Abstract
Mammalian peroxisomes are ubiquitous organelles that possess a comprehensive ensemble of more than 50 enzymes. Cells regulate the number of organelles through dynamic interplay between biogenesis and degradation. Under basal conditions, approximately 30% of the peroxisomal pool is turned over daily. Recycling of peroxisomes is necessary for preservation of their functional competence, and correctly functioning autophagic/lysosomal pathways play a central role. In this study, we investigated (1) how lipopolysaccharide (LPS) influences peroxisomal dynamics and functions; and (2) how a superimposed lysosomal dysfunction affects pexophagy and modifies peroxisomal responses to LPS. We demonstrated that a transiently increased autophagic degradation of peroxisomes, pexophagy, followed by increased proliferation of peroxisomes is a default response to endotoxic stress. Impairment of autophagy due to lysosomal dysfunction, however, abolishes the above peroxisomal dynamics and results in accumulation of functionally compromised peroxisomes. These exhibit an imbalance between preserved hydrogen peroxide (H2O2)-generating acyl-CoA oxidase (ACOX) and dysfunctional/inactivated catalase (CAT), which leads to intra-peroxisomal redox disequilibrium. This metabolic-oxidative mismatch causes further worsening of peroxisomal functions, peroxisomal burnout, with the consequence of enhanced oxidative stress and aggravated organ injury.
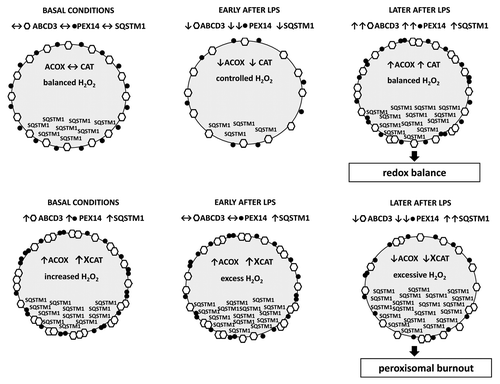
Peroxisomes were discovered more than 50 years ago and have been considered as “fossil organelles” for a long time afterwards. The importance of peroxisomes in human pathogenesis has been recognized by the discovery of disorders, like Zellweger syndrome, which are caused by specific defects of peroxisomes. In the maintenance of cell metabolic and redox homeostasis, peroxisomes closely cooperate and form a strategic partnership with mitochondria. Patients with peroxisomal disorders show major mitochondrial disturbances, which underline the relevance and implications of peroxisomal dysfunction. Among the numerous metabolic tasks performed, peroxisomal fatty acid β-oxidation (FAO) is one of the most important. Peroxisomal oxidases are unique due to the generation of large amounts of H2O2, which explains the highest cellular content of CAT in peroxisomes. Peroxisomes contain more than 30 specific peroxisomal proteins, peroxins. ABCD3/PMP70 is an integral membrane protein that, among other functions, participates in the transport of fatty acids across the peroxisomal membrane. PEX14, a peroxin, is a smaller protein with a crucial role in peroxisomal protein import (i.e., import of CAT). We showed that LPS temporarily induces pexophagy, as manifested by decreased ABCD3 and PEX14 levels, and a reduced density of peroxisomes in the vicinity of lysosomes. LPS exhibited a more profound effect on PEX14 than on ABCD3, thus compromising the protein import (e.g., of CAT) into peroxisomes. It is possible that ABCD3, as a large membrane-incorporated protein, resists LPS-induced stress better than the smaller membrane-associated PEX14. The initial activation of pexophagy with decline of peroxins after LPS is followed by increased peroxisomal proliferation and upregulation of ABCD3 and PEX14. In contrast to this, lysosomal dysfunction results in an accumulation of peroxisomes already under basal conditions. Stimulation with LPS does not significantly affect peroxisomal turnover, and the biphasic pattern observed in lysosome-competent cells is completely abolished in lysosome-defective cells. Moreover, in the presence of lysosomal dysfunction, 24 h after LPS application, ABCD3 and PEX14 levels progressively decrease, yet again with a more pronounced decline of PEX14. Under normal conditions, the SQSTM1/p62 protein shows a similar biphasic expression as peroxins, with an early decrease and subsequent upregulation after LPS. In lysosome-defective cells, the biphasic pattern disappears; however, in contrast with peroxins, 24 h after LPS, SQSTM1 levels progressively increase. This indicates that LPS exhibits a stimulatory