Abstract
IFNB1/interferon (IFN)-β belongs to the type I IFNs and exerts potent antiproliferative, proapoptotic, antiangiogenic and immunemodulatory functions. Despite the beneficial effects of IFNB1 in experimental breast cancers, clinical translation has been disappointing, possibly due to induction of survival pathways leading to treatment resistance. Defects in autophagy, a conserved cellular degradation pathway, are implicated in numerous cancer diseases. Autophagy is induced in response to cancer therapies and can contribute to treatment resistance. While the type II IFN, IFNG, which in many aspects differs significantly from type I IFNs, can induce autophagy, no such function for any type I IFN has been reported. We show here that IFNB1 induces autophagy in MCF-7, MDAMB231 and SKBR3 breast cancer cells by measuring the turnover of two autophagic markers, MAP1LC3B/LC3 and SQSTM1/p62. The induction of autophagy in MCF-7 cells occurred upstream of the negative regulator of autophagy MTORC1, and autophagosome formation was dependent on the known core autophagy molecule ATG7 and the IFNB1 signaling molecule STAT1. Using siRNA-mediated silencing of several core autophagy molecules and STAT1, we provide evidence that IFNB1 mediates its antiproliferative effects independent of autophagy, while the proapoptotic function of IFNB1 was strongly enhanced in the absence of autophagy. This suggests that autophagy induced by IFNB1 promoted survival, which might contribute to tumor resistance against IFNB1 treatment. It may therefore be clinically relevant to reconcile a role for IFNB1 in the treatment of breast cancer with concomitant inhibition of autophagy.
Introduction
Type I interferons (IFNs); IFNAs/IFN-αs and IFNB1/IFN-β and the type II IFN; IFNG/IFN-γ, trigger diverse biological responses in target cells, which include antiviral, immunemodulatory, antiangiogenic, antiproliferative and proapoptotic effects.Citation1-Citation4 All type I IFNs bind a common IFN receptor complex composed of two transmembrane proteins IFNAR1 and IFNAR2, which are constitutively associated with the nonreceptor protein-tyrosine kinases TYK2 and JAK1, respectively.Citation5 Upon binding of type I IFNs to their receptor, several intracellular signaling pathways are activated, which in addition to the classical JAK, signal transducer and activator of transcription (STATs) cascade that mediates gene transcription of the interferon stimulated genes (ISGs),Citation6 also include the phosphatidylinositol 3-kinase and MAPK11/12/13/14 mitogen-activated protein kinase pathways.Citation5 Together these signaling cascades coordinate the pleiotropic effects of type I IFNs. In contrast, IFNG binds to a distinct receptor complex, composed of IFNGR1 and IFNGR2 that associate with JAK1 and JAK2, respectively.Citation1 Importantly, although type I and type II IFNs execute some similar biological effects they also possess unique and even opposing properties. IFNG is primarily a proinflammatory cytokine produced by T cells and natural killer cells, while type I IFNs can be produced by virtually all cells and are considered largely antiinflammatory.Citation1 Accordingly, IFNG is involved in the pathogenesis of the inflammatory disease multiple sclerosis (MS), while IFNB1 is a major therapeutic agent used to treat MS.Citation7,Citation8 Moreover, the antiproliferative and proapoptotic responses to the different IFN species vary considerably among different cancer cell lines with IFNB1 in general being more potent than IFNAs and IFNG.Citation9-Citation14 Furthermore, IFNG may in some cases even enhance cancer progression.Citation15
Although IFNs have been used with success to treat some hematological malignancies and solid cancers, they have displayed poor and transient effects in breast cancer patients.Citation16 This is despite IFNB1’s cytotoxic effects on breast cancer cells in vitro,Citation13,Citation17-Citation21 and its ability to reduce both breast cancer growth and tumor-related angiogenesis in vivo.Citation19,Citation21,Citation22 The unsatisfactory clinical translation of type I IFN effects in some cancerous diseases could reflect activation of survival pathways leading to tumor resistance to IFN treatment.Citation2,Citation23
Macroautophagy (hereafter referred to as autophagy) is an evolutionarily conserved cellular survival mechanism, in which cytoplasmic constituents such as long-lived proteins, protein aggregates and entire organelles are targeted to lysosomes for degradation by means of double-membrane vesicles, called autophagosomes.Citation24 Autophagy occurs at a low basal level under normal conditions to maintain cellular homeostasis,Citation25 but is highly induced by many stimuli including starvation and metabolic stress.Citation26 Dysfunction in the autophagic pathway is implicated in a growing number of diseases, including cancer.Citation27,Citation28 However, the role of autophagy in relation to cancer and tumorigenesis is complex and highly context dependent.
Substantial evidence supports a role for autophagy in prevention of tumorigenesis by maintaining genomic integrity and decreasing DNA damage,Citation29,Citation30 restricting tissue necrosis and inflammation,Citation31 and degrading specific proteins or organelles that otherwise would lead to aberrant signaling pathways and increased oxidative stress.Citation32 It is also possible that autophagy-mediated cell death in some specific settings can contribute to tumor suppression.Citation33 In contrast, transformed cells often have elevated levels of autophagy, which promotes their metabolisms and is necessary for their continued proliferation and survival both in vitro and in vivo.Citation34-Citation36 In the hostile tumor microenvironment lacking nutrients and oxygen, cancer cells can also activate autophagy as an essential survival pathway to cope with periods of starvation and hypoxia.Citation27,Citation37 Genotoxic and metabolic stresses conferred by commonly used anticancer therapies may further enhance the autophagic activity of cancer cells, and it is believed that this response contributes to treatment resistance as inhibition of autophagy can potentiate the therapeutic efficiency of anticancer drugs.Citation37,Citation38 Thus, while autophagy can act as an initial barrier to tumorigenesis, it can also support progression and subsequent maintenance of already established cancers.
Several reports have shown that IFNG induces autophagy in various cell types with different biological outcomes.Citation39-Citation42 Interestingly, in hepatocellular carcinomas (HCC) IFNG-induced autophagy contributes to cell death rather than treatment resistance.Citation43 To our knowledge, direct regulation of autophagy by type I IFNs has not been reported previously.
In the present study we report for the first time that IFNB1 can induce autophagy in breast cancer cells using several different methods that measure steady-state autophagy levels or autophagic flow. We further show that IFNB1 has antiproliferative and proapoptotic effects in MCF-7 breast cancer cells. To elucidate the biological relevance of IFNB1-induced autophagy, we silenced several core autophagy molecules by siRNAs and showed that IFNB1-induced autophagy did not modulate its antiproliferative properties, but significantly reduced its proapoptotic capacity. We further showed that STAT1 was important for IFNB1-induced autophagy, thus also decreasing the proapoptotic effect of IFNB1. We moreover investigated molecular events underlying these biological effects of IFNB1.
Results
IFNB1 induced autophagy in MCF-7 breast cancer cells
In order to study the ability of IFNB1 to induce autophagy in breast cancer cells, we used MCF-7 breast carcinoma cells stably expressing a fusion protein consisting of enhanced green fluorescence protein (eGFP) and MAP1LC3B/LC3.Citation44 The MCF-7 cells responded to human recombinant IFNB1 treatment with a dose-dependent phosphorylation of tyrosine residue 701 in STAT1, which occurred immediately after activation of the IFN receptor complex ().Citation5 MAP1LC3B is a widely used marker in autophagy research, which upon autophagy induction is converted from a cytosolic MAP1LC3B-I form to an autophagosome-bound MAP1LC3B-II form by lipidation with phosphatidylethanolamine.Citation45,Citation46 We tested whether IFNB1 could induce MAP1LC3B conversion evidenced by further migration of the hydrophobic MAP1LC3B-II form in SDS-PAGE by western blotting. For this purpose, we treated the MCF-7-eGFP-MAP1LC3B cells for 24 h with IFNB1 or rapamycin, a well-characterized inducer of autophagy that inhibits the activity of mechanistic target of rapamycin complex 1 (MTORC1).Citation24 Notably, IFNB1 triggered an induction of MAP1LC3B-II levels comparable to that seen in rapamycin-treated cells (). The accumulation of MAP1LC3B-II was accompanied by the formation of eGFP-MAP1LC3B-positive puncta (autophagic vesicles) in the cytosol of IFNB1- and rapamycin-treated cells (). The number of cells with more than 5, 10 and 15 eGPF-MAP1LC3B puncta/cell was counted automatically. Upon IFNB1 treatment for 24 h, similar significant results were achieved under all investigated conditions ().
Figure 1. IFNB1 induced autophagy in MCF-7 breast cancer cells by mediating MAP1LC3B conversion. (A) MCF-7 cells were IFNB1 responsive. MCF-7 eGFP-MAP1LC3B cells were cultured for 24 h, serum starved 3 h and stimulated with control medium or 100 or 1000 U/ml of IFNB1 for 30 min. Western blot analysis was performed for p-STAT1 and STAT1 protein. (B and C) IFNB1 induced MAP1LC3B-I to MAP1LC3B-II conversion. (B) MCF-7 eGFP-MAP1LC3B cells were cultured for 24 h and then treated with control medium, 1000 U/ml IFNB1 or 1 μM rapamycin for further 24 h. Western blot analysis was performed for MAP1LC3B and ACTB/β-actin proteins. (C) Quantification of band intensities in (B). Data represent mean and SEM of three independent experiments. Statistical analysis was performed using one-way repeated measures ANOVA followed by Dunett’s post-test against the control sample, *p < 0.05 and **p < 0.01. (D–F) IFNB1 induced eGFP-MAP1LC3B translocation. MCF-7 eGFP-MAP1LC3B cells were cultured for 72 h and then treated for an additional 24 h with control medium or 100 U/ml IFNB1 or with 200 nM rapamycin 2 h prior to fixation. Percentage of eGFP-MAP1LC3B-positive cells was quantified automatically as described in Materials and Methods. (D) Representative pictures of control, IFNB1 and rapamycin-treated cells. (E) Quantification of eGFP-MAP1LC3B translocation after IFNB1 treatment. Three different thresholds, >5, >10 or >15 eGFP-MAP1LC3B puncta/cell, were used to define an eGFP-MAP1LC3B puncta-positive cell. Data represent mean and SEM of three independent experiments, each obtained from an average of five replicates. Statistical analysis was performed using Student’s paired t-test, *p < 0.05. (F) Quantification of eGFP-MAP1LC3B translocation after rapamycin treatment. A threshold of >5 eGFP-MAP1LC3B puncta/cell was used to define an eGFP-MAP1LC3B puncta-positive cell. Data represent mean and SD of three replicates. Statistical analysis was performed using Student’s unpaired t-test, ***p < 0.001. (G) IFNB1 induced autophagic flow. MCF-7 RLuc-MAP1LC3B WT and RLucMAP1LC3BG120A cells were cultured for 72 h and then treated with control medium, 100 or 1000 U/ml IFNB1 or 200 nM rapamycin. The autophagic flux was measured at 6, 12 and 24 h after treatment as described in Materials and Methods. Data represent mean and SEM of six replicates and is representative of two independent experiments. Statistical analysis using one-way repeated measures ANOVA followed by Dunett’s post test against the control sample was performed individually for each time point, *p < 0.05, **p < 0.01 and ***p < 0.001.
