Abstract
Acute and chronic ethanol administration increase autophagic vacuole (i.e., autophagosome; AV) content in liver cells. This enhancement depends on ethanol oxidation. Here, we used parental (nonmetabolizing) and recombinant (ethanol-metabolizing) Hep G2 cells to identify the ethanol metabolite that causes AV enhancement by quantifying AVs or their marker protein, microtubule-associated protein 1 light chain 3-II (LC3-II). The ethanol-elicited rise in LC3-II was dependent on ethanol dose, was seen only in cells that expressed alcohol dehydrogenase (ADH) and was augmented in cells that coexpressed cytochrome CYP2E1 (P450 2E1). Furthermore, the rise in LC3-II was inversely related to a decline in proteasome activity. AV flux measurements and colocalization of AVs with lysosomes or their marker protein Lysosomal-Associated Membrane Protein 1 (LAMP1) in ethanol-metabolizing VL-17A cells (ADH+/CYP2E1+) revealed that ethanol exposure not only enhanced LC3-II synthesis but also decreased its degradation. Ethanol-induced accumulation of LC3-II in these cells was similar to that induced by the microtubule inhibitor, nocodazole. After we treated cells with either 4-methylpyrazole to block ethanol oxidation or GSH-EE to scavenge reactive species, there was no enhancement of LC3-II by ethanol. Furthermore, regardless of their ethanol-metabolizing capacity, direct exposure of cells to acetaldehyde enhanced LC3-II content. We conclude that both ADH-generated acetaldehyde and CYP2E1-generated primary and secondary oxidants caused LC3-II accumulation, which rose not only from enhanced AV biogenesis, but also from decreased LC3 degradation by the proteasome and by lysosomes.
Introduction
After chronic ethanol consumption, there is significant liver enlargement, about 50% of which is protein accumulation.Citation1 The latter reflects impaired degradation of long-lived proteins, most of which are degraded in lysosomes.Citation2,Citation3 While we do not know the exact mechanism by which ethanol exposure impairs lysosomal protein degradation, we have shown that chronic ethanol administration to rats disrupts not only lysosomal proteolysis but also lysosome biogenesis.Citation4-Citation6 Lysosomes are crucial for the breakdown of all forms of cellular macromolecules. They participate in the catabolic phase of macroautophagy (autophagy), which begins with the formation of double-membrane-bound autophagic vacuoles (AVs or autophagosomes) that envelop and sequester soluble and particulate cytoplasmic components. AVs then fuse with lysosomes, forming autolysosomes that degrade the AV contents. A recent report showed that acute ethanol administration to mice (similar to binge drinking) increases the hepatic content of AVs and that this induction depends on ethanol metabolism.Citation7 These investigators concluded that ethanol oxidation generates reactive oxygen species (ROS) that inhibit the activity of the mechanistic target of rapamycin complex 1 (MTORC1), a large, 250 kDa complex with serine/threonine protein kinase activity that enhances cellular anabolic reactions, including protein synthesis, while it suppresses catabolic pathways, including macroautophagy. Inhibition of MTORC1 relieves autophagic suppression, thereby inducing AV formation. We have recently reported that ethanol-exposed rat liver slices and livers of mice subjected to chronic ethanol feeding, both exhibit elevated AVs and higher levels of the AV marker protein, LC3-II.Citation8 At the same time, ethanol exposure decreases 20S proteasome activity, which is inversely correlated with the rise in AVs and LC3-II. Thus, part of the ethanol-induced rise in LC3II (AVs) occurs because LC3-II is stabilized from degradation by the 20S proteasome, which is inhibited by products of ethanol oxidation.Citation8 The latter findings prompted the investigation described here. Using parental and recombinant Hep G2 cells, we sought to determine the mechanism of AV induction by ethanol and to identify the ethanol metabolite(s) that cause(s) it.
Results
AV induction increased with rising ethanol doses and depended on ethanol oxidation
We exposed VL-17A cells (ADH+/CYP2E1+) to zero, 5, 10, 25 and 50 mM ethanol for 24 h. No acetaldehyde was detected in the medium after treatment of cells with 5 and 10 mM ethanol. Exposure of cells to 25 and 50 mM ethanol generated acetaldehyde levels of 58 ± 4 and 81 ± 5 µM, respectively, indicating an acceleration of ethanol oxidation with increasing ethanol doses. The intracellular content of both LC3-I and LC3-II also increased with rising ethanol levels (). The dose response curve shown in is biphasic, with a more robust increase in LC3-II content after exposure to 25 and 50 mM ethanol than after treatment with lower ethanol doses. These data indicated that, at higher levels of ethanol, additional factors contributed to the rise in LC3-II, probably by its stabilization from proteolysis by the 20S proteasome, as previously published.Citation9 The chymotrypsin-like activity of the 20S proteasome in lysates of VL-17A cells was unaffected by prior exposure of cells to 5 and 10 mM ethanol, but exposure to 25 and 50 mM ethanol, decreased proteasome activity 20% and 46%, relative to controls, respectively (). These results were consistent with our recent findings that LC3-II (AVs) and proteasome are differentially sensitive to ethanol exposure and respond in a reciprocal manner.Citation8 They provided further evidence that proteasome inhibition at higher ethanol concentrations (), stabilized LC3-II, thereby augmenting its intracellular content.
Figure 1. Ethanol exposure increased LC3 content, decreased proteasome activity and enhanced AV content in VL-17A cells. (A) Mean densitometric ratios of LC3-I and LC3-II, each to β-actin (ACTB) in cells treated 24 h with the indicated doses of ethanol. (B) Proteasome activity in cells treated 24 h with the indicated doses of ethanol. Data are mean values of the LC3 protein bands from western blots (A) proteasome chymotrypsin-like activity (B) from quadruplicate culture flasks of each treatment group. (C) AV content in VL-17A cells: Cells were exposed to the conditions indicated in the figure for 24 h. After treatment, the cells were fixed in 4% paraformaldehyde and immuno-stained for AVs using anti LC3B. Images were captured by confocal microscopy. Green puncta are AVs. Nuclei stained with DAPI are shown in blue. Microscopy data (C) indicated are mean values from 400 to 1100 cell images. (D) AV numbers, size and its proportion to total cell area (fractional volume) in cells from (C). Letter superscripts or letters in the figures that are different from each other indicate that the data are significantly different from each other. Data with the same letter are not significantly different.
