to induce autophagy in many kinds of tumor cells, which confers resistance to apoptosis,Citation19,Citation20 it is still unknown whether it induces autophagy in the same manner in prostate cancer cells of different types.
In this study, we investigated the characteristics of autophagic responses to VPA treatment in androgen-sensitive prostate cancer LNCaP cells as well as in androgen-refractory DU145 and PC-3 cells. The results showed that VPA induced autophagy in both LNCaP and PC-3 cells as evidenced by LC3-II accumulation and LC3B immunofluorescent puncta formation. However, VPA did not induce autophagy in DU145 cells due to the absence of full-length ATG5 protein. Transfection of a wild-type ATG5 gene restored autophagy and decreased SQSTM1 accumulation in DU145 cells. Our results suggested that the genetically impaired autophagy pathway in DU145 cells should be taken into account when selecting this cell line as an in vitro advanced prostate cancer model.
Results
VPA induced autophagy in LNCaP and PC-3 cells, but not in DU145 cells
VPA is known as a class I HDAC inhibitor that has been shown to induce autophagy in a variety of tumor cells.Citation20-Citation22 In line with its HDAC-inhibitory property, VPA elevated the levels of acetylated histone H3 in LNCaP, DU145 and PC-3 cells (data not shown). As conversion of LC3-I to LC3-II (LC3-PE) and formation of LC3 puncta have been generally used as indicators of autophagy, we employed them to determine whether VPA treatment induced autophagy in the prostate cancer cells. There are three isoforms of LC3 in mammalian cells, LC3A, LC3B and LC3C, but LC3B is more frequently adopted as autophagy marker than the other two isoforms. By using western blot analysis, we found that VPA induced a dose-dependent increase of LC3B-II levels in both LNCaP and PC-3 cells (). This was further confirmed by inhibition of the “autophagic flux” with lysosomotropic chloroquine (CQ), which raises the pH within the lumen of lysosomes and/or autolysosomes and therefore compromises autophagic degradation, leading to a further accumulation of LC3B-II (). In addition, VPA-induced accumulation of LC3B-II was also time-dependent (). Unlike LC3B, LC3A was undetectable in untreated LNCaP cells (control), but was upregulated by VPA treatment and a small fraction of LC3A-I was converted into LC3A-II. In contrast, in PC-3 cells, both LC3A-I and LC3A-II basal levels were much higher, indicating a high flux of LC3A-I to LC3A-II, and VPA further enhanced their expression levels ().
Figure 1. Induction of autophagy by VPA treatment in LNCaP and PC-3 cells, but not in DU145 cells. Autophagy was measured by LC3-II western blot analysis (A–D) and LC3B immunofluorescence microscopy (E and F). Cells were treated with indicated concentrations of VPA for 24 h (A), or 10 mM VPA for 24 h in the presence or absence of 25 μM chloroquine (CQ) (B), or 10 mM VPA for indicated time lengths in the presence or absence of 25 μM CQ (C and D). Total proteins were extracted by 2× SDS-PAGE loading buffer. LC3A and LC3B were probed by western blotting, respectively. TUBB was used as loading control. The relative densitometry values under each LC3 blot is the ratio of LC3-II (16 kDa) densitometry to that of TUBB. A dash (−) indicates that no LC3-II band was observed. Data are from one of three independent experiments with similar results. (E and F) Cells were treated with 10 mM VPA and/or 25 μM CQ (E) or 2 μg/ml rapamycin (Rapa) (F) and then immunostained with LC3B antibody followed by CF568-conjugated second antibody. Fluorescent images were obtained by fluorescence microscopy with a 100 × oil objective lens. LC3B (red) fluorescent puncta were only observed in LNCaP and PC-3 cells. Rapamycin treatment was included to confirm that autophagy was deficient in DU145 cells (B and F). Nuclei (blue) were revealed by Hochest33342 staining. Arrowheads indicate 17-kDa bands of LC3B. Scale bar: 10 μm.
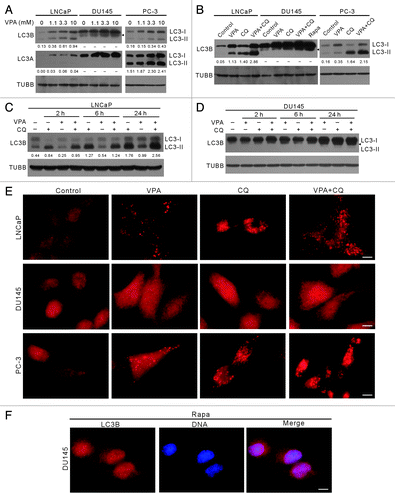
Surprisingly, in contrast to the situation in LNCaP and PC-3 cells, VPA did not induce autophagy in DU145 cells as evidenced by the absence of typical 16-kDa LC3A-II and LC3B-II immunoblot bands (). Moreover, no LC3-II was observed in DU145 cells after serum starvation (data not shown), CQ or even rapamycin treatment (), the latter being a mechanistic target of rapamycin (MTOR) inhibitor and a potent autophagy inducer.Citation23 Interestingly, a new band of LC3B with an MW of ~17 kDa was visible and was enhanced by VPA in DU145 cells in a dose-dependent manner (), suggesting a new modification form of LC3B that has not been reported before. Furthermore, VPA treatment could time-dependently elevate the amount of the 17-kDa LC3B band in DU145 cells, even though these bands were barely separated from the LC3B-I bands when the total amount of LC3B was upregulated (). Immunofluorescence microscopy analysis demonstrated that VPA induced the formation of LC3B puncta in both LNCaP and PC-3 cells (). However, such LC3B puncta were not observed in VPA-, CQ- or rapamycin-treated DU145 cells (). These results further confirmed that VPA induced autophagy in LNCaP and PC-3 cells, but not in DU145 cells. Altogether, these data indicated that the autophagy pathway was defective in DU145 cells.
ATG5 protein and ATG12–ATG5 conjugate were undetectable while SQSTM1 protein was highly expressed in DU145 cells
The post-translational modification of LC3-I during autophagy includes a series of catalytic steps. Several ATG gene products are involved in this process. Immediately after synthesis, LC3 is cleaved at its C-terminus to yield the cytosolic LC3-I form. LC3-I is then activated by E1-like ATG7, transferred to E2-like ATG3, and finally conjugated to PE. By such a ubiquitin-like modification, the 18-kDa LC3-I is converted into 16-kDa LC3-II (LC3–PE), allowing it to be associated with autophagosomes.Citation11 However, cup-shaped membrane isolation and targeting of LC3-II to phagophores needs another conjugation system, in which ATG12–ATG5 conjugates noncovalently associate with ATG16L1 to form a multimeric complex.Citation24 To investigate whether the expression of ATG genes was associated with the impaired LC3 processing in DU145 cells, we probed the expression of BECN1, ATG3, ATG5, ATG7, ATG12 and ATG16L1 in the three prostate cancer cell lines. Western blot analysis showed that all these proteins were expressed in LNCaP and PC-3 cells (). As expected, both free ATG12 and ATG12–ATG5 conjugates were detected in LNCaP and PC-3 cells. However, ATG5 was absent in DU145 cells, leading to a loss of ATG12–ATG5 conjugates either in the presence or absence of VPA (). Furthermore, ATG16L1 expression was very low in DU145 cells (), which could only be visualized after long exposure (see below). Our data suggested that the loss of ATG5 or low level of ATG16L1 contributed to the failure of LC3-I conversion into LC3-II and the absence of LC3 puncta formation in DU145 cells.