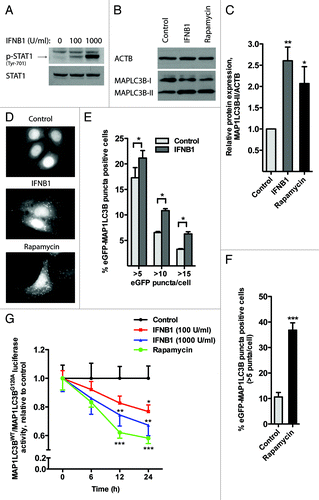
Autophagy is a dynamic process where MAP1LC3B is turned over even under basal conditions. Therefore, an increase in MAP1LC3B-II and accumulation of MAP1LC3B-positive puncta do not necessarily reflect an induction of autophagy, but can also result from impaired MAP1LC3B-II degradation.Citation47,Citation48 To clarify the cause of IFNB1-induced accumulation of MAP1LC3B-II and MAP1LC3B-positive puncta, we measured autophagic flow by a Renilla luciferase (RLuc) reporter-based assay for MAP1LC3B turnover.Citation49 This assay compares the rate of the MAP1LC3B degradation in MCF-7 cells expressing RLuc fused to either wild-type MAP1LC3B, which is degraded by autophagy, or to mutated MAP1LC3B (G120A), which cannot be lipidated or recruited to autophagosomal membranes.Citation49 We treated the MCF-7-RLuc-MAP1LC3BWT and MCF-7-RLuc-MAP1LC3BG120A cells in parallel with different concentrations of IFNB1 or rapamycin and measured luciferase activities 6, 12 and 24 h later. As shown in , IFNB1 induced autophagic flow in a dose- and time-dependent manner suggesting that the observed MAP1LC3B-II accumulation seen by western blot () and in the eGPF-MAP1LC3B translocation assay () indeed reflected an induction of autophagic flow by IFNB1.
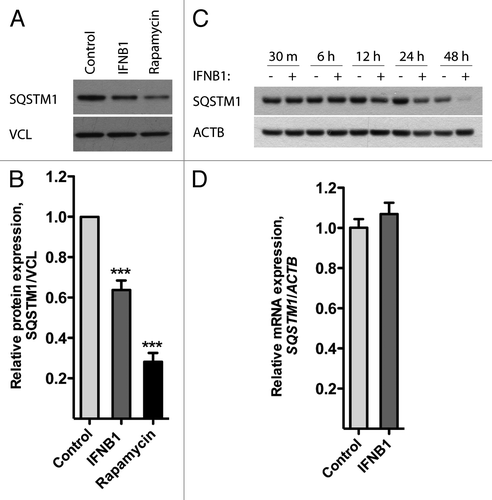
SQSTM1/p62 is another widely used autophagy marker. It binds directly to both MAP1LC3B and ubiquitin,Citation50 and drives the selective degradation of ubiquitinated cargo through the autophagic pathway.Citation51 The level of SQSTM1 is believed to reflect autophagosome turnover, since akin to MAP1LC3B, SQSTM1 is itself sequestered by the autophagosome during this process and degraded in the autolysosome, which is formed after fusion of the autophagosome with lysosomes.Citation52 As evident from , SQSTM1 levels were significantly decreased after 24 h treatment with IFNB1 or rapamycin. SQSTM1 degradation began after 12 h of IFNB1 treatment and further increased over 24 and 48 h () in accordance with the MAP1LC3B flow data (). The levels of SQSTM1 mRNA remained unchanged after 24 h of IFNB1 treatment, thus ruling out that the observed decrease in SQSTM1 protein levels was caused by transcriptional changes (). Collectively, the above data indicated that IFNB1 induced autophagic flow in MCF-7 cells.
Figure 2. IFNB1 induced autophagy in MCF-7 breast cancer cells as measured by SQSTM1 degradation. (A–C) IFNB1 treatment triggered SQSTM1 degradation. (A) MCF-7 eGFP-MAP1LC3B cells were cultured for 24 h and then treated with control medium, 1000 U/ml IFNB1 or 1 μM rapamycin for 24 h. Western blot analysis was performed for SQSTM1 and VCL/vinculin protein levels. (B) Quantification of band intensities in (A). Data represent mean and SEM of five independent experiments. Statistical analysis was performed using one-way repeated measures ANOVA followed by Dunett’s post test against the control sample, ***p < 0.001. (C) MCF-7 eGFP-MAP1LC3B cells were cultured for 24 h and then treated with control medium or 1000 U/ml IFNB1 for the indicated time intervals. Western blot analysis was performed for SQSTM1 and ACTB levels. (D) IFNB1 did not regulate SQSTM1 mRNA levels. MCF-7 eGFP-MAP1LC3B cells were cultured and treated as in (A) before RNA was extracted and qPCR used to analyze SQSTM1 and ACTB levels. Data represent mean and SEM of two independent experiments.
IFNB1 induced autophagy in MDAMB231 and SKBR3 breast cancer cells
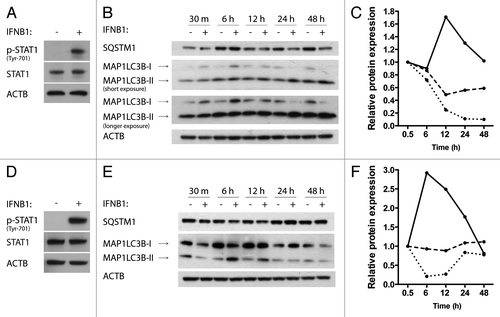
Breast cancer is a heterogenous disease and patients are treated differently depending on the hormone and ERBB2/HER2 receptor status of their cancers, among other features. MCF-7 cells are estrogen receptor (ER)-positive. We tested whether IFNB1 also induces autophagy, measured by MAP1LC3B conversion and SQSTM1 degradation, in two other breast cancer cell lines, namely the MDAMB231 cell line, which is ER recepetor negative, and the SKBR3 cell line, which is ER-negative but ERBB2 amplified.Citation53 Both cell lines were responsive to human recombinant IFNB1 measured as increased phosphorylation of STAT1 (). In MDAMB231 cells, IFNB1 induced autophagy in a time-dependent manner starting after 12 h of treatment, which increased over time (). In SKBR3 cells, IFNB1 induced a transient burst of autophagy that peaked after 6 to 12 h of IFNB1 treatment and was reduced after longer periods of treatment (). The effect was most pronounced at the MAP1LC3B levels, but with SQSTM1 being reduced to 86% of the corresponding time-point control at 6 h of treatment. These data showed that IFNB1 induced functional autophagy in various breast cancer cell lines.
Figure 3. IFNB1 induced autophagy in MDAMB231 and SKBR3 breast cancer cells. (A) MDAMB231 cells were IFNB1-responsive. MDAMB231 cells were cultured for 24 h, serum starved for 3 h and stimulated with control medium or 1000 U/ml of IFNB1 for 30 min. Western blot analysis was performed for p-STAT1, STAT1 and ACTB proteins. (B and C) IFNB1 induced autophagy in MDAMB231 cells. (B) MDAMB231 cells were cultured for 24 h and then treated with control medium or 1000 U/ml IFNB1 for the indicated time intervals. Western blot analysis was performed for SQSTM1, MAP1LC3B and ACTB protein levels. The blot is representative of two independent experiments. (C) Quantification of the results shown in (B). The values are normalized to the time-point control, and further to the 30 min time-point set to 1, and represent average values from two independent experiments. The full line shows relative MAP1LC3B-II/ACTB levels, the dotted line shows relative MAP1LC3B-I/MAP1LC3B-II levels and the dashed line shows relative SQSTM1/ACTB levels. (D) SKBR3 cells were IFNB1-responsive. SKBR3 cells were cultured for 24 h, serum starved for 3 h and stimulated with control medium or 1000 U/ml of IFNB1 for 30 min. Western blot analysis was performed for p-STAT1, STAT1 and ACTB protein levels. (E and F) IFNB1 induces autophagy in SKBR3 cells. (E) SKBR3 cells were cultured for 24 h and then treated with control medium or 1000 U/ml IFNB1 for the indicated time intervals. Western blot analysis was performed for SQSTM1, MAP1LC3B and ACTB levels. The blot is representative of two independent experiments. (F) Quantification of the results in (E), as described in (C).
IFNB1 affected MTORC1 activity in MCF-7 cells
MTORC1 is a key negative regulator of autophagy.Citation24 To understand the mechanism behind IFNB1-induced autophagy we therefore investigated how IFNB1 treatment affected the MTORC1 pathway using MCF7-cells. The activity of MTORC1 can be investigated indirectly by measuring the phosphorylation status of two downstream targets of MTORC1; EIF4EBP1/eukaryotic translation initiation factor 4E binding protein 1 and RPS6KB1/ribosomal protein S6 kinase, 70 kDa, polypeptide 1 or its downstream target RPS6/ribosomal protein S6.Citation48 As shown in , a decrease in phosphorylated EIF4EBP1 relative to total EIF4EBP1 protein levels was detected following IFNB1 treatment. The decrease was seen after 12 h of treatment and became more prominent after 24 to 48 h of treatment. Moreover, the decrease in phosphorylated EIF4EBP1 relative to total EIF4EBP1 protein levels was IFNB1 dose dependent, measured at 24 h of treatment (). These data indicated that long-term treatment with IFNB1 decreased MTORC1 activity. However, the changes in phosphorylated EIF4EBP1 were not concomitant with a decrease in phosphorylated RPS6 protein levels, except for a slight decrease observed after 48 h of treatment (), or phosphorylated RPS6KB1 levels (data not shown). This suggested that IFNB1 specifically inhibits the EIF4EBP1 branch of MTORC1 signaling at the concentrations and time points investigated.