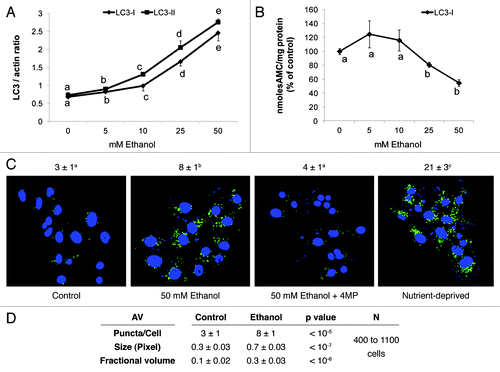
The LC3-II level in VL-17A cells was quantitatively associated with AV content, as judged by immunocytochemistry in VL-17A cells (). Untreated cells exhibited 3 ± 1 green puncta (dots) per cell, which increased 2.6-fold after we treated them with 50 mM ethanol. This treatment also increased the AV size and its proportion to total cell area by 2.3 and 3-fold respectively, over controls (). We included 4-methylpyrazole (4MP) to block ethanol oxidation, after which cells had AV puncta numbers and size equal to those in control cells, indicating that inhibition of ethanol oxidation prevented the rise in AVs. After cells were nutrient-starved in Earle’s Balanced Salt Solution (EBSS) they contained seven times more AV puncta than controls, demonstrating starvation-enhanced autophagosome content.
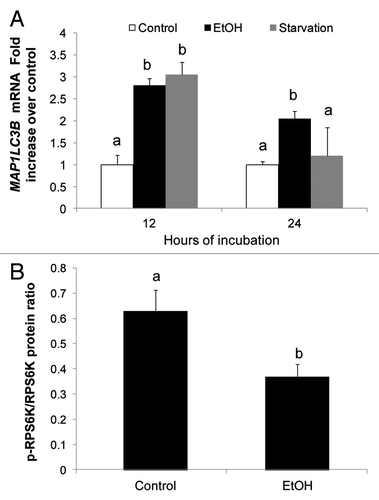
We assessed the effects of ethanol exposure and nutrient-deprivation on de novo AV biogenesis by measuring LC3 mRNA in VL-17A cells. After exposure of VL-17A cells to each condition for 12 h, they exhibited 3-fold higher levels of LC3 mRNA [MAP1LC3B] than untreated control cells. After 24 h, LC3B mRNA in ethanol-treated cells was 2-fold higher than controls while, in starved cells, it returned to basal levels (). These findings indicated that after 12 h of ethanol exposure or starvation, both conditions equally enhanced LC3B mRNA for eventual pro-LC3 (autophagosome) formation. Ethanol treatment also relieved autophagic suppression by MTORC1, as it decreased phosphorylation of the MTORC anabolic target, RPS6K/p70S6K (). However the same treatment did not affect the levels of BECN1, a key initiator protein of AV biogenesis, nor did it change the levels of phosphorylated AMPK, which regulates MTORC1 activity (data not shown). The data indicated that the apparent decrease in MTOR activity involved no change in AMPK activity but was itself related to ethanol oxidation as demonstrated previously.Citation7
Figure 2. Ethanol exposure enhanced LC3B mRNA and inhibited MTORC1. (A) LC3B mRNA levels in VL-17A cells after 12 and 24 h exposure to ethanol or nutrient deprivation. (B) Ethanol effect on RPS6K in VL-17A cells. Mean densitometric ratios of pRPS6K/RPS6K in cells treated 24 h with zero or 50 mM ethanol. Data are mean values (± SEM) from quadruplicate samples. Letters that are different from each other indicate that the data are significantly different from each other. Data with the same letter are not significantly different.
AV flux
To further assess the nature of the ethanol-induced rise in AVs, we measured LC3-II flux in VL-17A cells treated with or without 50 mM ethanol in the presence or absence of bafilomycin A1. Bafilomycin A1 inhibits the lysosome proton pump to prevent lysosome acidification, thereby blocking degradation of AV cargo, including LC3-II. Ethanol or bafilomycin alone each enhanced the level of LC3-II over untreated cells by 41 and 53%, respectively. The LC3-II content in cells exposed to both ethanol and bafilomycin A1, rose above those treated with either agent alone (), indicating that ethanol enhanced LC3 synthesis, and decreased its degradation.Citation10 The latter effect of ethanol on AV degradation was confirmed by measurements of the intracellular content of SQSTM1/p62, a signaling adaptor protein that is degraded by autophagy and the levels of which decrease when autophagy is enhanced. Exposure of VL-17A cells to 50 mM ethanol, bafilomycin A1, or both elevated SQSTM1 levels by 37% over unexposed controls. This increase was similar to that after bafilomycin A1 treatment alone or to combined treatment with ethanol and bafilomycin A1, to suggest that ethanol treatment blocked the degradation of SQSTM1 (). To verify this, we measured both LC3-II and SQSTM1 levels after 50 mM ethanol exposure and compared them to that after treatment with nocodazole (NOC), a microtubule inhibitor, and to rapamycin (Rap), a MTORC1 inhibitor that activates autophagy. Both ethanol and NOC treatments enhanced LC3-II and SQSTM1 contents over controls while Rap exposure decreased both proteins, indicating that it accelerated autophagic flux and enhanced lysosomal proteolysis. The ethanol-induced rise in LC3-II and SQSTM1 proteins, exhibited greater similarity to those of nocodazole treatment than that after rapamycin treatment to suggest that ethanol exposure decreased AV degradation by the same magnitude as that of the microtubule inhibitor ().
Figure 3. Ethanol exposure influenced LC3-II and SQSTM1 flux, mimicked the effects of nocodazole, and decreased AV-lysosome colocalization. (A) Flux measurements of LC3-II in VL-17A cells treated with or without 50 mM ethanol in the presence or absence of bafilomycin A1 (added during the last 4 h) and quantified as described in Methods. (B) Quantification of SQSTM1 under the same conditions as described in (A). (C) LC3-II levels in VL-17A cells after 24 h exposure to 50 mM ethanol, 10 µM nocodazole or 100 nM rapamycin. (D) SQSTM1 levels in VL-17A cells after 24 h exposure to 50 mM ethanol, 10 µM nocodazole or 100 nM rapamycin. Data are mean values (± SEM) from quadruplicate flasks per treatment. (E) AVs, lysosomes and AV-lysosome colocalization in VL-17A cells. Cells were transduced with adenovirus encoding GFP-LC3 and exposed to zero or 50 mM ethanol for 24 h. Images were captured by confocal microscopy. Green puncta are AVs and red dots are lysosomes. Nuclei stained with Hoechst are shown in blue. (F) Cells exposed to zero or 50 mM ethanol for 24 h and immunostained for AVs and LAMP1. Green puncta are AVs and red dots are LAMP1. Nuclei stained with DAPI are shown in blue. (G) Lysosome (LAMP1) number, size and its proportion to total cell area (Fractional volume) in cells from (F). Numerical data given above each image are mean AV, or lysosome, (represented as LysoTracker Red or LAMP1) puncta numbers or co-localized AVs with lysosomes (merged images) per nucleus obtained from 600 to 800 cell images per treatment group. Letter superscripts that are different from each other indicate that the data are significantly different from each other between the treatment groups. Data with the same letter superscript are not significantly different.