Figure 2. Differential expression profiles of autophagy-related proteins (ATG) in LNCaP, DU145 and PC-3 cells. (A) Cells were treated with indicated concentrations of VPA for 24 h and the expression profiles of ATG proteins were probed with indicated antibodies by western blotting. One experiment, representative of three independent experiments is shown. TUBB was used as a loading control. It is noteworthy that ATG5 proteins, as well as the ATG12–ATG5 conjugates, were absent while SQSTM1 levels were much higher in DU145 cells. (B) Quantitative analysis of SQSTM1 levels in LNCaP, DU145 and PC-3 cells. The relative levels of SQSTM1 were normalized to TUBB and the value of LNCaP control was set as 1.0. **p < 0.01 vs respective control; ##p < 0.01 vs the same dose of VPA-treated LNCaP; #p < 0.05 vs the same dose of VPA-treated PC-3. (C and D) Time-course analysis of SQSTM1 levels in LNCaP (C) and PC-3 (D) cells treated with 3.3 mM VPA and/or chloroquine (CQ) by western blotting. The relative levels of SQSTM1 were normalized to TUBB and the value of respective control (VPA–/CQ–) was set as 1.0. *p < 0.05; **p < 0.01 vs respective control.
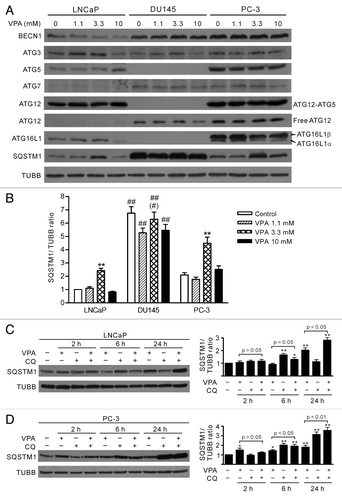
The level of SQSTM1 has also been used for monitoring autophagy flux, since it accumulates in cells when autophagy is blocked.Citation25,Citation26 As an autophagy adaptor, SQSTM1 binds directly to LC3-II and mediates the targeted degradation of ubiquitinated protein aggregates.Citation27,Citation28 We found that SQSTM1 protein was differentially expressed in the prostate cancer cell lines (). There was a much higher level of SQSTM1 in DU145 cells than in LNCaP and PC-3 cells. In LNCaP and PC-3 cells, SQSTM1 levels were increased after treatment with 3.3 mM VPA for 24 h but were still lower than that in DU145 cells (). Time-course analysis of SQSTM1 levels demonstrated that VPA treatment led to a slight reduction of SQSTM1 at 6 h but a significant increase at 24 h in LNCaP cells; while in PC-3 cells, VPA treatment increased SQSTM1 levels over 2 h to 24 h (). Upregulation of SQSTM1 by 3.3 mM VPA in these cells was likely due to enhanced protein synthesis, instead of blockade of lysosome-dependent protein degradation, since cotreatment of VPA and CQ resulted in even higher levels of SQSTM1 from 6 h to 24 h as compared with VPA treatment alone (). However, VPA treatment did not significantly change SQSTM1 levels in DU145 cells (). These results suggested that the accumulation of SQSTM1 protein may result from loss of autophagy in DU145 cells.
DU145 cells were lacking in canonical ATG5 transcript but had alternatively spliced variants
To assess whether ATG5 and ATG16L1 genes are not expressed or mutated in DU145 cells, we amplified the full-length coding sequences of these genes by reverse-transcription (RT)-PCR from total RNA extracts. The results showed that the ATG5 transcripts in DU145 cells were shorter than that in LNCaP and PC-3 cells, whereas the ATG16L1 transcripts were of the same length (~1800 bp, as expected) in the three cell lines ().
Figure 3. Analysis of transcripts of ATG5 and ATG16L1 genes in LNCaP and DU145 cells. Primers were designed to amplify ATG5 and ATG16L1 full-length encoding sequences by reverse transcription-polymerase chain reaction (RT-PCR). RT-PCR products were resolved by agarose gel electrophoresis (A). The RT-PCR products of ATG5 (B) and ATG16L1 (C) genes were cloned into the eukaryotic expression plasmid pEGFP-N1. Recombinant plasmids with expected inserts identified by restriction enzymes were analyzed by DNA sequencing. The results showed that LNCaP cells expressed a full-length ATG5 transcript (variant 1), while DU145 cells expressed two alternative spliced ATG5 variants (variant 2 lacking exon 6 and variant 3 lacking exon 2 plus exon 6) (B). However, LNCaP and DU145 cells were identified to express the same ATG16L1 transcript (variant 2 with an A898G mutation) (C). * indicates stop codon position.
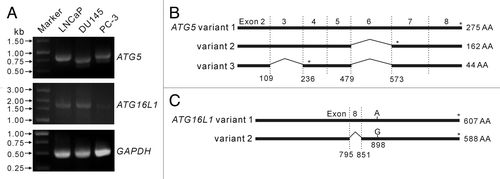
Next, the cDNA of ATG5 and ATG16L1 transcripts from LNCaP and DU145 cells were cloned into the eukaryotic expression plasmid pEGFP-N1 as non-fused genes. Sequencing analysis demonstrated that LNCaP cells expressed the canonical ATG5 transcript (referred to as ATG5 variant 1 in this study), whose sequence is identical to previously identified transcript encoding wild-type ATG5 protein (GenBank accession no. NM_004849). However, no canonical ATG5 transcript was found in DU145 cells. The ATG5 transcripts identified from DU145 cells lacked exon 6 (referred to as variant 2 in this study) or lacked both exon 2 and exon 6 (referred to as variant 3 in this study) (), which were likely to be generated by alternative splicing. Both variant 2 and variant 3 transcripts have a premature stop code, leading to truncated ATG5 products lacking the C-terminus sequence. The variant 2 transcript encodes a truncated ATG5 with 162 amino acid residues whereas the variant 3 transcript only encodes N-terminal 44 residues of ATG5 protein (). These sequence data have been submitted to the GenBank database under the accession numbers of JQ924061 (variant 1), JQ918353 (variant 2) and JQ918354 (variant 3). On the other hand, the ATG16L1 transcripts in LNCaP and DU145 cells were of the same sequence, which has been identified as ATG16L1 variant 2 (GenBank accession no. NM_017974). Remarkably, there was a point mutation (A841G, corresponding to site 898 of variant 1) in these ATG16L1 transcripts, resulting in a Thr→Ala mutation in their amino acid sequences (). This newly identified ATG16L1 sequence has also been submitted to the GenBank database under the accession no. JQ924062. These results suggest that the defective autophagy in DU145 cells is due to the loss of canonical transcript encoding wild-type ATG5 protein.