Figure 4. IFNB1 affected MTORC1 activity. (A and B) IFNB1 specifically decreased p-EIF4EBP1 levels. MCF-7 eGFP-MAP1LC3B cells were cultured for 24 h and then treated with (A) control medium or 1000 U/ml IFNB1 for the indicated time intervals, or (B) with control medium or 100 or 1000 U/ml IFNB1 for 24 h. Western blot analysis was performed for p-EIF4EBP1, EIF4EBP1, p-RPS6, RPS6, ACTB and VCL protein levels. (C) Rapamycin triggered SQSTM1 degradation in a dose-dependent manner. Cells were cultured as in (A) and stimulated with the indicated concentrations of rapamycin for 24 h in order to find a saturating and a nonsaturating concentration of rapamycin. Western blot analysis was performed for SQSTM1 and ACTB protein levels. (D) IFNB1 did not potentiate rapamycin-mediated SQSTM1 degradation. Cells were cultured as in (A) and stimulated with control medium, 1000 U/ml IFNB1 and/or 1 μM rapamycin for 24 h. Western blot analysis was performed for SQSTM1 and ACTB protein levels. Shown is a representative western blot for SQSTM1 and ACTB protein levels of two independent experiments. Band intensities were quantified and the average relative intensities shown below. (E) IFNB1 and rapamycin acted together to degrade SQSTM1 at subsaturated concentrations. Cells were cultured as in (A) and stimulated with 100 U/ml IFNB1 and/or 0.1 nM rapamycin for 24 h. Western blot analysis was performed for SQSTM1 and ACTB protein levels. Band intensities were quantified and the relative quantifications are indicated.
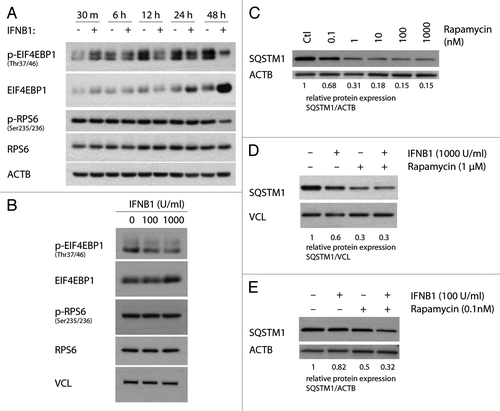
To get further evidence that IFNB1 induced autophagy by regulating the MTORC1 pathway we evaluated the effect of combined IFNB1 and rapamycin treatment on SQSTM1 degradation. When using a saturating concentration of rapamycin with regard to SQSTM1 degradation (), we could not detect any further increase in SQSTM1 degradation after cotreatment with IFNB1 relative to rapamycin treatment alone (), suggesting that IFNB1 acts upstream of MTORC1. Nevertheless when using a lower IFNB1 concentration and nonsaturated levels of rapamycin they collaborated to degrade SQSTM1 (). Collectively, these data suggested that IFNB1 mediated its effect on autophagy, at least partly, by inhibiting the MTORC1 pathway.
IFNB1 decreased proliferation and induced apoptosis in MCF-7 cells
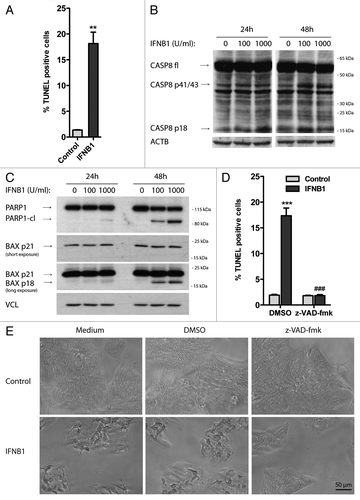
To understand the functional relevance of the induction of autophagy by IFNB1, we studied the biological effects of IFNB1 on MCF-7 cells. In accordance with earlier reports,Citation17-Citation20 we found that IFNB1 decreased the number of MCF-7 cells in a time- and dose-dependent manner as measured with a crystal violet assay (). We therefore addressed whether this was caused by an effect on proliferation or cell death. As measured by total BrdU incorporation, DNA synthesis was significantly reduced after 48 h of IFNB1 treatment compared with control cells (). This was supported by an accumulation of cells in the G1 phase of the cell cycle after IFNB1 treatment (). Moreover, IFNB1 treatment significantly increased the percentage of TUNEL-positive cells (), indicating that it can induce DNA fragmentation in MCF-7 cells—a phenomenon often associated with apoptotic cell death.Citation54 Apoptosis can be initiated by intrinsic or extrinsic cell cues, each associated with different molecular pathways. The intrinsic pathway is regulated at the mitochondrial level, where the balanced function of pro- and antiapoptotic members of the BCL2 family regulates mitochondrial membrane integrity. Loss of mitochondrial integrity leads to release of proapoptotic factors into the cytoplasm and activation of downstream caspase cascades contributing to apoptotic cell death.Citation55 The extrinsic pathway on the other hand is activated by death receptors at the cell surface leading to the activation of CASP8/caspase-8, that in turn activates downstream caspase cascades or crosstalk with the intrinsic pathway.Citation55 Supporting apoptosis induction by IFNB1 in MCF-7 cells, 48 h of IFNB1 treatment caused cleavage of CASP8 () and strong induction of a potent proapoptotic cleavage product of BAX, p18 BAX ().Citation56-Citation59 Furthermore, IFNB1 treatment triggered a time- and dose-dependent cleavage of PARP1/poly (ADP-ribose) polymerase 1 at a site corresponding to CASP3/caspase-3- and/or CAPSP7/caspase-7-dependent cleavage (giving rise to a 89-kDa C-terminal fragment) ().Citation60 We further treated cells with the pan-caspase inhibitor z-VAD-fmk during IFNB1 treatment, which completely prevented the induction of TUNEL-positive cells by IFNB1 treatment (), verifying that IFNB1 mediated cell death via apoptosis. This was supported by phase-contrast microscopy showing an increase in rounded and detached cells after IFNB1 treatment, which was absent in the presence of z-VAD-fmk treatment (). Interestingly, the antiproliferative effect of IFNB1 was sustained in the presence of z-VAD-fmk (), showing that the antiproliferative and proapoptotic functions of IFNB1 are controlled by separate molecular events. In combination, these results show that IFNB1 inhibits proliferation and induces apoptosis in MCF-7 cells.
Figure 5. IFNB1 inhibited cell proliferation. (A) IFNB1 decreased cell numbers. MCF-7 eGFP-MAP1LC3B cells were cultured for 24 h and then treated for 24 or 48 h with control medium or IFNB1 as indicated before a crystal violet assay was performed. Data represent mean and SEM of four replicates and is representative of two independent experiments. Statistical analysis was performed for each time point using one-way repeated measures ANOVA followed by Dunett’s post test against the control sample, ***p < 0.001. (B) IFNB1 inhibited proliferation. MCF-7 eGFP-MAP1LC3B cells were cultured for 24 h and then treated for further 48 h with control medium or 1000 U/ml IFNB1. Cell proliferation was measured by means of BrdU incorporation. Data represent mean and SEM in arbitrary units (A.U.) of three independent experiments each obtained from an average of four replicates. Statistical analysis was performed using Student’s paired t-test, **p < 0.01. (C) IFNB1 induced G1-phase accumulation. MCF-7 eGFP-MAP1LC3B cells were cultured and treated as in (B). Then cells were stained with PI and cell cycle was analyzed by FACS. Red line is control treatment, blue line is IFNB1 treatment. The experiment is representative of two independent experiments.
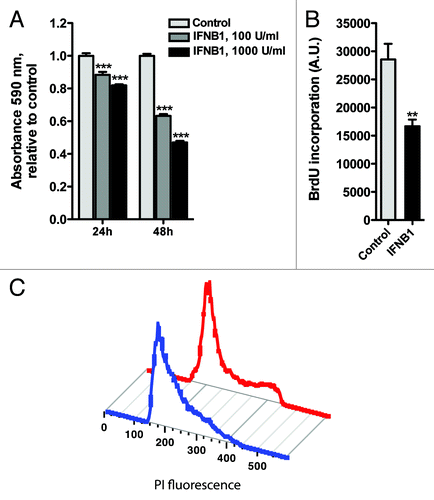
Figure 6. IFNB1 induced apoptosis. (A–E) IFNB1 induced apoptosis. (A) MCF-7 eGFP-MAP1LC3B cells were cultured for 24 h and then treated for further 48 h with control medium or 1000 U/ml IFNB1 and stained for DNA fragmentation using a TUNEL assay. TUNEL-positive cells were quantified as described in Materials and Methods. Data represent mean and SEM from three independent experiments. Statistical analysis was performed using Student’s paired t-test, **p < 0.01. (B and C) Representative western blots of cells cultured and treated as in (A) showing CASP8, PARP1, BAX and VCL protein levels. Blots are representative of three independent experiments. (D) MCF-7 eGFP-MAP1LC3B cells were cultured, treated and assayed as in (A), except that 30 μM z-VAD-fmk or DMSO was added at the time of IFNB1 addition. Data represent mean ± SEM from four replicates and is representative of two independent experiments. Statistical analysis was performed using one-way ANOVA followed by Bonferroni’s multiple comparison test, *** = p < 0.001 compares DMSO-treated samples in the presence or absence of IFNB1 and ###p < 0.001 compares samples treated with IFNB1 in the presence of either DMSO or z-VAD-fmk. (E) Phase-contrast pictures of cells treated as in (D).