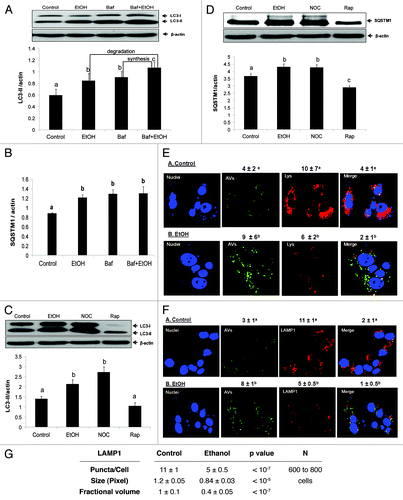
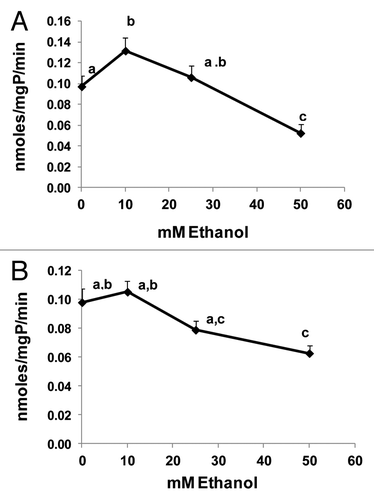
To further confirm that ethanol exposure, like nocodazole, affected AVs and lysosome trafficking, we examined AVs and their colocalization (fusion) with lysosomes after transducing VL-17A cells with an adenovirus bearing GFP-LC3 expression vector, exposing the cells to zero or 50 mM ethanol and staining them with LysoTracker red to measure AV-lysosome colocalization. These analyses revealed that, while ethanol exposure increased AVs by 2.2-fold, it simultaneously decreased lysosome numbers by 1.7-fold (). Moreover, ethanol exposure decreased AV-lysosome colocalization 2-fold () compared with controls, indicating that ethanol treatment likely affected AV-lysosome fusion. Decreased lysosome numbers and AV-lysosome fusion were further confirmed after immunostaining and colocalizing LAMP1 with AVs in VL-17A cells. Ethanol exposure elevated AV numbers 2.6-fold and, decreased LAMP1 (lysosome numbers) as well as AV-LAMP1 colocalization 2.2- and 2-fold, respectively, over untreated control cells (). Ethanol treatment also reduced lysosome (LAMP1) size 1.4-fold and its proportion to total cell area by 2.5-fold respectively, over untreated cells (). Twenty-four hours of ethanol exposure also caused a dose-dependent decrease in the specific activities of lysosomal CTSB and CTSL (cathepsins B and L) in VL-17A cells. Exposure to 10 or 25 mM ethanol either slightly increased or had no effect on either cathepsin activity, respectively. Compared with unexposed control cells, exposure to 50 mM ethanol decreased the activities of both proteases (), to suggest that such inhibition would also affect the catabolism of AV cargo.Citation11 Collectively, these data strongly suggest that, in addition to decreasing proteasome activity, 50 mM ethanol exposure decreases AV degradation by inhibiting the fusion of AVs with lysosomes and disrupting the activities of lysosomal proteases.
Figure 4. Ethanol dose-dependently affected cathepsin activity. (A) CTSB-specific activity, and (B) CTSL-specific activity in VL-17A cells after 24 h treatment with the indicated ethanol concentrations. Data are mean values (± SEM) from quadruplicate flasks per treatment. Letters that are different from each other indicate that the data are significantly different from each other. Data with the same letter are not significantly different.
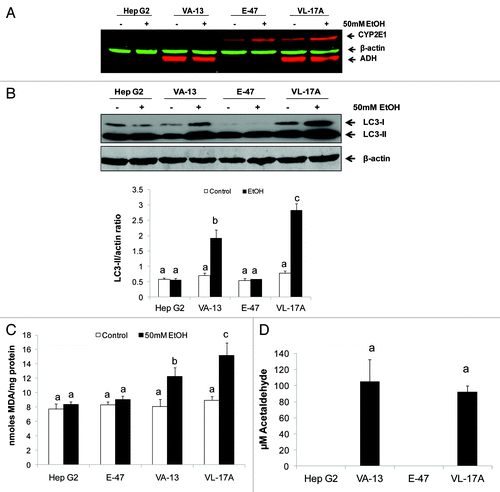
LC3-II content in four Hep G2 cell lines
We sought to identify the ethanol metabolite(s) that cause(s) the increase in AVs. To this end, parental, nonmetabolizing Hep G2 cells and their stable recombinant Hep G2 cell lines, VL-17A (ADH+/CYP2E1+), VA-13 (ADH+/ CYP2E1null) and E-47 (ADHnull/CYP2E1+) cells were exposed to zero or 50 mM ethanol for 24 h. The ADH and CYP2E1 phenotypes of the four cell lines were confirmed by western blot analyses, which also revealed that ethanol exposure enhanced the contents of CYP2E1 (2-fold vs controls; p < 0.05) and of ADH (1.4-fold vs controls; p < 0.05) in cells that expressed these enzymes (). Ethanol exposure increased LC3-II levels in ADH-expressing VL-17A and VA-13 cells, while Hep G2 and E-47 cells showed no such induction (). Ethanol-exposed VL-17A cells had 48% higher LC3-II content than identically treated ADH-expressing VA-13 cells (). The latter finding coincided with higher levels of lipid peroxides (MDA equivalents) in ethanol-exposed VL-17A cells, indicating elevated oxidant stress, compared with the other ethanol-treated cell lines (). Acetaldehyde was detectable only in the media from ADH-expressing cell lines (). Here, the acetaldehyde levels were ~100 µM, but we observed wide interexperimental variation, ranging from 81 to 400 µM, which we attribute, in part, to variations in the rate of acetaldehyde clearance by the cells. However, we observed no detectable acetaldehyde generation by nonexpressing HepG2 cells and only trace quantities of acetaldehyde generated by E-47 cells.