Autophagy was restored by transfection of a wild-type ATG5 gene in DU145 cells
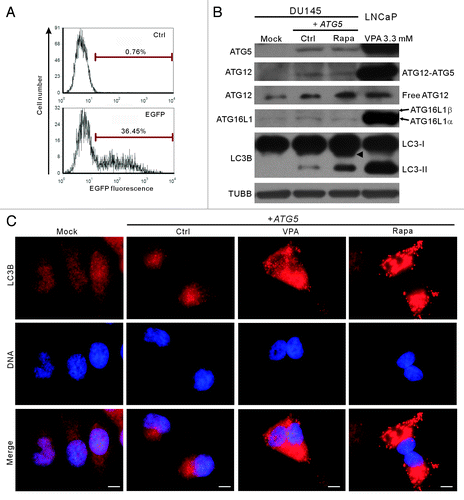
The wild-type ATG5 gene (cloned from LNCaP cells) was transfected into DU145 cells (referred to as DU145-ATG5WT cells hereafter). Transfection conditions were optimized by transfecting pEGFP-N1 plasmid into parental DU145 cells, which resulted in about 36% cells expressing EGFP, by flow cytometry analysis (). Western blot analysis showed that ATG5 protein was transiently expressed in DU145-ATG5WT cells (). ATG12–ATG5 conjugates were also observed after ATG5 rescue, although the levels of these conjugates in DU145-ATG5WT cells were much lower as compared with that in LNCaP cells (). Interestingly, the 16-kDa LC3B-II band and LC3B immunofluorescent puncta representing the conventional LC3 modification and autophagy were also restored in DU145-ATG5WT cells (). In addition, the 17-kDa LC3B band still existed in DU145-ATG5WT cells after rapamycin treatment. Although treatment of the DU145-ATG5WT cells with rapamycin did not increase the levels of ATG5, ATG16L1 and ATG12–ATG5, it significantly elevated the level of LC3B-II protein (). The rescued LC3B conjugation as well as formation of LC3B fluorescent puncta suggests that ATG16L1, though expressed at very low level, was functional in DU145-ATG5WT cells, since LNCaP cells transcribed the same variant mRNA of the ATG16L1 gene as DU145 cells. These results confirmed again that the loss of wild-type ATG5 protein led to the failure of autophagy in DU145 cells.
Figure 4. Transfection of the wild-type ATG5 gene rescued autophagy in DU145 cells. (A) Before transfection of the wild-type ATG5 gene, pEGFP-N1 plasmid with different ratios to transfection reagent was transfected into parental DU145 cells, and the percentages of cells expressing EGFP were analyzed by flow cytometry. Under optimal transfection condition, about 36% cells expressed EGFP (lower panel) compared with control. (B) The expression of ATG5 and other proteins was probed by western blotting. Cells transfected with the empty vector (mock) were recruited as a negative control. Remarkably, the conversion of LC3B-I into LC3B-II was restored with wild-type ATG5 expression. A sample of 3.3 mM-VPA-treated LNCaP cells was used as a positive control of LC3B-II and ATG5 expression. Arrowhead indicates 17-kDa bands of LC3B. (C) DU145 cells were transfected with mock or ATG5 plasmids, followed by treatment with 3.3 mM VPA or 2 μg/ml rapamycin for 24 h. The cells were then fixed and stained with LC3B antibody, followed by CF568-conjugated second antibody. Fluorescent images were obtained by fluorescence microscopy with a 100× oil objective lens (Scale bar: 10 μm). LC3B fluorescent puncta were only observed in ATG5-rescued samples, including the ATG5-transfected control with basal autophagy. Ctrl, control; Rapa, rapamycin.
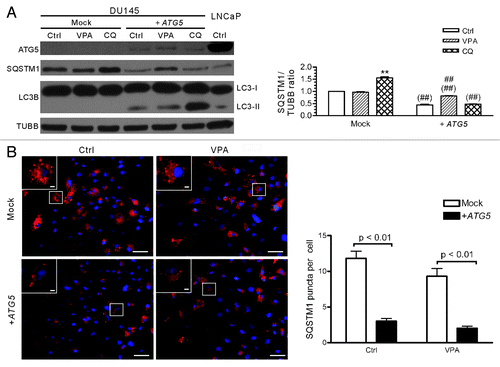
To further test whether autophagic flux was restored in DU145-ATG5WT cells, the cells were treated with CQ and the level of LC3B-II was evaluated. The result showed that CQ treatment increased the level of LC3B-II (), indicating that this lysosomotropic reagent had blocked the deconjugation and degradation of LC3B-II (LC3B-PE) as expected. Notably, the levels of SQSTM1 protein, which accumulated at a higher level in parental DU145 cells (), were sharply decreased after ATG5 transfection (). Similar to the situation in LNCaP and PC-3 cells (see ), VPA (3.3 mM) treatment increased the expression of SQSTM1 in DU145-ATG5WT cells as compared with the control cells (), which is in line with previous report indicating that induction of autophagy may upregulate the level of SQSTM1 protein.Citation29 In addition, immunofluorescent staining with SQSTM1 antibody demonstrated that SQSTM1 either diffusely distributed in the cytosol or formed aggregates in the so-called SQSTM1 bodies in both mock- and ATG5-transfected DU145 cells (), but the average number of SQSTM1 bodies/puncta per cell were significantly reduced in ATG5-transfected compared with mock-transfected cells (). These results further supported that ATG5 transfection restored the autophagy pathway in DU145 cells.
Figure 5. Analysis of SQSTM1 levels and body numbers in mock- or ATG5-transfected DU145 cells. (A) DU145 cells were transfected with mock or ATG5 plasmids, followed by treatment with VPA or chloroquine (CQ) for 24 h and western blot analysis were performed. A sample of LNCaP control cells was included in order to compare the SQSTM1 levels among DU145, LNCaP and PC-3 cells (referred to ). The relative levels of SQSTM1 were normalized to TUBB and the value of mock-treated control was set as 1.0 (right panel). **p < 0.01 vs mock control; ##p < 0.01 vs +ATG5 control; ##p < 0.01 vs the same group of mock. (B) DU145 cells were transfected with mock or ATG5 plasmids, followed by treatment with 3.3 mM VPA for 24 h. The cells were fixed and stained with SQSTM1 antibody, followed by CF568-conjugated second antibody. Fluorescent images were obtained by fluorescence microscopy with a 20× objective lens. Enlarged views of the boxed areas are shown as insets. Scale bar: 50 μm (2 μm in insets). The average numbers of SQSTM1 puncta were scored without prior knowledge of the experimental conditions (right panel). Ctrl, control.
Expression of wild-type ATG5 in DU145 or knockdown of ATG5 in LNCaP and PC-3 cells did not change the inhibitory effects of VPA on these cells
LNCaP cell line is generally used as an in vitro androgen-sensitive prostate cancer model, while DU145 and PC-3 cell lines are used as an androgen-refractory model. Previous studies have demonstrated that androgen-refractory prostate cancer cells, as compared with androgen-sensitive ones, are more resistant to chemotherapies.Citation30 Consistent with these works, our results also showed that VPA inhibited the proliferation of LNCaP, DU145 and PC-3 cells in a dose-dependent manner, but LNCaP cell line was more sensitive to VPA than the other two cell lines (). The 50% inhibitory concentration (IC50) values of VPA for these cell lines were 3.15 ± 0.50 mmol/L (LNCaP), 7.81 ± 0.07 mmol/L (DU145) and 15.08 ± 0.60 mmol/L (PC-3), respectively.