IFNB1 induced canonical autophagy in MCF-7 breast cancer cells and MEFs
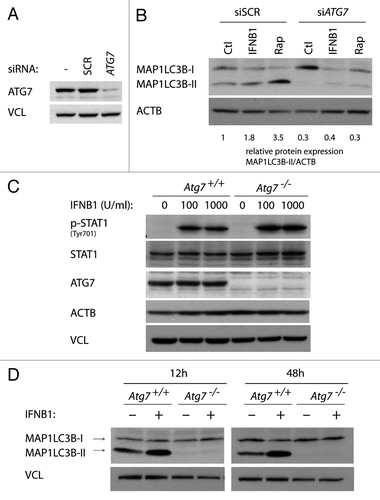
We investigated if IFNB1-mediated autophagosome formation was dependent on ATG7, which is considered one of the core autophagy molecules indispensable for canonical autophagy.Citation61 To evaluate the impact of ATG7 silencing on IFNB1-mediated autophagosome formation, we compared IFNB1- and rapamycin- induced autophagy in the face of ATG7 or nonsilencing scrambled control (SCR) siRNA transfection, by means of MAP1LC3B conversion. As shown in , a robust downregulation of ATG7 protein levels was obtained 48 h after ATG7 siRNA transfection relative to transfection with the SCR control. Steady-state MAP1LC3B-II levels were strongly inhibited after ATG7 depletion, as was the increase in MAP1LC3B-II after both IFNB1 and rapamycin treatment as compared with SCR transfected cells (). This indicated that IFNB1 is, at least to some extent, dependent on ATG7 for autophagosome formation, although still some MAP1LC3B-II conversion remained. However, this could reflect residual ATG7 after siRNA-mediated silencing. To verify the capacity of IFNB1 to mediate autophagy in a non-breast cancer cell type and to have a system where no residual ATG7 was present, we utilized wild-type mouse embryonic fibroblast (MEFs) (Atg7+/+) and the corresponding Atg7−/− MEFs generated from Atg7 knockout mice.Citation61 Both cell lines were found to be equally responsive to mouse recombinant IFNB1 measured as increased phosphorylation of STAT1 (). When MEFs were treated with IFNB1 for 12 or 48 h, MAP1LC3B-I was robustly converted to MAP1LC3B-II in Atg7+/+ MEFs, while no MAP1LC3B conversion was detected in atg7−/− cells (). These data indicated that the effect of IFNB1 to induce autophagy extended beyond breast cancer cells, and that IFNB1 induced autophagy through the canonical autophagosome formation pathway.
Figure 7. IFNB1 induced canonical autophagy in MCF-7 breast cancer cells and MEFs. (A) Knockdown efficiency of ATG7. MCF-7 eGFP-MAP1LC3B cells were cultured for 24 h and transfected the next day with siRNA targeting ATG7 or the nonsilencing SCR control. Seventy-two hours post-transfection cells were lysed and western blot analysis performed with antibodies against ATG7 and VCL. (B) Silencing of ATG7 reduced basal, as well as IFNB1- and rapamycin-induced MAP1LC3B conversion. Cells were cultured and transfected as in (A). Forty-eight hours post-transfection, control medium, 1000 U/ml IFNB1 or 100 nM rapamycin was added for 24 h, before western blot analysis was performed for MAP1LC3B and ACTB protein levels. Relative expression levels are shown below the blot, which are representative of two independent experiments. (C) Atg7+/+ and Atg7−/− MEFs were IFNB1-responsive. Atg7+/+ and Atg7−/− MEFs were cultured for 24 h, serum starved for 3 h and treated with control medium or 100 or 1000 U/ml of IFNB1 for 30 min. Western blot analysis was performed for p-STAT, STAT, ATG7, ACTB and VCL protein levels. (D) IFNB1 induced MAP1LC3B conversion in MEFs. Atg7+/+ and Atg7−/− MEFs were cultured for 24 h and treated with control medium or 1000 U/ml IFNB1 for further 12 or 48 h. Western blots were performed for MAP1LC3B and VCL.
Blocking autophagy increased the proapoptotic activity of IFNB1 in MCF-7 cells
To investigate the function of IFNB1-induced autophagy in MCF-7 cells, we inhibited autophagy by siRNA-mediated silencing of several autophagy-promoting genes, ATG5, ATG7, or ULK1/2, whose knockout or knockdown previously have been shown to effectively impair the autophagic pathway.Citation61-Citation64 Efficient silencing of ATG7 (), ATG5, and ULK1/2 () was accompanied by inhibition of autophagy, measured as a decrease in MAP1LC3B-II levels ( and ) and an increase in SQSTM1 levels (), relative to SCR transfected cells.
Figure 8. Blocking autophagy increased the proapoptotic activity of IFNB1. (A) Knockdown efficiency of core autophagy proteins. MCF-7 eGFP-MAP1LC3B cells were cultured for 24 h and transfected the next day with siRNAs targeting ATG5, ULK1/2 or ATG7 as indicated. SCR was used as a nonsilencing control. Seventy-two hours post-transfection cells were lysed and western blot analysis performed with antibodies against ULK1, ATG5 and VCL. (B) Knockdown of core autophagy proteins reduced MAP1LC3B-II levels. MCF-7 eGFP-MAP1LC3B cells were cultured and transfected as in (A), but western blot analysis was performed for MAP1LC3B and ACTB levels. Relative expression levels are shown below the blot. (C) Knockdown of core autophagy proteins increased SQSTM1 levels. Cells were cultured and transfected as in (A), but western blot was performed for SQSTM1 and ACTB levels. Relative expression levels are shown below the blot. (D) IFNB1-induced autophagy did not modulate its antiproliferative response. MCF-7 eGFP-MAP1LC3B cells were cultured for 24 h and transfected the next day with siRNAs targeting different core-autophagy proteins as indicated. Forty-eight hours post-transfection, control medium or 1000 U/ml IFNB1 was added for another 48 h before cell proliferation was measured by BrdU incorporation. Data represent mean and SEM of three independent experiments, each obtained from an average of four replicates. Statistical analysis was performed using one-way repeated measures ANOVA followed by Dunett’s post test against the SCR transfected sample, #p < 0.05 and ##p < 0.01 in the absence of IFNB1, while **p < 0.01 in the presence of IFNB1. Note the lack of significant interaction between IFNB1 treatment and any of the siRNA transfections using two-way repeated measures ANOVA. (E) IFNB1-induced autophagy counteracted its proapoptotic function. Cells were cultured, transfected and treated as in (A) and stained for DNA fragmentation with a TUNEL assay. Data represent mean and SEM of three independent experiments, where each value is an average of three replicates. The results were analyzed as in (A). #p < 0.05 and ***p < 0.001 using Dunett’s post test against the SCR transfected sample in the absence or presence of IFNB1 respectively. ¤p < 0.05, ¤¤p < 0.01 and ¤¤¤p < 0.001 indicate significant interaction between IFNB1 treatment and the indicated siRNA transfection using two-way repeated measures ANOVA. Significant interaction suggests a relatively larger effect of autophagy on the amount of TUNEL-positive cells in IFNB1-treated samples compared with control-treated samples.
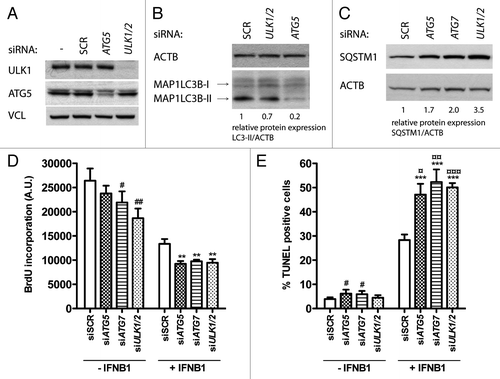
We next investigated how inhibition of autophagy would impact on the antiproliferative and proapoptotic effects of IFNB1. In the absence of IFNB1 treatment, silencing of ATG7 and ULK1/2 led to a small, but still significant inhibition of proliferation compared with SCR-transfected cells as measured by BrdU incorporation (). This indicated a positive regulatory function for constitutive autophagy in proliferation of MCF-7 breast cancer cells as shown previously.Citation65 In the presence of IFNB1 the same tendency was observed, suggesting that autophagy was not responsible for the antiproliferative effect of IFNB1 seen in these cells ().
Using a similar experimental setup, we next performed a TUNEL assay. In the absence of IFNB1 there were in general only few TUNEL-positive cells. However, a slight but significant increase in TUNEL-positive cells were seen after ATG5 and ATG7 silencing relative to SCR transfected cells, which is in line with the well-established prosurvival function of constitutive autophagy (). In contrast, this was not the case for ULK1/2 silencing (). Interestingly, after IFNB1 treatment there was a robust induction of TUNEL-positive cells in ATG5, ATG7 and ULK1/2 siRNA transfected cells relative to the SCR controls (). These data strongly suggested that IFNB1-induced autophagy promoted survival and thereby reduced IFNB1’s proapoptotic capacity.
STAT1 was a positive regulator of IFNB1-induced autophagy
IFNB1 signals through the JAK-STAT pathway to regulate transcription of many target genes.Citation1 In accordance, STAT1 was phosphorylated upon IFNB1 treatment in the breast cancer cell lines we examined (; ). To investigate whether STAT1 was influencing IFNB1-induced autophagy, we silenced STAT1 using siRNA in MCF-7 cells () in the absence or presence of IFNB1. In the absence of IFNB1, STAT1 silencing led to a significant increase in SQSTM1 levels (), which was not accompanied by increased SQSTM1 mRNA levels (data not shown), indicating that basal autophagy could be affected by STAT1. However, we could not detect a concomitant decrease in MAP1LC3B-II following STAT1 silencing (), and thus the role of STAT1 in regulation of basal autophagy in MCF-7 cells requires further investigation. Nevertheless, when cells were treated with IFNB1 after STAT1 silencing, autophagy induction was significantly inhibited, measured as decreased MAP1LC3B-II levels and increased SQSTM1 levels, relative to SCR-transfected cells treated with IFNB1 (). These data suggested that STAT1 was required for IFNB1-induced autophagy.
Figure 9. STAT1 was a positive regulator of IFNB1-induced autophagy. (A) Knockdown efficiency of STAT1. MCF-7 eGFP-MAP1LC3B cells were cultured for 24 h and transfected the next day with siRNAs targeting STAT1. SCR siRNA was used as a nonsilencing control. Seventy-two hours post-transfection cells were lysed and western blot analysis performed with antibodies against STAT1 and ACTB. (B) Silencing of STAT1 reduces IFNB1-induced autophagy. MCF-7 eGFP-MAP1LC3B cells were cultured and transfected as in (A). Forty-eight hours post-transfection control medium or 1000 U/ml IFNB1 was added for additional 24 h before cell lysis. Western blot analysis was performed with antibodies against SQSTM1, MAP1LC3B and ACTB. (C) Quantification of band intensities in (B). Data represent mean and SEM of three independent experiments. Statistical analysis was performed using Student’s t-test, *p < 0.05 or **p < 0.01 relative to SCR transfected samples in the absence of IFNB1; #p < 0.05 or ##p < 0.01 relative to SCR transfected samples in the presence of IFNB1. (D) STAT1-dependent autophagy did not affect IFNB1’s antiproliferative capacity. MCF-7 eGFP-MAP1LC3B cells were cultured and transfected as in (B), and 48 h post-transfection control medium or 1000 U/ml IFNB1 was added for additional 48 h before cell proliferation was measured by BrdU incorporation. Data represent mean and SEM of three independent experiments, each obtained from an average of three replicates. Statistical analysis was performed using Student’s paired t-test comparing SCR and STAT1 siRNA transfected samples, *p < 0.05 in the presence of IFNB1. Note the lack of a significant interaction between IFNB1 treatment and STAT1 siRNA silencing using two-way repeated measures ANOVA. (E) STAT1-dependent autophagy counteracted IFNB1’s proapoptotic function. MCF-7 eGFP-MAP1LC3B cells were cultured, transfected and treated as in (D) but stained for DNA fragmentation with a TUNEL assay. Data represent mean and SEM of four replicates and are representative of two independent experiments. Statistical analysis was performed using one-way ANOVA followed by Dunett’s post test against the SCR transfected sample, ##p < 0.01 and ###p < 0.001 in the absence of IFNB1, while ***p < 0.01 in the presence of IFNB1. ¤¤¤p < 0.001 indicates significant interaction between IFNB1 treatment and STAT1 silencing using two-way ANOVA. Significant interaction suggests a relatively larger effect of autophagy on the amount of TUNEL-positive cells in IFNB1-treated samples when compared with control-treated samples.