Figure 5. Ethanol exposure differentially affected LC3-II levels, MDA and acetaldehyde generation in four Hep G2 cell lines that differentially express ethanol metabolizing enzymes. (A) Representative western blot showing ADH and CYP2E1 protein phenotypes in the four cell lines indicated. (B) Representative western blot and mean densitometric ratios of LC3-II to ACTB in the four Hep G2 cell lines after 24 h exposure to zero or 50 mM ethanol. (C) Malondialdehyde levels in the four cell lines after exposure to zero or 50 mM ethanol. (D) Mean acetaldehyde levels in media from the Hep G2 cells lines after 24 h exposure to 50 mM ethanol. Data are mean values (± SEM) from 4 to16 culture flasks of each treatment group. Letters that are different from each other indicate that the data are significantly different from each other. Data with the same letter are not significantly different.
Acetaldehyde and AV induction
To confirm the identity of the ethanol metabolite responsible for AV induction we exposed VL-17A cells to compounds that either block the generation of reactive species or neutralize their effects. All agents were added either alone or with 50 mM ethanol to the culture medium. shows only the results of VL-17A cells treated with 50 mM ethanol plus the compound of interest (see figure legend). When we treated VL-17A cells with 4MP to block ethanol oxidation, or with glutathione ethyl-ester (GSH-EE) to scavenge reactive species, each compound completely prevented the ethanol-induced rise in LC3-II. Chlormethiazole (CMZ), a specific CYP2E1 inhibitor and trolox, a soluble vitamin E derivative that suppresses lipid peroxidation, each partially blocked induction of LC3-II by 33% and 18%, respectively. Together, these results further supported the notion that the product of ADH oxidation is the primary inducer of AV content in liver cells. The product(s) of CYP2E1 catalysis participated in this induction but did not initiate it.
Figure 6. Ethanol and acetaldehyde exposures enhanced LC3-II levels in ethanol-metabolizing and nonmetabolizing Hep G2 cells. (A) LC3-II content after 24 h treatment with 50 mM ethanol with or without 5 mM 4-methylpyrazole (4MP), 5mM glutathione ethyl ester (GSH-EE), 100 µM chlormethiazole (CMZ) or 20 µM trolox. (B) LC3II content after 24 h exposure of VL-17A cells or VA-13 cells to 50 mM ethanol or to 100 µM acetaldehyde. (C) LC3-II levels in Hep G2 cells after treatment for 1 h with 300 µM acetaldehyde. Data are mean densitometric ratios of LC3-II to ACTB from quadruplicate culture flasks. Letters that are different from each other indicate that the data are significantly different from each other. Data with the same letter are not significantly different.
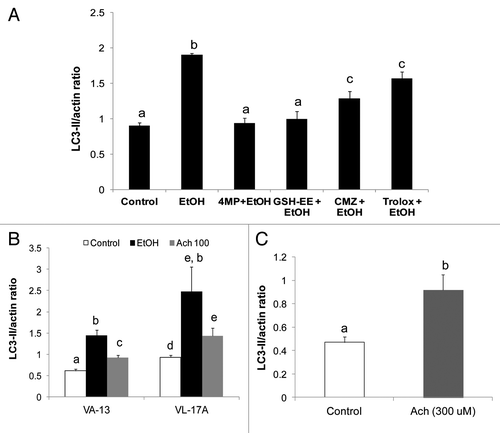
Because, acetaldehyde was implicated as the primary ethanol metabolite that initiated AV accumulation (), we exposed VL-17A and VA-13 cells directly to 100 µM acetaldehyde or to 50 mM ethanol for 24 h and detected significantly higher LC3-II levels in acetaldehyde-treated VL-17A cells compared with untreated controls. However, the LC3-II level was lower than that in 50 mM ethanol-exposed cells (). The attenuated response to direct acetaldehyde treatment reflected its rapid clearance by the cells, as it was undetectable 3 h after its addition (data not shown). In contrast, 50 mM ethanol steadily generated acetaldehyde levels as high as 300 µM or more in these experiments over the 24-h period. Inclusion of 10 mM acetate, the oxidation product of acetaldehyde had no effect on LC3-II content (data not shown), indicating that acetaldehyde formation, but not its oxidation is essential for LC3-II elevation in ethanol-metabolizing Hep G2 cells. When we exposed ethanol nonmetabolizing HepG2 cells directly to 300 µM acetaldehyde for 1 h, it elevated LC3-II content 1.9-fold over that in untreated controls ().
Discussion
When VL17A cells, which stably express ADH and CYP2E1, were exposed to increasing ethanol concentrations, there was a dose-dependent rise in LC3-II content. At lower (5 to10 mM) concentrations, ethanol was principally oxidized by ADH to acetaldehyde, which was undetectable because it was rapidly oxidized to acetate or it covalently bound to cellular proteins.Citation12 Exposure of cells to 25 and 50 mM ethanol accelerated ethanol oxidation, causing a rise in acetaldehyde production. While exposure to all ethanol concentrations tested here enhanced cellular LC3-II content over controls, only exposure to higher (25 and 50 mM) ethanol concentrations inhibited proteasome activity, which, in turn, augmented LC3-II content. The ethanol-induced rise in AVs was caused initially by an increase in AV biogenesis, as judged by higher levels of LC3B mRNA, which likely resulted via a release from MTOR suppression, as judged by decreased phosphorylation of the MTOR target, RPS6K, thereby inducing alterations in AV flux. The latter measurement also implied decreased LC3-II and SQSTM1 degradation, which was verified when we quantified LC3-II and SQSTM1 levels after exposing VL-17A cells to ethanol, nocodazole or rapamycin. Ethanol or nocodazole treatment enhanced the levels of both LC3-II and SQSTM1 proteins, to suggest that treatment with each agent had the same effect on the degradative phase of autophagy. In contrast, rapamycin, an autophagy inducer, enhanced the disappearance of LC3-II and SQSTM1 indicating that it accelerated autophagic flux, as expected. Our microscopy analyses confirmed that ethanol exposure increased AV number and size while it decreased these parameters in lysosomes. Increased AV size likely reflects ballooning of these organelles, due to decreased degradation of their cargo. Conversely, the reduction in lysosome number and size indicates that ethanol exposure decreased lysosome biogenesis, which is consistent with our earlier studies in rats.Citation6 The ethanol-elicited reduction in lysosome size is consistent with lower CTSB and CTSL activities () and leads us to suggest that ethanol oxidation caused not only decreased lysosome production but also generated faulty lysosomes.