Figure 6. The inhibitory effect of VPA on cell proliferation was not changed by ATG5 rescue in DU145 cells or ATG5 knockdown in LNCaP and PC-3 cells (A) Valproic acid (VPA) dose-dependently inhibited the proliferation of LNCaP, DU145 and PC-3 cells. Logarithmic phase cells were incubated with indicated concentrations of VPA for 48 h. (B) DU145 cells were transfected with a plasmid containing the wild-type ATG5 gene and then treated with indicated concentrations of VPA for 48 h. (C and D) LNCaP and PC-3 cells were transfected with ATG5 siRNA or negative control (NC) siRNA for 48 h, and then treated with indicated concentrations of VPA for another 48 h. Cell proliferation was evaluated using MTS assay (A–D). One representative data of three independent experiments with similar results are shown as mean ± SD. The efficient knockdown of ATG5 protein in LNCaP and PC-3 cells was confirmed by western blotting (C and D, lower panels; VPA = 3.3 mM). The relative levels of ATG5 were normalized to TUBB and the value of NC siRNA-treated control of each group was set as 1.00, respectively.
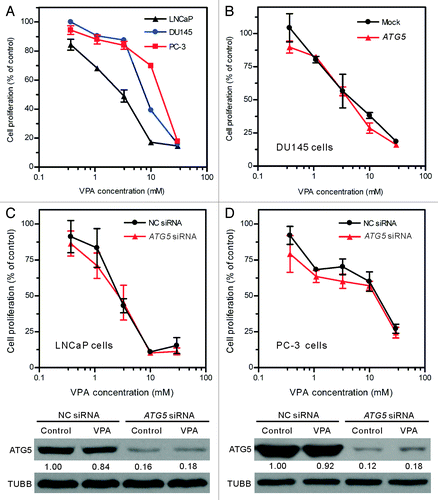
To further investigate whether ATG5 was associated with the differential inhibitory effects of VPA on these prostate cancer cell lines, we examined the proliferation of these cell lines after restoration of ATG5 in DU145 cells or knockdown of ATG5 with siRNA in LNCaP and PC-3 cells. The restored ATG5 expression in DU145 cells () did not alter the inhibitory effect of VPA (). Similarly, ATG5 siRNA knockdown in LNCaP and PC-3 cells, which reduced ~80% of ATG5 protein levels (, lower panels), also had no significant effect on their VPA sensitivity as compared with respective mock-treated control (, upper panels). These results suggest that the distinct splicing profiles of ATG5 may not have contributed to the differential inhibitory effects of VPA on these prostate cancer cell lines.
Discussion
In the present study, we analyzed the autophagic responses of human prostate cancer cells to VPA treatment by measuring accumulation of LC3-PE conjugates and formation of LC3 puncta. The results showed that autophagy was induced in LNCaP and PC-3 cells, but was impaired in DU145 cells. One critical autophagy-related protein, ATG5 (and its ATG12–ATG5 conjugate form), was detected in LNCaP and PC-3 cells, but was deficient in DU145 cells due to alternative splicing of ATG5 mRNA leading to nonfunctional truncated products. Transfection of the wild-type ATG5 gene in DU145 cells restored autophagy in the cells. Thus, the differential autophagic responses in human LNCaP, DU145 and PC-3 cells are mediated by distinct splicing profiles of the ATG5 gene.
One major finding of this study is that no stimuli used could induce conversion of LC3-I into LC3-II (LC3-PE) and formation of LC3 puncta in DU145 cells, indicating a defect in autophagy in this cell line. Interestingly, a new 17-kDa band of LC3B was upregulated by VPA or other stimuli in DU145 cells. We have previously observed a similar immunoblot band in VPA-treated B16F10 cells, but such a band disappears when the cells are cotreated with VPA and cucurbitacin B.Citation22 The possibility of a contaminant of protein degradation, resulting in the 17-kDa band of LC3B, could be excluded, since the total protein samples were directly extracted by 2 × SDS-PAGE loading buffer and the band always appeared upon various stimuli in DU145 cells, instead of in LNCaP and PC-3 cells. To our knowledge, the defect in autophagy in DU145 cells upon various stimuli has not been reported previously. We provided solid evidence to show that this was due to the deficiency of a critical autophagy-related gene product, the ATG5 protein and thus the loss of ATG12–ATG5 conjugates. This is consistent with previous studies that LC3 lipidation and membrane targeting after cell starvation is impaired in Atg5-deficient mouse embryonic stem cells.Citation12 ATG5 is critical for conversion of LC3-I to lipidated LC3-II (LC3-PE) since it is covalently conjugated to ATG12 and the conjugate shows E3-like activity in transfer of LC3 from LC3-ATG3 high energy conjugate to PE. ATG16L1, by interacting with ATG12–ATG5 conjugates, specifies the site at autophagosome for LC3 lipidation and membrane association.Citation24,Citation31 Although PE is the only in vivo target of LC3 that has been currently confirmed, other lipids such as phosphatidylserine (PS) can also be targets of LC3 in vitro.Citation32 We speculate that the ~17-kDa band observed in DU145 cells may be another LC3 modification form yet unidentified. On the other hand, transfection with wild-type ATG5 restored the conjugation of LC3–PE and formation of LC3 puncta in DU145 cells. Therefore, blockage of LC3–PE conjugation and LC3 puncta formation in DU145 cells was due to lack of full-length ATG5 protein.
It is worth noting that the 17 kDa LC3B band still existed in DU145-ATG5WT cells after rapamycin treatment (). This may be due to the low transfection efficiency (about 30 ~40% of the pEGFP-N1-transfected cells produced green fluorescence), and thus a fraction of the cells did not express the wild-type ATG5. At present, we are unable to determine whether the 17-kDa derivative of LC3B was coexisting with LC3B-II in DU145-ATG5WT cells at single cell level. Although autophagy (in terms of bulk degradation of subcellular constituents) may be alternatively induced in ATG5−/− cells, without induction of LC3 processing,Citation33 it needs more evidence to clarify whether DU145 cells undergo such an alternative autophagy in the presence of autophagy stimuli.