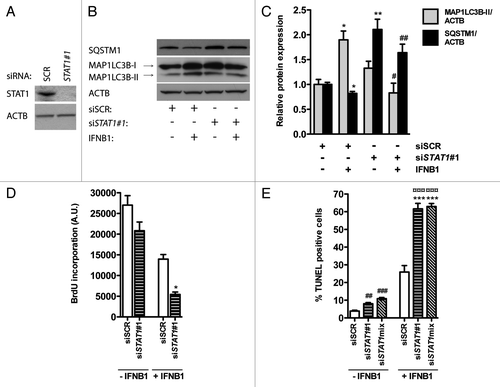
Identical to the already known core autophagy molecules, silencing of STAT1 led to a small reduction in proliferation and a slight, but significant, increase in apoptosis in the absence of IFNB1 (). Likewise, silencing of STAT1 affected proliferation similarly with or without IFNB1 (), while in the presence of IFNB1 there was a significant increase in TUNEL-positive cells (). The latter indicated that STAT1 was important for IFNB1-induced autophagy, which served to promote survival in MCF-7 cells, thereby reducing IFNB1’s proapoptotic capacity.
Discussion
Pharmacological usage of IFNs in cancer therapy has received much attention in the past due to their potent growth inhibitory and proapoptotic functions. However, widespread use has been hampered due to severe side effects and dosing issues,Citation66 and the ability of tumor cells to escape the effects of IFNs by inducing survival pathways thereby causing tumor resistance to IFN treatment.Citation2 These include the EGFR and STAT3 pathways.Citation67,Citation68 Recently IFI6/GIP3, an ISG heavily induced by IFNA, has been furthermore shown to contribute to tamoxifen resistance and poor outcomes in breast cancer.Citation23,Citation69 Autophagy is another survival pathway that has caused treatment resistance in MCF-7 cells.Citation70-Citation73 IFNG can induce autophagy in various biological settings, which interestingly contributes to cell death in HCC cells.Citation43 A direct function for type I IFNs in inducing autophagy has not previously been reported, and we therefore investigated whether IFNB1 could induce autophagy and how this would impact on IFNB1’s biological functions.
We report here for the first time that IFNB1 directly induced autophagy in MCF-7, MDAMB231 and SKBR3 breast cancer cells. In addition, we showed that IFNB1 induced MAP1LC3B conversion in MEFs, suggesting that the capacity of IFNB1 to induce autophagy extends beyond breast cancer cells. Few other reports have addressed the role of IFNB1 in relation to induction of autophagy.
A very recent study shows that IFNB1 induces autophagy in pancreatic tumor cells only when given together with the PPARG agonist troglitazone, an effect not observed with IFNB1 treatment alone.Citation67 However, pancreatic tumor cells have a very high level of autophagic flux relative to MCF-7 cells,Citation36 which potentially could mask an ability of IFNB1 to induce autophagy in these cells. Moreover, STAT2, which is specifically activated in type I IFN signaling, in contrast to STAT1, which is also activated during type II IFN signaling,Citation5 has been recently identified in a genome-wide screen for its ability to positively regulate two forms of selective autophagy, virophagy and mitophagy.Citation74 This further supports a positive regulatory function for type I IFN signaling in autophagy.
MTORC1 is a key negative regulator of autophagy, and many upstream stimuli regulate autophagy through the MTORC1 pathway.Citation24 To uncover molecular events involved in IFNB1-induced autophagy, we therefore studied the relationship between IFNB1, autophagy and the MTORC1 pathway. We found that treatment with IFNB1 specifically inhibited the EIF4EBP1 branch downstream of MTORC1 without affecting RPS6 phosphorylation, suggesting a negative effect of IFNB1 on MTORC1 activity. The mechanisms that determine the selectivity of MTORC1 for its substrates are not clear, but it has been recently shown that rapamycin, a natural inhibitor of MTORC1, reduces the phosphorylation of EIF4EBP1and RPS6 with different sensitivity.Citation75 It is possible that a likewise differential sensitivity is imposed by IFNB1 treatment. We further found that IFNB1 was unable to potentiate rapamycin-induced autophagy, which supports that IFNB1 acts in the MTORC1 pathway to induce autophagy. Importantly, rapamycin-induced autophagy at saturating rapamycin concentrations can be potentiated by activation of MTORC1-independent autophagy.Citation76,Citation77
We further showed that IFNB1 strongly inhibited cell proliferation in MCF-7 cells, in accordance with earlier reports showing reduced growth after IFNB1 treatment.Citation17,Citation19,Citation20 Our results in addition suggested that IFNB1 affects autophagy and cell proliferation independently. In agreement, MTORC1 is thought to regulate cell growth/proliferation and autophagy antagonistically.Citation78
IFNB1 potently induced apoptosis in MCF-7 cells. Previous reports have described cytotoxic effects of IFNB1 on MCF-7 cells,Citation18-Citation20 however little is known about the molecular mechanisms mediating this effect. Hundreds of ISGs are upregulated in response to IFN treatment in different cells, among which are several proapoptotic genes such as TNFSF10/TRAIL, FAS and CASP8, which regulate the extrinsic apoptotic pathway.Citation3 IFNA can also downregulate antiapoptotic and activate proapoptotic BCL2 family members in hematopoietic cells,Citation79,Citation80 thereby activating the intrinsic apoptotic pathway. We found that IFNB1 triggered an increase in CASP8 activation and the presence of a proapoptotic BAX fragment (p18BAX) and a CASP3/7 cleaved PARP fragment.Citation60 As MCF-7 cells are devoid of CASP3,Citation81 IFNB1 most likely induces CASP7 activity in these cells. Collectively, these data indicated that IFNB1 treatment activated both the intrinsic and extrinsic apoptotic pathway in MCF-7 cells. Interestingly, silencing of autophagy in the presence of IFNB1 strongly enhanced its proapoptotic function, suggesting that IFNB1-induced autophagy promoted survival and reduced the proapoptotic capacity of IFNB1. The phenotype was importantly reproduced when targeting three different components of the canonical autophagy core machinery. Among these were ATG7, which we also found to be necessary for IFNB1-induced autophagosome formation.
Of note, we additionally identified STAT1 as an important mediator of IFNB1-induced autophagy in MCF-7 breast cancer cells. STAT1 depletion consequently increased induction of IFNB1-induced apoptosis to the same extent as the known core autophagy molecules suggesting similar mechanisms of action. STAT1 activity has been recently described as a positive regulator of IFNG-mediated autophagy in melanoma cells, causing autophagy-related cell death and thereby restricting melanoma lung metastasis.Citation82 Moreover, IFNG-induced autophagy, which is dependent on the STAT1-induced transcription factor IRF1, also contributes to cell death in HCC cells.Citation43 STAT1 therefore seems necessary for both IFNG- and IFNB1-induced autophagy, but whereas autophagy induction by IFNG appears to promote cell death, we report here that IFNB1-induced autophagy promoted survival. IFNB1 is incapable of inducing IRF1 in MCF-7 cells,Citation83 which underscores that IFNG and IFNB1 can induce autophagy by different pathways downstream of STAT1. As IRF1 is a tumor suppressor known to induce apoptosis after IFNG-STAT1 activation,Citation83 this could potentially also explain the differential biological outcomes of IFNG- and IFNB1-induced autophagy.
It is well established that chemotherapeutics, endocrine therapy, as well as ERBB2 targeted therapy induce autophagy in breast cancer cells, which appears to have a prosurvival function likely contributing to treatment resistance.Citation70-Citation73 It could be argued that part of the autophagy induction we detected after IFNB1 treatment is a compensatory stress response to the cytotoxic effect of IFNB1. However, because the autophagic flow was induced already after 12 h following IFNB1 treatment, we believe that autophagy induction preceded the proapoptotic effect of IFNB1. Interestingly, we also observed that IFNB1 treatment caused MAP1LC3B conversion in MEFs after 12 and 48 h without a concomitant increase in cell death (data not shown). This supports that autophagy triggered by IFNB1 was a primary effect rather than a secondary stress-induced survival response.
Collectively, our data supported that autophagy might be yet another survival pathway activated by IFNB1, which could contribute to treatment resistance. Clinical trials are currently ongoing which explore the combination of anti-autophagy strategies with standard therapies in human cancers.Citation37 It might therefore be clinically relevant to reconcile a role for IFNB1 in the treatment of breast cancer with concomitant use of autophagy inhibitors, which possibly could enhance the proapoptotic effect of IFNB1 at doses well tolerated by patients.
Materials and Methods
Cell culture and treatments
MCF-7 eGFP-MAP1LC3B cellsCitation44 and MCF-7 RLuc-MAP1LC3B WT and MCF-7 RLuc-MAP1LC3B G120A cellsCitation49 were propagated in RPMI 1640 (Invitrogen, 21875) supplemented with 6% fetal bovine serum (FBS) (Sigma, F7524), 100 units/ml penicillin and 100 μg/ml streptomycin (Invitrogen, 15070063). Mouse embryonic fibroblasts (MEFs) from Atg7+/+ or Atg7−/− miceCitation61 were propagated in DMEM (Invitrogen, 31966) supplemented with 10% FBS, 100 units/ml penicillin, 100 μg/ml streptomycin and 1% nonessential amino acids (Invitrogen, 11140050). MDAMB231 (ATCC, HTB-26) and SKBR3 (ATCC, HTB-30) cells were propagated in DMEM (Invitrogen, 31966) supplemented with 10% FBS. All cells were incubated at 37°C in 5% CO2. All treatment of cells was done by adding 10 x solutions of control medium (0.1% BSA in PBS), human IFNB1 (PBL InterferonSource, 11420-1), mouse IFNB1 (Sigma, I-9032), rapamycin (Sigma, R0395) or carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone (z-VAD-fmk, Bachem, N-1560). All treatments were done in full medium, except for short-term stimulations with IFNB1 (30 min) for evaluation of STAT1 phosphorylation, where cells were serum starved for 3 h prior to IFNB1 stimulation.