Our confocal microscopy studies revealed that ethanol exposure decreased AV-lysosome colocalization (fusion). These results strongly implied that ethanol metabolism impaired AV trafficking to lysosomes. The latter finding supported the notion that ethanol metabolism likely affected the microtubule network and had effects similar to nocodazole to decrease AV degradation. Further evidence for decreased AV degradation was again indicated by decreased CTSB- and CTSL-specific activities in ethanol-treated cells. SQSTM1 protein is degraded by the proteasome and is also elevated by higher levels of its mRNA in response to oxidative stress through the NFE2L2/NRF2 transcription factor.Citation13 Even though ethanol oxidation induces oxidant stress and downregulates proteasome activity, we associate a significant part of the rise in SQSTM1 content to its decreased degradation due to defects in the distal phases of autophagy. However, we recognize that SQSTM1, like LC3-II, is a proteasome substrateCitation14 and we could not exclude this possibility under our experimental conditions.
The involvement of acetaldehyde in ethanol-elicited AV induction was clearly illustrated by the differential effects of ethanol exposure in native and recombinant HepG2 cell lines. Ethanol exposure increased LC3-II content only in VA-13 and VL-17A cells. Each cell line produces acetaldehyde from ADH catalysis. Acetaldehyde production from ethanol oxidation proved to be essential for AV induction in cells. This was strengthened by the finding that direct exposure of cells to acetaldehyde induced LC3-II not only in ADH-expressing cells but also in nonexpressing Hep G2 cells, which do not oxidize ethanol to acetaldehyde (). Further supporting this notion were data showing that the blockade of ethanol oxidation by 4MP completely prevented the rise in LC3-II as did, GSH-EE. The latter compound likely obstructed the ethanol-elicited increase in LC3-II by binding to and neutralizing metabolically-generated acetaldehyde, thereby blocking its ability to enhance LC3 synthesis or disrupt autophagic flux. Ding et al. similarly demonstrated that treatment of hepatocytes with N-acetylcysteine (NAC), a glutathione precursor, blocks ethanol-elicited AV induction, to suggest that NAC acts by a mechanism similar to that of GSH-EE.Citation7
Following ethanol treatment, LC3-II content in VL-17A cells was 48% higher than in VA-13 cells, indicating that combined catalysis by ADH and CYP2E1 in VL-17A cells increased LC3-II levels over those induced by ADH alone in VA-13 cells. Further, we showed that CMZ or trolox treatment of ethanol-exposed VL-17A cells only partially blocked alcohol-induced LC3-II content, to suggest that CYP2E1-generated metabolites augment LC3-II levels during ethanol exposure, but only after initiation of such induction by acetaldehyde. Acetaldehyde’s ability to form adducts with proteinsCitation15 has been implicated in both the negative and positive regulation of the transcription factors PPARA and SREBPF1, respectively.Citation16,Citation17 There is also recent evidence that another transcription factor, TFEB, regulates autophagy and lysosome biogenesis through its interaction with the CLEAR (Coordinated Lysosomal Expression and Regulation) gene network.Citation18 Therefore, it is plausible that acetaldehyde binding to autophagy-related proteins or to TFEB alters their activities by activating autophagy-related genes, especially during the early stages of ethanol exposure when LC3B mRNA was higher than at later times ().
Acetaldehyde generation by ethanol-treated VL-17A and VA-13 cells was associated with higher LC3-II levels, which, in part, reflects faulty AV trafficking to lysosomes. Acetaldehyde binds to a highly reactive lysine residue on the α tubulin subunit, thereby slowing its association with the tubulin β subunit and their subsequent polymerization into microtubules.Citation19 The latter comprise the principal cellular transit system by which AVs fuse with lysosomes for subsequent AV degradation. Our findings suggest that acetaldehyde generation via ADH initiated this process and its action was augmented by reactive species derived from enhanced CYP2E1 catalysis. Such reactive products include malondialdehyde (MDA), a product of lipid peroxidation and which was clearly elevated by ethanol oxidation in VA-13 cells and further increased in VL-17A cells (). MDA reacts with acetaldehyde to form distinct species called malondialdehyde-acetaldehyde, also known as MAA.Citation20,Citation21 The latter compound rapidly reacts with proteins to form covalent adducts on proteins that are more bulky than those with acetaldehyde alone. MAA adducts are also immunochemically detectable in livers of ethanol-fed animalsCitation20 and antibodies against such adducts are detectable in patients with alcoholic liver disease.Citation21 Our observations here were consistent with a scenario in which MDA could augment acetaldehyde-induced formation of AVs by reacting with acetaldehyde to form MAA, which, in turn, may similarly react with tubulin to prevent AV trafficking to lysosomes, resulting in enhanced AV content. This hypothesis, however, must be rigorously tested.
Of equal importance are reports of ethanol-induced hyper-acetylation (addition of acetyl groups to susceptible lysine residues) of tubulin as well as other cellular proteins. Such chemical modifications seem to explain ethanol-induced alterations in protein trafficking.Citation22-Citation24 There appears to be a close association between autophagy and protein acetylation, although the results from different experimental systems are mixed.Citation25-Citation27 Our preliminary analyses of autophagy using one de-acetylation inhibitor, indicate that this compound enhances LC3 content to a degree that is similar to that caused by ethanol exposure (unpublished data). Thus protein acetylation is an alternative post-translational mechanism that may have a role in ethanol-induced disruption of autophagy.
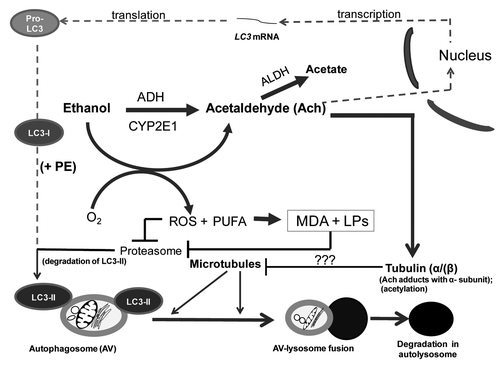
In summary, ADH-catalyzed ethanol oxidation generated acetaldehyde, which enhanced AV content in ethanol-metabolizing VL-17A and VA-13 cells. Such induction occurred at multiple levels: There was enhancement of AV (LC3-II) formation brought about by MTOR suppression due to ethanol oxidation, confirming previous findings.Citation7 There was increased LC3B mRNA, to suggest enhanced LC3B gene expression. Ethanol-induced inhibition of proteasome activity further contributed to LC3-II elevation by stabilizing the protein from degradation. Finally, there was disruption of autophagic flux due, in part to downstream defects in the degradative phase of autophagy. Our current studies are focused on the temporal sequence of these events, as we have made similar observations in animal studies.Citation8 We postulate that after acute ethanol consumption (binge drinking) there is enhanced AV biogenesis, while long-term continuous (chronic) drinking causes a downstream blockade of AV degradation due to decreased proteasome activity, reduced lysosome biogenesis, compromised lysosomal hydrolytic function and faulty AV trafficking. From the data presented here, depicts our current view of how short- and long-term ethanol oxidation affects autophagy in liver cells.