We also found that lack of wild-type ATG5 was associated with SQSTM1 accumulation in DU145 cells. SQSTM1 is a specific autophagy adaptor involved in selective autophagy of cellular components by binding to LC3 proteins via its LIR (LC3-interacting region) motif.Citation28 It is degradable in autophagy and may accumulate when autophagy is blocked, thus becoming an indicator for monitoring autophagy flux.Citation25,Citation26,Citation34 In line with this notion, there was a higher level of SQSTM1 protein in DU145 cells than in LNCaP and PC-3 cells. Moreover, VPA treatment could not significantly change the level of SQSTM1 protein in DU145 cells, suggesting that autophagy-dependent SQSTM1 degradation was impaired in this cell line. As expected, ATG5 gene rescue in DU145 cells significantly reduced the expression of SQSTM1 protein to a level comparable to that of LNCaP and PC-3 cells ( and ) and the average number of SQSTM1 puncta per cell was also reduced after ATG5 transfection (), consistent with the restoration of autophagy in this cell line. Considering that SQSTM1 is degradable through the autophagic pathway, accumulation of SQSTM1 level and SQSTM1 aggregates in parental DU145 cells also supported that conventional autophagy was impaired and that the so-called alternative autophagic flux, if present, should be low in this cell line. Taken together, our data demonstrated that the deficiency of full-length ATG5 protein in DU145 cells impaired autophagy (at least the conventional one) in this cell line.
As a ubiquitin-binding protein, SQSTM1 may recruit ubiquitinated protein inclusions or nonubiquitinated protein substrates into phagophores for autophagic degradation.Citation27,Citation35,Citation36 In addition, SQSTM1 protein is also required for the formation of sequestosomes containing ubiquitinated protein aggregates, serving as a shuttle protein in targeting them for proteasome degradation.Citation37,Citation38 Therefore, maintenance of high level of SQSTM1 in autophagy-null DU145 cells may be beneficial to the degradation of protein aggregates. Indeed, large inclusion-like aggregates (so-called sequestosomes or SQSTM1 bodies) were observed in DU145 cells (). However, the precise role of SQSTM1 accumulation in the prostate cancer cells remains largely unknown.
Another issue concerns whether ATG5 expression correlated with VPA sensitivity in the prostate cancer cells. While a large body of literature has revealed that induction of autophagy confers tumor cells drug resistance,Citation15,Citation16 our study demonstrated that autophagy-positive LNCaP and PC-3 cells showed differential sensitivity to VPA treatment, and LNCaP cells were even more sensitive to VPA than autophagy-null DU145 cells. Moreover, neither ATG5 transfection into DU145 cells nor siRNA knockdown of ATG5 in LNCaP and PC-3 cells changed the inhibitory effects of VPA on cell proliferation. Therefore, the distinct splicing profiles of ATG5 may not have contributed to the differential inhibitory effects of VPA on these prostate cancer cell lines, but rather other mechanism(s) may have been involved.
One possible mechanism is their differential expression of other genes critical for cell survival and DNA repair, such as TP53.Citation39,Citation40 It is well known that LNCaP cells express a wild-type TP53, while PC-3 cells are null for TP53, and DU145 cells have a mutated TP53 gene.Citation41 HDAC inhibitors, including VPA, have been reported to increase the acetylation of TP53 and so stabilize the tumor suppressor protein, leading to an increase in its proapoptotic function and thus sensitizing tumor cells to cell death stimuli.Citation42-Citation44 Since VPA causes DNA instability in tumor cells,Citation22,Citation45,Citation46 expression of a wild-type TP53 in LNCaP cells may make them more sensitive than the other two cell lines. Yet, more investigations are warranted to clarify this issue, which is out of the scope of current study.
Interestingly, there was a point mutation (A898G) in ATG16L1 genes in both LNCaP and DU145 cells, leading to a T300A conversion in amino acid sequence. To our knowledge, this is the first time that such a T300A conversion has been reported in prostate cancer cells. A large number of studies have demonstrated that the T300A conversion in ATG16L1 protein contributes to Crohn disease, a chronic inflammatory disease of the gastrointestinal tract.Citation47,Citation48 T300A conversion in ATG16L1 may confine the innate immune cells to induce autophagy via pattern-recognition receptor NOD2 and enhance NOD2-mediated proinflammatory cytokine production.Citation49,Citation50 However, the exact function of the ATG16L1 T300A conversion during the course of Crohn disease and prostate cancer development is largely unknown.
In summary, we demonstrated in this study that autophagy was differently induced in human prostate cancer LNCaP, DU145 and PC-3 cells due to distinct splicing profiles of the ATG5 gene. In particular, autophagy was genetically impaired in DU145 cells, which produced a 17-kDa LC3B band, instead of the conventional 16-kDa LC3-II, in response to VPA as well as other stimuli. The differences of these prostate cancer cell lines in autophagy induction should be taken into account when choosing them as in vitro prostate cancer models. Further clinical investigations of ATG5 defect and ATG16L1 T/A conversion in human subjects will facilitate our understanding of the role of autophagy in prostate cancer development.
Materials and Methods
Reagents
Valproic acid sodium salt (VPA) (P4543), chloroquine (CQ) (C6628) and rapamycin (R8781) were bought from Sigma-Aldrich. Rapamycin was dissolved in dimethyl sulfoxide (DMSO) (Sigma, D4540) and kept at −20°C if necessary (but no more than 1 week). The final concentration of DMSO never exceeded 0.2%, which had no cytotoxicity in this study (data not shown). The catalog numbers of antibodies used in this study are listed as: Acetyl-histone H3 (Lys9) (9649), histone H3 (9717), BECN1 (3495), LC3B (3868), LC3A (4599), ATG3 (3415), ATG5 (8540), ATG7 (2631), ATG12 (4180), ATG16L1 (8089), SQSTM1 (5114, for western blot), SQSTM1 (7695, for immunofluorescent analysis), and TUBB/β-tubulin (2128) (Cell Signaling Technology). ATG5 siRNA I (6345) and negative control siRNA (6568) were also obtained from Cell Signaling Technology.
Cell culture and treatment
Human prostate cancer DU145, PC-3 and LNCaP cell lines were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). DU145 and PC-3 Cells were maintained in DMEM (Invitrogen, C11995) supplemented with 10% fetal bovine serum (Gibco/Invitrogen, 10099-141), 100 U/ml penicillin G and 100 μg/ml streptomycin (Invitrogen, 15070-063) at 37°C in a humidified incubator with 5% CO2 and subcultured every 2–3 d. LNCaP cells were maintained in DMEM/F12 (Invitrogen, C11330) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin G and 100 μg/ml streptomycin. Before LNCaP cells were seeded, the culture surface was precoated with poly-l-lysine (Sigma-Aldrich, P1274). In all experiments of this study, cells in log phase were subcultured for 24 h and the medium was replaced with fresh medium with or without reagents.
Cell proliferation assay
Cells were seeded in 96-well plates (3300 cells/well) for 24 h and then treated with indicated concentrations of reagents. Cell proliferation was determined using a CellTiter96 AQueous One Solution Cell Proliferation Assay (MTS) kit (Promega, G3580) as previously described.Citation22 Briefly, 20 μl of MTS reagent was added to each well and the plates were incubated for 2–4 h at 37°C. The absorbance at 490 nm was measured by a microplate reader (Model 680; Bio-Rad, Richmond, CA, USA), and the 50% inhibiting concentration (IC50) was determined from the dose-response curve.