Cell growth assay
MCF-7 eGFP-MAP1LC3B cells were seeded in 24-well plates (NUNC, 142475) and added control medium or IFNB1 (100 or 1000 U/ml) the next day. After 24 or 48 h the medium was removed and cells were fixed in 4% formaldehyde. The cells were stained with 0.1% w/v crystal violet (Sigma, C3886) for 30 min at room temperature, excess dye was washed away with water and the remaining dye extracted in 10% acetic acid. Sample absorbance was measured at 590 nm.
BrdU cell proliferation assay
MCF-7 eGFP-MAP1LC3B cells were seeded in 96-well plates (PerkinElmer, 6005070) and added control medium or 1000 U/ml of IFNB1 the next day. After additional 48 h cells were pulsed with BrdU for 3 h, before DNA incorporation was measured using the DELFIA Cell Proliferation kit (PerkinElmer, AD0200).
TUNEL assay
MCF-7 eGFP-MAP1LC3B cells were seeded in 96-well plates (NUNC, 167008) and control medium or 1000 U/ml of IFNB1 was added the next day. Forty-eight hours later cells were fixed in by adding a 1:1 volume of 8% formaldehyde with 0.6% glutaraldehyde to the cell culture medium. Cells were permeabilized in 0.25% triton-X-100 and stained with a terminal-deoxynucleotidyl transferase-dUTP nick end labeling (TUNEL) assay (Click-iT® TUNEL Alexa Fluor® 594 Imaging Assay, Invitrogen, C10246) to identify cells with fragmented DNA. Nuclei were counterstained with Hoechst 33342 (Molecular Probes). Image acquisition was done using an Amersham InCell1000 High throughput microscope (GE Healthcare) equipped with a 20× Nikon (GE Healthcare) objective. For each well 30 pictures (counting ~5000 cells/well) were taken from randomly placed positions and analyzed automatically using the InCell1000 workstation 3.5 software package (GE Healthcare). Nuclei were segmented based on the Hoechst signal, upon which a 2 μm cellular collar derived from the 594 nm channel was superimposed. TUNEL-positive cells were scored as having a signal/background ratio greater than 1.1 based on initial manual inspection of the images. Each cell per well was scored based on this ratio.
eGFP-MAP1LC3B translocation
MCF-7 eGFP-MAP1LC3B cells were seeded in 96-well plates (NUNC, 167008) and cultured for 72 h before treated with control medium or 100 U/ml of IFNB1 for an additional 24 h period, whereas rapamycin (200 nM) was added 2 h prior to fixation in 4% formaldehyde. Nuclei were stained with Hoechst 33342 and image acquisition was done using an Amersham InCell1000 High throughput microscope equipped with a 40× Nikon objective (GE Healthcare). For each well 15 images were acquired (counting ~100 cells/well) from randomly placed positions within each well and analyzed using the InCell1000 workstation 3.5 software package. Nuclei were identified and segmented based on the Hoechst signal and cells were segmented from the GFP channel. MAP1LC3B was evaluated in the GFP channel. Cells with >5, >10 or >15 eGFP-MAP1LC3B puncta were considered positive for eGFP-MAP1LC3B translocation after IFNB1 or rapamycin treatment, as indicated.
qPCR analysis
Total RNA was isolated using the RNeasy kit (Qiagen, 74106) and reverse transcribed into cDNA with the iScript cDNA Synthesis kit (Biorad, 170-8891). qPCR was performed with Maxima SYBR Green/ROX qPCR Master Mix (Fermentas, K0221). PCR primers used were as follows: SQSTM1 FW ACCCGTCTACAGGTGAACTC and RV GGATGCTTTGAATACTGGATGG, ACTB FW CATCCACGAAACTACCTTCAACTC and RV ATACTCCTGCTTGCTGATCCA. The data were analyzed according to the 2-ΔΔCt method.Citation84
Autophagic flux assay
MCF-7 RLuc-MAP1LC3BWT and MCF-7 Rluc-MAP1LC3BG120A cells were plated side by side in white 96-well plates (NUNC, 136101).Citation49 72 h later 50 nM of Enduren substrate (Promega, E6481) was added and RLuc activity was measured 2 h later followed by immediate addition of control medium, 100 or 1000 U/ml IFNB1 or 200 nM rapamycin. Luciferase measurements were done using the Glomax Multi+ luminometer 6, 12 and 24 h after IFNB1 treatment.Citation85 The readout was obtained by calculating the ratio of MCF-7 RLuc-MAP1LC3BWT/MCF-7 Rluc-MAP1LC3BG120A luminescence in neighboring wells exposed to the same treatment. Values measured after 6, 12 and 24 h were normalized to time point “0 h” (before treatment) for a given treatment and in addition to the value of the corresponding time point from control medium.
Antibodies and western blot analysis
MCF-7 eGFP-MAP1LC3B cells or Atg7+/+ or Atg7−/− MEFs were seeded in 6-well plates (NUNC, 140675) and treated the next day with control medium, IFNB1 or rapamycin as indicated. At given time points after treatment, cells were washed once in PBS and breast cancer cells were lysed in RIPA buffer (25 mM TRIS-HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS), while MEFs were lysed in 3% SDS, 10% glycerol, 10 mM Na2HPO4 both containing 1× Complete Mini Protease Inhibitor Cocktail (Roche Applied Science, 11836153001) and 1× Phosphatase Inhibitor Cocktail (Roche Applied Science, 4906845001). Samples from MEFs were sonicated. Protein concentrations were measured using the bicinchoninic acid assay (Pierce, 23227) and 20 to 40 µg of reduced (100 mM dithiothreitol) protein sample was separated on 4–12% NuPAGE Bis-Tris gels (Invitrogen, NP0336BOX) or 16% Tris-Glycine gels (Invitrogen, EC6498BOX) and transferred to PVDF membranes (#IPVH00010, Millipore). Membranes were blocked 1 h at RT in either 5% skim milk powder or 5% BSA in PBS-Tween 0.1% (PBS-T) and incubated overnight with primary antibodies at 4°C. The primary antibodies used were against ACTB/β-actin (4967), ULK1 (4776), p-EIF4EBP1/p-4E-BP1 (2855), EIF4EBP1/4E-BP1 (9644), RPS6/S6 (2217), p-RPS6/p-S6 (2211), BAX (2744), STAT1 (9172), p-STAT1 (9171), PARP1/PARP (9532), CASP8/caspase-8 (9746) all from Cell Signaling, ATG5 (TMD-PH-AT5, Cosmo Bio), SQSTM1/p62 (MBL International, PM045), MAP1LC3B (Nanotools, 0231-100/LC3-5F10), ATG7 (ProSci, Inc., 3617) and VCL/vinculin (Sigma, V9131). The next day membranes were incubated with horse-anti-mouse-horse radish peroxidase (HRP) (Vector Laboratories, PI-2000) or donkey-anti-rabbit-HRP (GE Healthcare, NA934-1ML) 1 h at RT and developed with the enhanced chemiluminiscence detection system (PerkinElmer, NEL104001EA). Band intensities were quantified with ImageJ (v.1.45p).
Propidium iodide cell cycle staining
Cells were harvested, washed twice in PBS and fixed for 30 min on ice in 70% ice-cold ethanol. Then cells were spun down and 1 ml of propidium iodide (PI)/RNase Staining Buffer (BD Biosciences, 550825) was added to the cell pellet and thoroughly mixed before analysis by FACS. Data were analyzed using FlowJo vs 8.8.6.
siRNA transfection
Cells were seeded in relevant tissue plates and transfected the next day with 20 to 50 nM siRNA using Lipofectamine 2000 (Invitrogen, 11668027). Forty-eight hours post-transfection cells were treated with control medium, IFNB1 or rapamycin for 24–48 h as indicated, and the relevant assays performed. The siRNAs used were all synthesized by Sigma including the universal negative control 1 (SCR). The sequences were as follows: ATG7: 5′ CAGUUUGGCACAAUCAAUA 3′, ULK1/2a: 5′ CGCGGUACCUCCAGAGCAATT 3′, ULK1/2b: 5′ CCCUUUGCGUUAUAUUGUATT 3′, ATG5a: 5′ CAACUUGUUUCACGCUAUATT 3′, ATG5b: 5′ CCUUUGGCCUAAGAAGAAATT 3′, STAT1 #1: 5′ CUGUGAAGUUGAGACUGUU 3′, STAT1 #2: 5′ CUCAUUCCGUGGACGAGGU 3′, STAT1 #3: 5′ CCUGAUUAAUGAUGAACUA and STAT1 #4: 5′ CGUAAUCUUCAGGAUAAUU 3′. For ULK1/2 and ATG5 a mixture of 10 nM of each of the two siRNAs was used, while STAT1 mix was a mix of 10 nM of each of STAT1 #1–4.
Statistical analysis
All statistical analysis was performed on raw data before normalization. For data comparing treatments to a control treatment, Student’s paired t-test (if two groups) or one-way ANOVA followed by Dunett’s post test against the control sample (if more than two groups) was used. Further, to evaluate the contribution of autophagy to IFNB1 effects in the BrdU and TUNEL assays, two-way ANOVAs were performed to test for interaction between IFNB1 treatment and the effect of each of the specific siRNA transfections.
| Abbreviations: | ||
| IFN | = | interferon |
| MAP1LC3B | = | microtubule-associated protein 1 light chain 3 beta |
| ATG | = | AuTophaGy-related |
| ULK1 | = | unc-51-like kinase 1 (C. elegans) |
| STAT | = | signal transducer and activator of transcription |
| eGFP | = | enhanced green fluorescence protein |
| ER | = | estrogen receptor |
| MTORC1 | = | mechanistic target of rapamycin complex 1 |
| EIF4EBP1 | = | eukaryotic translation initiation factor 4E binding protein 1 |
| RPS6 | = | ribosomal protein S6 |
| RPS6KB1 | = | ribosomal protein S6 kinase, 70 kDa, polypeptide 1 |
| TUNEL | = | terminal-deoxynucleotidyl transferase-dUTP nick end labeling |
| BAX | = | BCL2-associated X protein |
| PARP1 | = | poly (ADP-ribose) polymerase 1 |
| ISGs | = | interferon stimulated genes |
| IRF1 | = | interferon regulatory factor 1 |
Acknowledgments
We would like to acknowledge Masaaki Komatsu for kindly providing the Atg7+/+ or Atg7−/− MEFs. We are also thankful to Lisa Frankel for valuable help with the luciferase based autophagic flow assay and careful reading of the manuscript.