Figure 7. Multilevel regulation of autophagosome content by ethanol oxidation liver cells. During acute (or early) ethanol administration, metabolically-derived acetaldehyde (Ach) enhances AV formation in liver cells by increasing the level of LC3B mRNA (presumably by enhanced transcription). LC3B mRNA is then translated into pro-LC3, which matures to LC3-I. The latter is lipidated with phosphatidylethanolamine (PE) to form LC3-II, which attaches to the AV membrane. The AV is trafficked by microtubules to the lysosome for degradation. During habitual (chronic) ethanol exposure, CYP2E1 is induced, generating MDA and other lipid peroxides (LPs), which inhibit proteasome activity and stabilize LC3-II from degradation. Acetaldehyde (Ach) generated by ethanol oxidation forms adducts with proteins, including the α tubulin subunit.Citation19 We hypothesize that formation of Ach-α-tubulin adducts or tubulin acetylation block or stabilize the polymerization of microtubules (see “???” in lower half of figure). Either change will prevent the fusion of AVs with lysosomes, thereby causing ethanol-induced proteopathy and steatosis in liver cells.
Our findings also provide a unique perspective in understanding how metabolism of toxic substances such as ethanol substantially alters the intracellular catabolism of macromolecular species. Accumulation of undegraded molecules is the pathological basis for the hereditary lysosomal storage diseases, as well as normal aging.Citation28,Citation29 Here, an acquired defect caused by ethanol oxidation, contributes to macromolecular accumulation, thereby increasing the likelihood for intracellular aggregate formation that likely hastens cell injury and death after years of heavy drinking.
Materials and Methods
Reagents
Antibodies directed against LC3 and to the phosphorylated and unphosphorylated forms of RPS6K/p70S6 kinase were from Cell Signaling Technology, Inc. (9205 and 9202, respectively). Anti-SQSTM1 was purchased from Medical and Biological Laboratories LTD (PM045). Anti-CYP2E1 (C9095-15M) and anti-ACTB (A-5316) were from Calbiochem and Sigma Chemical Co., respectively. Anti-LAMP1 (ab25630) was from Abcam. Alexa Fluor 568 (A-11031) and 488 (A11070) was from Invitrogen. Anti-ADH was a gift from Dr. Michael Felder, (University of South Carolina). Protease inhibitor cocktail (P2714), 4-methylpyrazole (4MP) (M-1387), glutathione ethyl-ester (GSH-EE; G-1404), chlormethiazole (CMZ; C-1240), trolox (6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid; 238813) and other specialized reagents were purchased from Sigma-Aldrich.
Cell lines
Parental Hep G2 (ADHnull/CYP2E1null) hepatoblastoma cells were purchased from the American Type Culture Collection (HB-8065). Dr. Arthur Cederbaum (Mt. Sinai Medical Center) provided us with E-47 cells that are stably-transfected Hep G2 cells that constitutively express human CYP2E1.Citation30 VA-13 and VL-17A cells are both recombinant Hep G2 cells that stably express either ADH alone (VA-13) or both CYP2E1 and ADH (VL-17A). Both the latter two cell lines were established at this VA Medical Center and previously characterized.Citation31,Citation32
Cell treatments
We cultured Hep G2 cells and their recombinant cell lines in high glucose Dulbecco’s modified Eagle’s medium (DMEM, 12800-017) supplemented with 10% FBS, 100 units penicillin/ml and 100 µg streptomycin/ml (Life Technologies, 15140-122). E-47cells were grown in the presence of the selective antibiotic, G418 sulfate (400 µg/ml; Gemini Bio-Products, 400-113). VA-13 cells were grown in the presence of zeocin (400 µg/ml; Life Technologies, R25001). VL-17A cells were cultured with both antibiotics at the same concentrations. For experiments, we seeded cells into T-12.5 or T-25 flasks at densities of 6 × 105 or 1 × 106 cells per flask, respectively. Before treatment, we washed the cells and replaced the medium with serum-free DMEM with or without ethanol and/or other agents as described in the figures and text. At harvest, we removed aliquots of culture media and subjected each to head-space gas chromatographyCitation33 to quantify ethanol and acetaldehyde. After washing, we scraped the cells into PBS, pelleted them by centrifugation and resuspended them in PBS containing 1× protease inhibitor cocktail. Cells were then sonically disrupted and then frozen as lysates at −70°C.
Cell transfections and microscopy
Cells were transduced with adenovirus that carried GFP fused to the human LC3B gene (courtesy of Dr. Xiao-Min Yin, Indiana University). Untransduced and transduced cells were grown on coverslips or a chambered cover glass and treated as described in results. After treatment, cells were stained for lysosomes and nuclei using lysotracker red and Hoechst respectively in the chambered cover glass. For AVs and LAMP1, cells were stained with rabbit anti-LC3 and mouse anti-LAMP1 respectively, followed by incubation of goat anti-rabbit (Alexa Fluor 488) and goat anti-mouse secondary antibody (Alexa Fluor 568). We digitally acquired images with a Zeiss confocal fluorescent microscope (Carl Zeiss, LSM 510 Meta Confocal Laser Scanning Microscope) and quantified the number of AVs as GFP-LC3 puncta, lysosome and LAMP1 numbers in multiple images using NIH Image J analyzer software. Particles were quantified with the ‘analyze particles’ function in thresholded images with size (pixel2) settings from 0.1 to 10 and circularity from 0 to 1.Citation34 Numerical data presented above each image are expressed as puncta per nucleus.
Immunochemical protein quantification
After subjecting cell lysates to SDS-PAGE and transferring proteins onto nitrocellulose membranes, we quantified specific proteins by incubating with primary antibodies. Membranes were then incubated with either HRP-conjugated or fluorescent-tagged secondary antibodies and blots were developed using enhanced chemiluminescence (ECL) (Thermo Scientific, 34080) or by the Odyssey® infrared imaging system (Li-Cor, Inc.). Each immunoreactive protein band was quantified by densitometry using either Quantity One software from Bio-Rad or the Li-Cor software (LI-COR Biosciences). Each protein of interest was corrected for its protein load by calculating the densitometric ratio of each protein of interest to that of β-actin (ACTB).