Immunofluorescence microscopy
After incubation with indicated regents, cells were fixed in 4% paraformaldehyde prepared in phosphate-buffered saline (PBS), permeabilized with ice-cold 100% methanol, and immunostained with rabbit anti-LC3B antibody or anti-SQSTM1 antibody, followed by CF568-conjugated goat-anti-rabbit IgG (Biotium, 20103-1). Nuclei were revealed by Hoechst 33342 (Sigma, B2261) staining. Fluorescence images were observed and collected under a Leica DMIRB fluorescent microscope (Leica).
RNA isolation and cDNA synthesis
Total RNA samples were extracted from LNCaP, DU145 and PC-3 cells in logarithm using Trizol reagent (Invitrogen, 15596026) according to the manufacturer’s instruction. The total RNA products were immediately transcribed into cDNA using a PrimeScript 1st strand cDNA synthesis kit (Takara, D6110A).
Cloning and sequencing of ATG5 and ATG16L1 genes
ATG5 and ATG16L1 sequences were amplified by polymerase chain reaction (PCR) using high-fidelity PrimeSTAR HS DNA polymerase (Takara, DR010A) according to the manufacturer's protocol. Primers synthesized by Invitrogen are listed as following: ATG5: sense: 5′-ATA GAA GCT TGC CAC CAT GAC AGA TGA CAA AGA TGT GCT TCG-3′; anti-sense: 5′-ATA GGG ATC CTC AAT CTG TTG GCT GTG GGA TGA TAC-3′; ATG16L1: sense: 5′-ATA GCT CGA GGC CAC CAT GTC GTC GGG CCT CCG CGC-3′; anti-sense: 5′-ATA GGA ATT CTC AGT ACT GTG CCC ACA GCA CAG C-3′. PCR products were excised after agarose gel electrophoresis and purified using the QIAquick gel extraction kit (Qiagen, 28706). Purified ATG5 and ATG16L1 products were digested with Hind III (Takara, D1060A) plus BamH I (Takara, D1010A) (for ATG5 products), or Xho I (Takara, D1094A) plus EcoR I (Takara, D1040A) (for ATG16L1) and then inserted into eukaryotic expression vector pEGFP-N1 (ClonTech, 6085–1). The clones harboring the plasmid with inserts of expected lengths were identified by indicated restriction enzymes and then sequenced by Invitrogen (Guangzhou) from both orientations.
Transient expression of wild-type ATG5 gene in DU145 cells
Wild-type ATG5 sequences from LNCaP cells were cloned into pEGFP-N1 as a nonfused gene as described above. Transfection of recombinant plasmid into DU145 cells was mediated by Trans-EZ reagent (Shanghai Sunbio Medical Biotechnology, STP07006) as previously reported.Citation51 Transfection efficiency was monitored by transfecting a pEGFP-N1 empty vector and green fluorescence were observed under a fluorescent microscope. After 48 h of culture, transient gene expression was determined by western blotting, because the inserted sequences were ended with a terminal codon before the EGFP gene. The cells were further stimulated with rapamycin (2 μg/ml) for 4 h to probe the function of the ATG5 gene product on LC3 conversion.
Knockdown of ATG5 gene
LNCaP and PC-3 cells were seeded in 96-well plates (Corning, 3599) (for MTS assay), 6-well plates (Corning, 3516) (for western blotting) or glass bottom culture dishes (NEST, GBD-35-15) (for immunofluorescent staining) for 24 h. Knockdown of ATG5 was performed according to the instructions provided by the manufacturers. Briefly, transfection regent LipofectamineTM RNAiMAX (Invitrogen, 13778-075) and ATG5 siRNA or control siRNA was diluted in Opti-MEM reduced serum medium (Invitrogen, 31985-070), respectively. Then the diluted siRNA and transfection reagent was mixed thoroughly, followed by incubation at room temperature for 15 min. Cell culture medium was replaced with Opti-MEM reduced serum medium and the siRNA mixture was added to each well at a final siRNA concentration of 20 nM. Six hours later, cells were cultured in normal medium containing 10% FBS. After being cultured for another 48 h, cells were treated with indicated doses of VPA or other reagents for cell proliferation assay or western blot analysis.
Protein extraction
Cells were washed thoroughly with ice-cold PBS and lysed with 2× sodium dodecyl sulfate PAGE (SDS-PAGE) loading buffer. Lysates were sonicated, boiled and clarified by centrifugation at 12, 000× g for 20 min at 4°C. Protein concentration was measured by SDS-PAGE with a known sample determined by a BCA protein assay kit (Pierce, 23227) according to the manufacturer’s instructions. Samples were quickly frozen in liquid nitrogen and then stored at −70°C until use.
Western blotting
Samples containing 30 μg of total protein were separated by SDS-PAGE and transferred onto a PVDF membrane (Amersham, RPN303F). After incubation in blocking buffer (50 mM Tris-buffered saline (pH7.4) containing 5% nonfat milk and 0.1% Tween-20), the membranes were probed with indicated antibodies, followed by a horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (Jackson ImmunoResearch, 111-035-003) or goat anti-mouse IgG (Jackson ImmunoResearch, 115-035-003). Bands were revealed by a BeyoECL Plus kit (Beyotime, P0018) and recorded on X-ray films (Kodak, 6535873). The densitometry of each band was quantified by FluorChem 8000 (AlphaInnotech).
Statistical analysis
Data were collected from experiments repeated in triplicate except where indicated otherwise. Values were analyzed by Prism software 4.0 (GraphPad). One-way ANOVA followed by Newman-Keuls post-test were used to compare between treatment groups and a p value < 0.05 was considered as significant.