The project has received support from the Danish Cancer Society, The Danish Medical Research Council and the Lundbeck Foundation to S.I.-N.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Borden EC, Sen GC, Uze G, Silverman RH, Ransohoff RM, Foster GR, et al. Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov 2007; 6:975 - 90; http://dx.doi.org/10.1038/nrd2422; PMID: 18049472
- Caraglia M, Marra M, Tagliaferri P, Lamberts SW, Zappavigna S, Misso G, et al. Emerging strategies to strengthen the anti-tumour activity of type I interferons: overcoming survival pathways. Curr Cancer Drug Targets 2009; 9:690 - 704; http://dx.doi.org/10.2174/156800909789056980; PMID: 19508175
- Chawla-Sarkar M, Lindner DJ, Liu YF, Williams BR, Sen GC, Silverman RH, et al. Apoptosis and interferons: role of interferon-stimulated genes as mediators of apoptosis. Apoptosis 2003; 8:237 - 49; http://dx.doi.org/10.1023/A:1023668705040; PMID: 12766484
- Sangfelt O, Erickson S, Grander D. Mechanisms of interferon-induced cell cycle arrest. Front Biosci 2000; 5:D479 - 87; http://dx.doi.org/10.2741/Sangfelt; PMID: 10762599
- Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol 2005; 5:375 - 86; http://dx.doi.org/10.1038/nri1604; PMID: 15864272
- Darnell JE Jr., Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994; 264:1415 - 21; http://dx.doi.org/10.1126/science.8197455; PMID: 8197455
- Codarri L, Fontana A, Becher B. Cytokine networks in multiple sclerosis: lost in translation. Curr Opin Neurol 2010; 23:205 - 11; http://dx.doi.org/10.1097/WCO.0b013e3283391feb; PMID: 20442570
- Kieseier BC. The mechanism of action of interferon-β in relapsing multiple sclerosis. CNS Drugs 2011; 25:491 - 502; http://dx.doi.org/10.2165/11591110-000000000-00000; PMID: 21649449
- Rosenblum MG, Yung WK, Kelleher PJ, Ruzicka F, Steck PA, Borden EC. Growth inhibitory effects of interferon-beta but not interferon-alpha on human glioma cells: correlation of receptor binding, 2′,5′-oligoadenylate synthetase and protein kinase activity. J Interferon Res 1990; 10:141 - 51; http://dx.doi.org/10.1089/jir.1990.10.141; PMID: 2140395
- Damdinsuren B, Nagano H, Sakon M, Kondo M, Yamamoto T, Umeshita K, et al. Interferon-beta is more potent than interferon-alpha in inhibition of human hepatocellular carcinoma cell growth when used alone and in combination with anticancer drugs. Ann Surg Oncol 2003; 10:1184 - 90; http://dx.doi.org/10.1245/ASO.2003.03.010; PMID: 14654475
- Vitale G, de Herder WW, van Koetsveld PM, Waaijers M, Schoordijk W, Croze E, et al. IFN-beta is a highly potent inhibitor of gastroenteropancreatic neuroendocrine tumor cell growth in vitro. Cancer Res 2006; 66:554 - 62; http://dx.doi.org/10.1158/0008-5472.CAN-05-3043; PMID: 16397272
- Vitale G, van Eijck CH, van Koetsveld Ing PM, Erdmann JI, Speel EJ, van der Wansem Ing K, et al. Type I interferons in the treatment of pancreatic cancer: mechanisms of action and role of related receptors. Ann Surg 2007; 246:259 - 68; http://dx.doi.org/10.1097/01.sla.0000261460.07110.f2; PMID: 17667505
- Coradini D, Biffi A, Pirronello E, Di Fronzo G. The effect of alpha-, beta- and gamma-interferon on the growth of breast cancer cell lines. Anticancer Res 1994; 14:5A 1779 - 84; PMID: 7531412
- Horikoshi T, Fukuzawa K, Hanada N, Ezoe K, Eguchi H, Hamaoka S, et al. In vitro comparative study of the antitumor effects of human interferon-alpha, beta and gamma on the growth and invasive potential of human melanoma cells. J Dermatol 1995; 22:631 - 6; PMID: 8537547
- Zaidi MR, Merlino G. The two faces of interferon-γ in cancer. Clin Cancer Res 2011; 17:6118 - 24; http://dx.doi.org/10.1158/1078-0432.CCR-11-0482; PMID: 21705455
- Carpi A, Nicolini A, Antonelli A, Ferrari P, Rossi G. Cytokines in the management of high risk or advanced breast cancer: an update and expectation. Curr Cancer Drug Targets 2009; 9:888 - 903; http://dx.doi.org/10.2174/156800909790192392; PMID: 20025599
- Schmidberger H, Rave-Fränk M, Lehmann J, Schweinfurth S, Rehring E, Henckel K, et al. The combined effect of interferon beta and radiation on five human tumor cell lines and embryonal lung fibroblasts. Int J Radiat Oncol Biol Phys 1999; 43:405 - 12; http://dx.doi.org/10.1016/S0360-3016(98)00411-8; PMID: 10030269
- Kang JX, Liu J, Wang J, He C, Li FP. The extract of huanglian, a medicinal herb, induces cell growth arrest and apoptosis by upregulation of interferon-beta and TNF-alpha in human breast cancer cells. Carcinogenesis 2005; 26:1934 - 9; http://dx.doi.org/10.1093/carcin/bgi154; PMID: 15958519
- Lindner DJ, Borden EC. Synergistic antitumor effects of a combination of interferon and tamoxifen on estrogen receptor-positive and receptor-negative human tumor cell lines in vivo and in vitro. J Interferon Cytokine Res 1997; 17:681 - 93; PMID: 9402106
- Lindner DJ, Kolla V, Kalvakolanu DV, Borden EC. Tamoxifen enhances interferon-regulated gene expression in breast cancer cells. Mol Cell Biochem 1997; 167:169 - 77; http://dx.doi.org/10.1023/A:1006854110122; PMID: 9059994
- Kalie E, Jaitin DA, Abramovich R, Schreiber G. An interferon alpha2 mutant optimized by phage display for IFNAR1 binding confers specifically enhanced antitumor activities. J Biol Chem 2007; 282:11602 - 11; http://dx.doi.org/10.1074/jbc.M610115200; PMID: 17310065
- Lindner DJ, Borden EC. Effects of tamoxifen and interferon-beta or the combination on tumor-induced angiogenesis. Int J Cancer 1997; 71:456 - 61; http://dx.doi.org/10.1002/(SICI)1097-0215(19970502)71:3<456::AID-IJC25>3.0.CO;2-C; PMID: 9139884
- Cheriyath V, Kuhns MA, Jacobs BS, Evangelista P, Elson P, Downs-Kelly E, et al. G1P3, an interferon- and estrogen-induced survival protein contributes to hyperplasia, tamoxifen resistance and poor outcomes in breast cancer. Oncogene 2012; 31:2222 - 36; http://dx.doi.org/10.1038/onc.2011.393; PMID: 21996729
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009; 43:67 - 93; http://dx.doi.org/10.1146/annurev-genet-102808-114910; PMID: 19653858
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27 - 42; http://dx.doi.org/10.1016/j.cell.2007.12.018; PMID: 18191218
- Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell 2010; 40:280 - 93; http://dx.doi.org/10.1016/j.molcel.2010.09.023; PMID: 20965422
- Kimmelman AC. The dynamic nature of autophagy in cancer. Genes Dev 2011; 25:1999 - 2010; http://dx.doi.org/10.1101/gad.17558811; PMID: 21979913
- Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 2010; 90:1383 - 435; http://dx.doi.org/10.1152/physrev.00030.2009; PMID: 20959619
- Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, et al. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev 2007; 21:1367 - 81; http://dx.doi.org/10.1101/gad.1545107; PMID: 17510285
- Karantza-Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, Jin S, et al. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev 2007; 21:1621 - 35; http://dx.doi.org/10.1101/gad.1565707; PMID: 17606641
- Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006; 10:51 - 64; http://dx.doi.org/10.1016/j.ccr.2006.06.001; PMID: 16843265
- Dikic I, Johansen T, Kirkin V. Selective autophagy in cancer development and therapy. Cancer Res 2010; 70:3431 - 4; http://dx.doi.org/10.1158/0008-5472.CAN-09-4027; PMID: 20424122
- Denton D, Nicolson S, Kumar S. Cell death by autophagy: facts and apparent artefacts. Cell Death Differ 2012; 19:87 - 95; http://dx.doi.org/10.1038/cdd.2011.146; PMID: 22052193
- Lock R, Roy S, Kenific CM, Su JS, Salas E, Ronen SM, et al. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell 2011; 22:165 - 78; http://dx.doi.org/10.1091/mbc.E10-06-0500; PMID: 21119005
- Wei H, Wei S, Gan B, Peng X, Zou W, Guan JL. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev 2011; 25:1510 - 27; http://dx.doi.org/10.1101/gad.2051011; PMID: 21764854
- Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev 2011; 25:717 - 29; http://dx.doi.org/10.1101/gad.2016111; PMID: 21406549
- Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther 2011; 10:1533 - 41; http://dx.doi.org/10.1158/1535-7163.MCT-11-0047; PMID: 21878654
- Cook KL, Shajahan AN, Clarke R. Autophagy and endocrine resistance in breast cancer. Expert Rev Anticancer Ther 2011; 11:1283 - 94; http://dx.doi.org/10.1586/era.11.111; PMID: 21916582
- Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 2004; 119:753 - 66; http://dx.doi.org/10.1016/j.cell.2004.11.038; PMID: 15607973
- Inbal B, Bialik S, Sabanay I, Shani G, Kimchi A. DAP kinase and DRP-1 mediate membrane blebbing and the formation of autophagic vesicles during programmed cell death. J Cell Biol 2002; 157:455 - 68; http://dx.doi.org/10.1083/jcb.200109094; PMID: 11980920
- Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science 2006; 313:1438 - 41; http://dx.doi.org/10.1126/science.1129577; PMID: 16888103
- Tu SP, Quante M, Bhagat G, Takaishi S, Cui G, Yang XD, et al. IFN-γ inhibits gastric carcinogenesis by inducing epithelial cell autophagy and T-cell apoptosis. Cancer Res 2011; 71:4247 - 59; http://dx.doi.org/10.1158/0008-5472.CAN-10-4009; PMID: 21512143