AV flux
We quantified LC3-II as the marker for AVs by exposing cells to zero or 50 mM ethanol for 24 h. Four h before cell harvest, we added bafilomycin A1, a lysosome inhibitor, to half the culture flasks (final concentration: 100 nM; Sigma-Aldrich, B1793). The other half of the culture flasks received no bafilomycin A1. Following cell harvest, we quantified LC3-II and the protein, SQSTM1 in cell lysates by immunoblot analysis.
Lipid peroxides
These were quantified in cell lysates as thiobarbituric acid reactive material and quantified spectrophotometrically as malondialdehyde (MDA) equivalents, using a MDA standard.Citation35
Proteasome and lysosomal cathepsin assays
The chymotrypsin-like activity of the 20S proteasome was assayed as described previously.Citation36 Activities of CTSB and CTSL were assayed fluorometrically as published.Citation37
Statistical analysis
All data are presented as mean values ± SEM. We determined statistical significance between two groups by Student’s t-test and among multiple groups by one way analysis of variance, with a Newman-Keuls post-hoc analysis. A probability value p ≤ 0.05 is statistically significant.
| Abbreviations: | ||
| LC3 | = | microtubule-associated protein 1 light chain 3 |
| ADH | = | alcohol dehydrogenase |
| CYP2E1 | = | cytochrome P450 2E1 |
| SDS-PAGE | = | sodium dodecyl sulfate |
| 4-MP | = | 4-methylpyrazole |
| DAPI | = | (4’,6-Diamidine-2’-phenylindole dihydrochloride) EBSS, Earle’s Balanced Salt Solution |
| CMZ | = | chlormethiazole |
| GSH-EE | = | glutathione ethyl-ester |
| RNA | = | ribonucleic acid |
| LAMP1 | = | lysosomal-associated membrane protein 1 |
| MTORC1 | = | mechanistic target of rapamycin complex 1 |
| GFP | = | green fluorescent protein |
| PPARA | = | peroxisome proliferator-activated receptor-α |
| SREBF1 | = | sterol regulatory element binding transcription factor 1 |
| MDA | = | malondialdehyde |
| MAA | = | malondialdehyde-acetaldehyde |
Acknowledgments
Resources, Facilities and Grant Support: Financial support for this project was provided by institutional funds from the Section of Gastroenterology and Hepatology, Department of Internal Medicine, and a Dean's Reviewed Research Grant from the University of Nebraska Medical Center. The data presented are results of work supported with resources and use of facilities at the Nebraska-Western Iowa Health Care System of the Department of Veterans Affairs Veterans Health Administration, Office of Research and Development.
Disclaimer: The contents of this paper do not represent the views of the Department of Veterans Affairs or the United States Government.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Baraona E, Leo MA, Borowsky SA, Lieber CS. Alcoholic hepatomegaly: accumulation of protein in the liver. Science 1975; 190:794 - 5; http://dx.doi.org/10.1126/science.1198096; PMID: 1198096
- Donohue TM Jr., Zetterman RK, Tuma DJ. Effect of chronic ethanol administration on protein catabolism in rat liver. Alcohol Clin Exp Res 1989; 13:49 - 57; http://dx.doi.org/10.1111/j.1530-0277.1989.tb00283.x; PMID: 2646978
- Donohue TM Jr., McVicker DL, Kharbanda KK, Chaisson ML, Zetterman RK. Ethanol administration alters the proteolytic activity of hepatic lysosomes. Alcohol Clin Exp Res 1994; 18:536 - 41; http://dx.doi.org/10.1111/j.1530-0277.1994.tb00906.x; PMID: 7943651
- Kharbanda KK, McVicker DL, Zetterman RK, MacDonald RG, Donohue TM Jr.. Flow cytometric analysis of vesicular pH in rat hepatocytes after ethanol administration. Hepatology 1997; 26:929 - 34; http://dx.doi.org/10.1002/hep.510260419; PMID: 9328315
- Kharbanda KK, McVicker DL, Zetterman RK, Donohue TM Jr.. Ethanol consumption reduces the proteolytic capacity and protease activities of hepatic lysosomes. Biochim Biophys Acta 1995; 1245:421 - 9; http://dx.doi.org/10.1016/0304-4165(95)00121-2; PMID: 8541322
- Kharbanda KK, McVicker DL, Zetterman RK, Donohue TM Jr.. Ethanol consumption alters trafficking of lysosomal enzymes and affects the processing of procathepsin L in rat liver. Biochim Biophys Acta 1996; 1291:45 - 52; http://dx.doi.org/10.1016/0304-4165(96)00043-8; PMID: 8781524
- Ding WX, Li M, Chen X, Ni HM, Lin CW, Gao W, et al. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology 2010; 139:1740 - 52; http://dx.doi.org/10.1053/j.gastro.2010.07.041; PMID: 20659474
- Thomes PG, Trambly CS, Thiele GM, Duryee MJ, Fox HS, Haorah J, et al. Proteasome activity and autophagosome content in liver are reciprocally regulated by ethanol treatment. Biochem Biophys Res Commun 2012; 417:262 - 7; http://dx.doi.org/10.1016/j.bbrc.2011.11.097; PMID: 22142844
- Gao Z, Gammoh N, Wong PM, Erdjument-Bromage H, Tempst P, Jiang X. Processing of autophagic protein LC3 by the 20S proteasome. Autophagy 2010; 6:126 - 37; http://dx.doi.org/10.4161/auto.6.1.10928; PMID: 20061800
- Rubinsztein DC, Cuervo AM, Ravikumar B, Sarkar S, Korolchuk V, Kaushik S, et al. In search of an “autophagomometer”. Autophagy 2009; 5:585 - 9; http://dx.doi.org/10.4161/auto.5.5.8823; PMID: 19411822
- Repnik U, Stoka V, Turk V, Turk B. Lysosomes and lysosomal cathepsins in cell death. Biochim Biophys Acta 2012; 1824:22 - 33; http://dx.doi.org/10.1016/j.bbapap.2011.08.016; PMID: 21914490
- Donohue TM Jr., Tuma DJ, Sorrell MF. Binding of metabolically derived acetaldehyde to hepatic proteins in vitro. Lab Invest 1983; 49:226 - 9; PMID: 6876749
- Fujita K, Maeda D, Xiao Q, Srinivasula SM. Nrf2-mediated induction of p62 controls Toll-like receptor-4-driven aggresome-like induced structure formation and autophagic degradation. Proc Natl Acad Sci U S A 2011; 108:1427 - 32; http://dx.doi.org/10.1073/pnas.1014156108; PMID: 21220332