| Abbreviations: | ||
| ATG3 | = | autophagy-related 3 |
| ATG5 | = | autophagy-related 5 |
| ATG7 | = | autophagy-related 7 |
| ATG12 | = | autophagy-related 12 |
| ATG16L1 | = | autophagy-related 16-like 1 |
| BECN1 | = | Beclin 1 |
| CQ | = | chloroquine |
| HDAC | = | histone deacetylase |
| EGFP | = | enhanced green fluorescent protein |
| LC3A | = | microtubule-associated protein 1 light chain 3 alpha (MAP1LC3A) |
| LC3B | = | microtubule-associated protein 1 light chain 3 beta (MAP1LC3B) |
| LIR | = | LC3-interacting region |
| MTOR | = | mechanistic target of rapamycin |
| NOD2 | = | nucleotide-binding oligomerization domain containing 2 |
| PE | = | phosphotidylethanolamine |
| PCR | = | polymerase chain reaction |
| PS | = | phosphatidylserine |
| SQSTM1 | = | sequestosome 1 |
| SDS-PAGE | = | sodium dodecyl sulfate−polyacrylamide gel electrophoresis |
| TP53 | = | tumor protein p53 |
| TUBB | = | β-tubulin |
| VPA | = | valproic acid |
Acknowledgments
This work is supported by grants from the National Natural Science Foundation of China (no. 81173604), the Specialized Research Program of “Twelfth Five-Year Plan” of China (no. 2011ZX09307-303-03), the Major State Basic Research Development Program of China (973 Program) (no. 2010CB833603), and the Fundamental Research Funds for the Central Universities (no. 21612411).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Abbas A, Gupta S. The role of histone deacetylases in prostate cancer. Epigenetics 2008; 3:300 - 9; http://dx.doi.org/10.4161/epi.3.6.7273; PMID: 19029799
- Zhao X, Yang W, Shi C, Ma W, Liu J, Wang Y, et al. The G1 phase arrest and apoptosis by intrinsic pathway induced by valproic acid inhibit proliferation of BGC-823 gastric carcinoma cells. Tumour Biol 2011; 32:335 - 46; http://dx.doi.org/10.1007/s13277-010-0126-5; PMID: 21113745
- Kuendgen A, Schmid M, Schlenk R, Knipp S, Hildebrandt B, Steidl C, et al. The histone deacetylase (HDAC) inhibitor valproic acid as monotherapy or in combination with all-trans retinoic acid in patients with acute myeloid leukemia. Cancer 2006; 106:112 - 9; http://dx.doi.org/10.1002/cncr.21552; PMID: 16323176
- Weichert W, Röske A, Gekeler V, Beckers T, Stephan C, Jung K, et al. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br J Cancer 2008; 98:604 - 10; http://dx.doi.org/10.1038/sj.bjc.6604199; PMID: 18212746
- Xia Q, Sung J, Chowdhury W, Chen CL, Höti N, Shabbeer S, et al. Chronic administration of valproic acid inhibits prostate cancer cell growth in vitro and in vivo. Cancer Res 2006; 66:7237 - 44; http://dx.doi.org/10.1158/0008-5472.CAN-05-0487; PMID: 16849572
- Shabbeer S, Kortenhorst MS, Kachhap S, Galloway N, Rodriguez R, Carducci MA. Multiple Molecular pathways explain the anti-proliferative effect of valproic acid on prostate cancer cells in vitro and in vivo. Prostate 2007; 67:1099 - 110; http://dx.doi.org/10.1002/pros.20587; PMID: 17477369
- Valentini A, Biancolella M, Amati F, Gravina P, Miano R, Chillemi G, et al. Valproic acid induces neuroendocrine differentiation and UGT2B7 up-regulation in human prostate carcinoma cell line. Drug Metab Dispos 2007; 35:968 - 72; http://dx.doi.org/10.1124/dmd.107.014662; PMID: 17371798
- Sharma S, Symanowski J, Wong B, Dino P, Manno P, Vogelzang N. A phase II clinical trial of oral valproic acid in patients with castration-resistant prostate cancers using an intensive biomarker sampling strategy. Transl Oncol 2008; 1:141 - 7; PMID: 18795124
- Long J, Zhao J, Yan Z, Liu Z, Wang N. Antitumor effects of a novel sulfur-containing hydroxamate histone deacetylase inhibitor H40. Int J Cancer 2009; 124:1235 - 44; http://dx.doi.org/10.1002/ijc.24074; PMID: 19058176
- Iacopino F, Urbano R, Graziani G, Muzi A, Navarra P, Sica G. Valproic acid activity in androgen-sensitive and -insensitive human prostate cancer cells. Int J Oncol 2008; 32:1293 - 303; PMID: 18497991
- Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol 2004; 36:2503 - 18; http://dx.doi.org/10.1016/j.biocel.2004.05.009; PMID: 15325588
- Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, et al. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol 2001; 152:657 - 68; http://dx.doi.org/10.1083/jcb.152.4.657; PMID: 11266458
- Lomonaco SL, Finniss S, Xiang C, Decarvalho A, Umansky F, Kalkanis SN, et al. The induction of autophagy by gamma-radiation contributes to the radioresistance of glioma stem cells. Int J Cancer 2009; 125:717 - 22; http://dx.doi.org/10.1002/ijc.24402; PMID: 19431142
- Turner LS, Cheng JC, Beckham TH, Keane TE, Norris JS, Liu X. Autophagy is increased in prostate cancer cells overexpressing acid ceramidase and enhances resistance to C6 ceramide. Prostate Cancer Prostatic Dis 2011; 14:30 - 7; http://dx.doi.org/10.1038/pcan.2010.47; PMID: 21116286
- Dalby KN, Tekedereli I, Lopez-Berestein G, Ozpolat B. Targeting the prodeath and prosurvival functions of autophagy as novel therapeutic strategies in cancer. Autophagy 2010; 6:322 - 9; http://dx.doi.org/10.4161/auto.6.3.11625; PMID: 20224296
- Carew JS, Nawrocki ST, Kahue CN, Zhang H, Yang C, Chung L, et al. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance. Blood 2007; 110:313 - 22; http://dx.doi.org/10.1182/blood-2006-10-050260; PMID: 17363733
- Ko H, Kim YJ, Park JS, Park JH, Yang HO. Autophagy inhibition enhances apoptosis induced by ginsenoside Rk1 in hepatocellular carcinoma cells. Biosci Biotechnol Biochem 2009; 73:2183 - 9; http://dx.doi.org/10.1271/bbb.90250; PMID: 19809182
- Wu Z, Chang PC, Yang JC, Chu CY, Wang LY, Chen NT, et al. Autophagy blockade sensitizes prostate cancer cells towards Src family kinase inhibitors. Genes Cancer 2010; 1:40 - 9; http://dx.doi.org/10.1177/1947601909358324; PMID: 20811583
- Michaelis M, Suhan T, Michaelis UR, Beek K, Rothweiler F, Tausch L, et al. Valproic acid induces extracellular signal-regulated kinase 1/2 activation and inhibits apoptosis in endothelial cells. Cell Death Differ 2006; 13:446 - 53; http://dx.doi.org/10.1038/sj.cdd.4401759; PMID: 16167071
- Fu J, Shao CJ, Chen FR, Ng HK, Chen ZP. Autophagy induced by valproic acid is associated with oxidative stress in glioma cell lines. Neuro Oncol 2010; 12:328 - 40; http://dx.doi.org/10.1093/neuonc/nop005; PMID: 20308311
- Xiong N, Jia M, Chen C, Xiong J, Zhang Z, Huang J, et al. Potential autophagy enhancers attenuate rotenone-induced toxicity in SH-SY5Y. Neuroscience 2011; 199:292 - 302; http://dx.doi.org/10.1016/j.neuroscience.2011.10.031; PMID: 22056603