- Li P, Du Q, Cao Z, Guo Z, Evankovich J, Yan W, et al. Interferon-γ induces autophagy with growth inhibition and cell death in human hepatocellular carcinoma (HCC) cells through interferon-regulatory factor-1 (IRF-1). Cancer Lett 2012; 314:213 - 22; http://dx.doi.org/10.1016/j.canlet.2011.09.031; PMID: 22056812
- Høyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell 2007; 25:193 - 205; http://dx.doi.org/10.1016/j.molcel.2006.12.009; PMID: 17244528
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19:5720 - 8; http://dx.doi.org/10.1093/emboj/19.21.5720; PMID: 11060023
- Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci 2004; 117:2805 - 12; http://dx.doi.org/10.1242/jcs.01131; PMID: 15169837
- Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy 2007; 3:542 - 5; PMID: 17611390
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008; 4:151 - 75; PMID: 18188003
- Farkas T, Høyer-Hansen M, Jäättelä M. Identification of novel autophagy regulators by a luciferase-based assay for the kinetics of autophagic flux. Autophagy 2009; 5:1018 - 25; http://dx.doi.org/10.4161/auto.5.7.9443; PMID: 19652534
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131 - 45; http://dx.doi.org/10.1074/jbc.M702824200; PMID: 17580304
- Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005; 171:603 - 14; http://dx.doi.org/10.1083/jcb.200507002; PMID: 16286508
- Bjørkøy G, Lamark T, Pankiv S, Øvervatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol 2009; 452:181 - 97; http://dx.doi.org/10.1016/S0076-6879(08)03612-4; PMID: 19200883
- Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006; 10:515 - 27; http://dx.doi.org/10.1016/j.ccr.2006.10.008; PMID: 17157791
- Samejima K, Earnshaw WC. Trashing the genome: the role of nucleases during apoptosis. Nat Rev Mol Cell Biol 2005; 6:677 - 88; http://dx.doi.org/10.1038/nrm1715; PMID: 16103871
- Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ 2012; 19:107 - 20; http://dx.doi.org/10.1038/cdd.2011.96; PMID: 21760595
- Gao G, Dou QP. N-terminal cleavage of bax by calpain generates a potent proapoptotic 18-kDa fragment that promotes bcl-2-independent cytochrome C release and apoptotic cell death. J Cell Biochem 2000; 80:53 - 72; http://dx.doi.org/10.1002/1097-4644(20010101)80:1<53::AID-JCB60>3.0.CO;2-E; PMID: 11029754
- Yanase N, Ohshima K, Ikegami H, Mizuguchi J. Cytochrome c release, mitochondrial membrane depolarization, caspase-3 activation, and Bax-alpha cleavage during IFN-alpha-induced apoptosis in Daudi B lymphoma cells. J Interferon Cytokine Res 2000; 20:1121 - 9; http://dx.doi.org/10.1089/107999000750053799; PMID: 11152579
- Wood DE, Newcomb EW. Caspase-dependent activation of calpain during drug-induced apoptosis. J Biol Chem 1999; 274:8309 - 15; http://dx.doi.org/10.1074/jbc.274.12.8309; PMID: 10075737
- Wood DE, Thomas A, Devi LA, Berman Y, Beavis RC, Reed JC, et al. Bax cleavage is mediated by calpain during drug-induced apoptosis. Oncogene 1998; 17:1069 - 78; http://dx.doi.org/10.1038/sj.onc.1202034; PMID: 9764817
- Chaitanya GV, Steven AJ, Babu PP. PARP-1 cleavage fragments: signatures of cell-death proteases in neurodegeneration. Cell Commun Signal 2010; 8:31; http://dx.doi.org/10.1186/1478-811X-8-31; PMID: 21176168
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 2005; 169:425 - 34; http://dx.doi.org/10.1083/jcb.200412022; PMID: 15866887
- Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, et al. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol 2001; 152:657 - 68; http://dx.doi.org/10.1083/jcb.152.4.657; PMID: 11266458
- Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 2003; 112:1809 - 20; PMID: 14638851
- Chan EY, Kir S, Tooze SA. siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J Biol Chem 2007; 282:25464 - 74; http://dx.doi.org/10.1074/jbc.M703663200; PMID: 17595159
- Høyer-Hansen M, Bastholm L, Mathiasen IS, Elling F, Jäättelä M. Vitamin D analog EB1089 triggers dramatic lysosomal changes and Beclin 1-mediated autophagic cell death. Cell Death Differ 2005; 12:1297 - 309; http://dx.doi.org/10.1038/sj.cdd.4401651; PMID: 15905882
- Pestka S. The interferons: 50 years after their discovery, there is much more to learn. J Biol Chem 2007; 282:20047 - 51; http://dx.doi.org/10.1074/jbc.R700004200; PMID: 17502369
- Vitale G, Zappavigna S, Marra M, Dicitore A, Meschini S, Condello M, et al. The PPAR-γ agonist troglitazone antagonizes survival pathways induced by STAT-3 in recombinant interferon-β treated pancreatic cancer cells. Biotechnol Adv 2012; 30:169 - 84; http://dx.doi.org/10.1016/j.biotechadv.2011.08.001; PMID: 21871555
- Caraglia M, Tagliaferri P, Marra M, Giuberti G, Budillon A, Gennaro ED, et al. EGF activates an inducible survival response via the RAS-> Erk-1/2 pathway to counteract interferon-alpha-mediated apoptosis in epidermoid cancer cells. Cell Death Differ 2003; 10:218 - 29; http://dx.doi.org/10.1038/sj.cdd.4401131; PMID: 12700650
- Cheriyath V, Glaser KB, Waring JF, Baz R, Hussein MA, Borden EC. G1P3, an IFN-induced survival factor, antagonizes TRAIL-induced apoptosis in human myeloma cells. J Clin Invest 2007; 117:3107 - 17; http://dx.doi.org/10.1172/JCI31122; PMID: 17823654
- Sun WL, Chen J, Wang YP, Zheng H. Autophagy protects breast cancer cells from epirubicin-induced apoptosis and facilitates epirubicin-resistance development. Autophagy 2011; 7:1035 - 44; http://dx.doi.org/10.4161/auto.7.9.16521; PMID: 21646864
- Samaddar JS, Gaddy VT, Duplantier J, Thandavan SP, Shah M, Smith MJ, et al. A role for macroautophagy in protection against 4-hydroxytamoxifen-induced cell death and the development of antiestrogen resistance. Mol Cancer Ther 2008; 7:2977 - 87; http://dx.doi.org/10.1158/1535-7163.MCT-08-0447; PMID: 18790778
- Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. Autophagy facilitates the development of breast cancer resistance to the anti-HER2 monoclonal antibody trastuzumab. PLoS One 2009; 4:e6251; http://dx.doi.org/10.1371/journal.pone.0006251; PMID: 19606230
- Abedin MJ, Wang D, McDonnell MA, Lehmann U, Kelekar A. Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ 2007; 14:500 - 10; http://dx.doi.org/10.1038/sj.cdd.4402039; PMID: 16990848
- Orvedahl A, Sumpter R Jr., Xiao G, Ng A, Zou Z, Tang Y, et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature 2011; 480:113 - 7; http://dx.doi.org/10.1038/nature10546; PMID: 22020285
- Yellen P, Saqcena M, Salloum D, Feng J, Preda A, Xu L, et al. High-dose rapamycin induces apoptosis in human cancer cells by dissociating mTOR complex 1 and suppressing phosphorylation of 4E-BP1. Cell Cycle 2011; 10:3948 - 56; http://dx.doi.org/10.4161/cc.10.22.18124; PMID: 22071574
- Sarkar S, Krishna G, Imarisio S, Saiki S, O’Kane CJ, Rubinsztein DC. A rational mechanism for combination treatment of Huntington’s disease using lithium and rapamycin. Hum Mol Genet 2008; 17:170 - 8; http://dx.doi.org/10.1093/hmg/ddm294; PMID: 17921520
- Sarkar S, Ravikumar B, Floto RA, Rubinsztein DC. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ 2009; 16:46 - 56; http://dx.doi.org/10.1038/cdd.2008.110; PMID: 18636076
- Wang RC, Levine B. Autophagy in cellular growth control. FEBS Lett 2010; 584:1417 - 26; http://dx.doi.org/10.1016/j.febslet.2010.01.009; PMID: 20096689
- Chen Q, Gong B, Mahmoud-Ahmed AS, Zhou A, Hsi ED, Hussein M, et al. Apo2L/TRAIL and Bcl-2-related proteins regulate type I interferon-induced apoptosis in multiple myeloma. Blood 2001; 98:2183 - 92; http://dx.doi.org/10.1182/blood.V98.7.2183; PMID: 11568006
- Panaretakis T, Pokrovskaja K, Shoshan MC, Grandér D. Interferon-alpha-induced apoptosis in U266 cells is associated with activation of the proapoptotic Bcl-2 family members Bak and Bax. Oncogene 2003; 22:4543 - 56; http://dx.doi.org/10.1038/sj.onc.1206503; PMID: 12881711
- Jänicke RU. MCF-7 breast carcinoma cells do not express caspase-3. Breast Cancer Res Treat 2009; 117:219 - 21; http://dx.doi.org/10.1007/s10549-008-0217-9; PMID: 18853248
- Yan J, Wang ZY, Yang HZ, Liu HZ, Mi S, Lv XX, et al. Timing is critical for an effective anti-metastatic immunotherapy: the decisive role of IFNγ/STAT1-mediated activation of autophagy. PLoS One 2011; 6:e24705; http://dx.doi.org/10.1371/journal.pone.0024705; PMID: 21931823
- Ning Y, Riggins RB, Mulla JE, Chung H, Zwart A, Clarke R. IFNgamma restores breast cancer sensitivity to fulvestrant by regulating STAT1, IFN regulatory factor 1, NF-kappaB, BCL2 family members, and signaling to caspase-dependent apoptosis. Mol Cancer Ther 2010; 9:1274 - 85; http://dx.doi.org/10.1158/1535-7163.MCT-09-1169; PMID: 20457620
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001; 25:402 - 8; http://dx.doi.org/10.1006/meth.2001.1262; PMID: 11846609
- Frankel LB, Wen J, Lees M, Høyer-Hansen M, Farkas T, Krogh A, et al. microRNA-101 is a potent inhibitor of autophagy. EMBO J 2011; 30:4628 - 41; http://dx.doi.org/10.1038/emboj.2011.331; PMID: 21915098