- Fusco C, Micale L, Egorov M, Monti M, D’Addetta EV, Augello B, et al. The E3-ubiquitin ligase TRIM50 interacts with HDAC6 and p62, and promotes the sequestration and clearance of ubiquitinated proteins into the aggresome. PLoS One 2012; 7:e40440; http://dx.doi.org/10.1371/journal.pone.0040440; PMID: 22792322
- Tuma DJ, Hoffman T, Sorrell MF. The chemistry of acetaldehyde-protein adducts. Alcohol Alcohol Suppl 1991; 1:271 - 6; PMID: 1845549
- Galli A, Pinaire J, Fischer M, Dorris R, Crabb DW. The transcriptional and DNA binding activity of peroxisome proliferator-activated receptor alpha is inhibited by ethanol metabolism. A novel mechanism for the development of ethanol-induced fatty liver. J Biol Chem 2001; 276:68 - 75; http://dx.doi.org/10.1074/jbc.M008791200; PMID: 11022051
- You M, Fischer M, Deeg MA, Crabb DW. Ethanol induces fatty acid synthesis pathways by activation of sterol regulatory element-binding protein (SREBP). J Biol Chem 2002; 277:29342 - 7; http://dx.doi.org/10.1074/jbc.M202411200; PMID: 12036955
- Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, et al. TFEB links autophagy to lysosomal biogenesis. Science 2011; 332:1429 - 33; http://dx.doi.org/10.1126/science.1204592; PMID: 21617040
- Smith SL, Jennett RB, Sorrell MF, Tuma DJ. Acetaldehyde substoichiometrically inhibits bovine neurotubulin polymerization. J Clin Invest 1989; 84:337 - 41; http://dx.doi.org/10.1172/JCI114159; PMID: 2500458
- Tuma DJ, Thiele GM, Xu D, Klassen LW, Sorrell MF. Acetaldehyde and malondialdehyde react together to generate distinct protein adducts in the liver during long-term ethanol administration. Hepatology 1996; 23:872 - 80; http://dx.doi.org/10.1002/hep.510230431; PMID: 8666344
- Tuma DJ. Role of malondialdehyde-acetaldehyde adducts in liver injury. Free Radic Biol Med 2002; 32:303 - 8; http://dx.doi.org/10.1016/S0891-5849(01)00742-0; PMID: 11841919
- Shepard BD, Tuma DJ, Tuma PL. Chronic ethanol consumption induces global hepatic protein hyperacetylation. Alcohol Clin Exp Res 2010; 34:280 - 91; http://dx.doi.org/10.1111/j.1530-0277.2009.01091.x; PMID: 19951295
- Shepard BD, Tuma DJ, Tuma PL. Lysine acetylation induced by chronic ethanol consumption impairs dynamin-mediated clathrin-coated vesicle release. Hepatology 2012; 55:1260 - 70; http://dx.doi.org/10.1002/hep.24785; PMID: 22095875
- Shepard BD, Tuma PL. Alcohol-induced protein hyperacetylation: mechanisms and consequences. World J Gastroenterol 2009; 15:1219 - 30; http://dx.doi.org/10.3748/wjg.15.1219; PMID: 19291822
- Cao DJ, Wang ZV, Battiprolu PK, Jiang N, Morales CR, Kong Y, et al. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc Natl Acad Sci U S A 2011; 108:4123 - 8; http://dx.doi.org/10.1073/pnas.1015081108; PMID: 21367693
- Chen MY, Liao WS, Lu Z, Bornmann WG, Hennessey V, Washington MN, et al. Decitabine and suberoylanilide hydroxamic acid (SAHA) inhibit growth of ovarian cancer cell lines and xenografts while inducing expression of imprinted tumor suppressor genes, apoptosis, G2/M arrest, and autophagy. Cancer 2011; 117:4424 - 38; http://dx.doi.org/10.1002/cncr.26073; PMID: 21491416
- Yi C, Yu L. How does acetylation regulate autophagy?. Autophagy 2012; 8:1529 - 30; http://dx.doi.org/10.4161/auto.21156; PMID: 22732483
- Bargal R, Bach G. Mucolipidosis type IV: abnormal transport of lipids to lysosomes. J Inherit Metab Dis 1997; 20:625 - 32; http://dx.doi.org/10.1023/A:1005362123443; PMID: 9323557
- Cuervo AM, Dice JF. When lysosomes get old. Exp Gerontol 2000; 35:119 - 31; http://dx.doi.org/10.1016/S0531-5565(00)00075-9; PMID: 10767573
- Marí M, Cederbaum AI. CYP2E1 overexpression in HepG2 cells induces glutathione synthesis by transcriptional activation of gamma-glutamylcysteine synthetase. J Biol Chem 2000; 275:15563 - 71; http://dx.doi.org/10.1074/jbc.M907022199; PMID: 10748080
- Clemens DL, Forman A, Jerrells TR, Sorrell MF, Tuma DJ. Relationship between acetaldehyde levels and cell survival in ethanol-metabolizing hepatoma cells. Hepatology 2002; 35:1196 - 204; http://dx.doi.org/10.1053/jhep.2002.32668; PMID: 11981770
- Donohue TM, Osna NA, Clemens DL. Recombinant Hep G2 cells that express alcohol dehydrogenase and cytochrome P450 2E1 as a model of ethanol-elicited cytotoxicity. Int J Biochem Cell Biol 2006; 38:92 - 101; http://dx.doi.org/10.1016/j.biocel.2005.07.010; PMID: 16181800
- Eriksson CJ, Sippel HW, Forsander OA. The occurrence of acetaldehyde binding in rat blood but not in human blood. FEBS Lett 1977; 75:205 - 8; http://dx.doi.org/10.1016/0014-5793(77)80087-2; PMID: 852583
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature 2009; 458:1131 - 5; http://dx.doi.org/10.1038/nature07976; PMID: 19339967
- Buttkus H, Rose R. Amine malondialdehyde condensation products and their relative color contribution in the thiobarbituric acid test. J Am Oil Chem Soc 1972; 49:440 - 3; http://dx.doi.org/10.1007/BF02582530
- Osna NA, White RL, Todero S, McVicker BL, Thiele GM, Clemens DL, et al. Ethanol-induced oxidative stress suppresses generation of peptides for antigen presentation by hepatoma cells. Hepatology 2007; 45:53 - 61; http://dx.doi.org/10.1002/hep.21442; PMID: 17187415
- Curry-McCoy TV, Osna NA, Nanji AA, Donohue TM Jr.. Chronic ethanol consumption results in atypical liver injury in copper/zinc superoxide dismutase deficient mice. Alcohol Clin Exp Res 2010; 34:251 - 61; http://dx.doi.org/10.1111/j.1530-0277.2009.01088.x; PMID: 19951287