- Ouyang D, Zhang Y, Xu L, Li J, Zha Q, He X. Histone deacetylase inhibitor valproic acid sensitizes B16F10 melanoma cells to cucurbitacin B treatment. Acta Biochim Biophys Sin (Shanghai) 2011; 43:487 - 95; http://dx.doi.org/10.1093/abbs/gmr032; PMID: 21628505
- Cao C, Subhawong T, Albert JM, Kim KW, Geng L, Sekhar KR, et al. Inhibition of mammalian target of rapamycin or apoptotic pathway induces autophagy and radiosensitizes PTEN null prostate cancer cells. Cancer Res 2006; 66:10040 - 7; http://dx.doi.org/10.1158/0008-5472.CAN-06-0802; PMID: 17047067
- Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, et al. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem 2007; 282:37298 - 302; http://dx.doi.org/10.1074/jbc.C700195200; PMID: 17986448
- Korolchuk VI, Menzies FM, Rubinsztein DC. A novel link between autophagy and the ubiquitin-proteasome system. Autophagy 2009; 5:862 - 3; PMID: 19458478
- Larsen KB, Lamark T, Øvervatn A, Harneshaug I, Johansen T, Bjørkøy G. A reporter cell system to monitor autophagy based on p62/SQSTM1. Autophagy 2010; 6:784 - 93; http://dx.doi.org/10.4161/auto.6.6.12510; PMID: 20574168
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131 - 45; http://dx.doi.org/10.1074/jbc.M702824200; PMID: 17580304
- Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011; 7:279 - 96; http://dx.doi.org/10.4161/auto.7.3.14487; PMID: 21189453
- Fujita K, Maeda D, Xiao Q, Srinivasula SM. Nrf2-mediated induction of p62 controls Toll-like receptor-4-driven aggresome-like induced structure formation and autophagic degradation. Proc Natl Acad Sci U S A 2011; 108:1427 - 32; http://dx.doi.org/10.1073/pnas.1014156108; PMID: 21220332
- van Brussel JP, van Steenbrugge GJ, Romijn JC, Schröder FH, Mickisch GH. Chemosensitivity of prostate cancer cell lines and expression of multidrug resistance-related proteins. Eur J Cancer 1999; 35:664 - 71; http://dx.doi.org/10.1016/S0959-8049(98)00435-3; PMID: 10492644
- Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell 2008; 19:2092 - 100; http://dx.doi.org/10.1091/mbc.E07-12-1257; PMID: 18321988
- Sou YS, Tanida I, Komatsu M, Ueno T, Kominami E. Phosphatidylserine in addition to phosphatidylethanolamine is an in vitro target of the mammalian Atg8 modifiers, LC3, GABARAP, and GATE-16. J Biol Chem 2006; 281:3017 - 24; http://dx.doi.org/10.1074/jbc.M505888200; PMID: 16303767
- Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, et al. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009; 461:654 - 8; http://dx.doi.org/10.1038/nature08455; PMID: 19794493
- Barth S, Glick D, Macleod KF. Autophagy: assays and artifacts. J Pathol 2010; 221:117 - 24; http://dx.doi.org/10.1002/path.2694; PMID: 20225337
- Vadlamudi RK, Joung I, Strominger JL, Shin J. p62, a phosphotyrosine-independent ligand of the SH2 domain of p56lck, belongs to a new class of ubiquitin-binding proteins. J Biol Chem 1996; 271:20235 - 7; http://dx.doi.org/10.1074/jbc.271.34.20235; PMID: 8702753
- Watanabe Y, Tanaka M. p62/SQSTM1 in autophagic clearance of a non-ubiquitylated substrate. J Cell Sci 2011; 124:2692 - 701; http://dx.doi.org/10.1242/jcs.081232; PMID: 21771882
- Seibenhener ML, Babu JR, Geetha T, Wong HC, Krishna NR, Wooten MW. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol Cell Biol 2004; 24:8055 - 68; http://dx.doi.org/10.1128/MCB.24.18.8055-8068.2004; PMID: 15340068
- Shvets E, Elazar Z. Autophagy-independent incorporation of GFP-LC3 into protein aggregates is dependent on its interaction with p62/SQSTM1. Autophagy 2008; 4:1054 - 6; PMID: 18776740
- Nelson WG, Kastan MB. DNA strand breaks: the DNA template alterations that trigger p53-dependent DNA damage response pathways. Mol Cell Biol 1994; 14:1815 - 23; PMID: 8114714
- Schneider E, Montenarh M, Wagner P. Regulation of CAK kinase activity by p53. Oncogene 1998; 17:2733 - 41; http://dx.doi.org/10.1038/sj.onc.1202504; PMID: 9840937
- van Bokhoven A, Varella-Garcia M, Korch C, Johannes WU, Smith EE, Miller HL, et al. Molecular characterization of human prostate carcinoma cell lines. Prostate 2003; 57:205 - 25; http://dx.doi.org/10.1002/pros.10290; PMID: 14518029
- Chen X, Wong JY, Wong P, Radany EH. Low-dose valproic acid enhances radiosensitivity of prostate cancer through acetylated p53-dependent modulation of mitochondrial membrane potential and apoptosis. Mol Cancer Res 2011; 9:448 - 61; http://dx.doi.org/10.1158/1541-7786.MCR-10-0471; PMID: 21303901
- Fortson WS, Kayarthodi S, Fujimura Y, Xu H, Matthews R, Grizzle WE, et al. Histone deacetylase inhibitors, valproic acid and trichostatin-A induce apoptosis and affect acetylation status of p53 in ERG-positive prostate cancer cells. Int J Oncol 2011; 39:111 - 9; PMID: 21519790
- Roy S, Packman K, Jeffrey R, Tenniswood M. Histone deacetylase inhibitors differentially stabilize acetylated p53 and induce cell cycle arrest or apoptosis in prostate cancer cells. Cell Death Differ 2005; 12:482 - 91; http://dx.doi.org/10.1038/sj.cdd.4401581; PMID: 15746940
- Catalano MG, Fortunati N, Pugliese M, Poli R, Bosco O, Mastrocola R, et al. Valproic acid, a histone deacetylase inhibitor, enhances sensitivity to doxorubicin in anaplastic thyroid cancer cells. J Endocrinol 2006; 191:465 - 72; http://dx.doi.org/10.1677/joe.1.06970; PMID: 17088416
- Marchion DC, Bicaku E, Daud AI, Sullivan DM, Munster PN. Valproic acid alters chromatin structure by regulation of chromatin modulation proteins. Cancer Res 2005; 65:3815 - 22; http://dx.doi.org/10.1158/0008-5472.CAN-04-2478; PMID: 15867379
- Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet 2007; 39:596 - 604; http://dx.doi.org/10.1038/ng2032; PMID: 17435756
- Naser SA, Arce M, Khaja A, Fernandez M, Naser N, Elwasila S, et al. Role of ATG16L, NOD2 and IL23R in Crohn’s disease pathogenesis. World J Gastroenterol 2012; 18:412 - 24; http://dx.doi.org/10.3748/wjg.v18.i5.412; PMID: 22346247
- Plantinga TS, Crisan TO, Oosting M, van de Veerdonk FL, de Jong DJ, Philpott DJ, et al. Crohn’s disease-associated ATG16L1 polymorphism modulates pro-inflammatory cytokine responses selectively upon activation of NOD2. Gut 2011; 60:1229 - 35; http://dx.doi.org/10.1136/gut.2010.228908; PMID: 21406388
- Plantinga TS, Joosten LA, Netea MG. ATG16L1 polymorphisms are associated with NOD2-induced hyperinflammation. Autophagy 2011; 7:1074 - 5; http://dx.doi.org/10.4161/auto.7.9.15867; PMID: 21673517
- Ouyang D, He X, Xu L, Wang X, Gao Q, Guo H. Differential cell surface expression of rhesus macaque’s major histocompatibility complex class I alleles Mamu-B*1703 and Mamu-B*0101. Acta Biochim Biophys Sin (Shanghai) 2010; 42:281 - 7; http://dx.doi.org/10.1093/abbs/gmq019; PMID: 20383467