effect on SQSTM1 expression (despite an early temporary decrease through activated autophagy in lysosome-competent cells). We propose that the functional significance of elevated SQSTM1 levels is different under normal conditions (mainly newly synthesized SQSTM1) and under conditions of lysosomal dysfunction (predominantly accumulated and potentially damaged SQSTM1). Cytotoxic effects of increased SQSTM1 levels due to defective autophagy have been previously reported. The distinct mechanisms of SQSTM1 upregulation could probably explain the controversies in the literature surrounding the role of SQSTM1. We also demonstrated that lysosome-defective cells exhibit higher levels of oxidative stress and proinflammatory cytokines. Toward this end, we examined the expression of the H2O2 producing and rate-limiting peroxisomal FAO enzyme ACOX, as well as CAT in vivo using lysosome-defective mice (LYS). Early after LPS injection, wild-type (WT) mice show decreased ACOX levels/activity and decreased CAT protein levels with preserved activity of the enzyme. Later, this primary decrease is followed by an increase and restoration of both ACOX and CAT levels. Similarly, ABCD3 and PEX14 show biphasic expression in WT mice. LPS-treated LYS mice show initially higher ACOX levels/activity and higher but inactivated CAT compared with WT mice. The consequence of the imbalance between active ACOX and dysfunctional CAT in LYS mice is excessive H2O2 generation, deterioration of peroxisomal homeostasis, and decline of both ACOX and CAT 24 h after LPS injection. These distinct metabolic-oxidative patterns detected in WT- and LYS mice are additionally supported by analysis of free fatty acid and lipid peroxide tissue levels in these animals. Peroxisome proliferator-activated receptor α (PPARA), the principal inducer of peroxisomes, demonstrates an abnormally increased activity in LYS mice (contrary to repressed activity in WT mice) that potentially contributes to the preservation of ACOX. The factors involved in PPARA dysregulation in impaired autophagy remain elusive, although some authors indicate a causal role of unmetabolized phytanic acid, a peroxisomal substrate. On the other hand, PPARA upregulation can represent a compensatory mechanism to activate nonresponding peroxisomes, which in the case of ACOX-CAT functional imbalance produces an opposite effect. Similar metabolic–oxidative patterns were described in patients with hereditary peroxisomal disorders. We demonstrated that these inherited patterns are recapitulated during pathogenesis of acquired nonperoxisomal diseases. Our model of LPS-mediated acute injury represents an extreme example, and we think that the described pathogenetic constellations occur at much lower intensities under chronic conditions. For example, studies showing positive effects of PPARA stimulation in early diabetic nephropathy were unable to reproduce these effects in advanced disease. Some degree of lysosomal dysfunction is a common companion of chronic diseases, which may result in subtle but ongoing impairment of autophagy/pexophagy, and modify the progression and therapeutic response of the disease ().
Figure 1. (A) Lysosome-competent cells. Under normal conditions, LPS activates pexophagy and induces a biphasic expression response, with an early decrease followed by later upregulation of ABCD3 and PEX14, and increased proliferation of peroxisomes. Both, H2O2-generating ACOX and H2O2-decomposing CAT are decreased, thus preserving peroxisomal H2O2 equilibrium. (B) Lysosome-defective cells. Lysosomal dysfunction inhibits pexophagy, which results in a higher number of peroxisomes under basal conditions. LPS stimulation of lysosome-defective cells does not affect peroxisomal turnover. Cells with accumulated dysfunctional peroxisomes exhibit impaired enzymatic activity of CAT, which together with functioning ACOX creates H2O2 disequilibrium and enhances oxidative stress. Later, this metabolic-oxidative imbalance results in a global deterioration of peroxisomal functions— peroxisomal burnout—and in an aggravated organ injury.
| Abbreviations: | ||
| ABCD3 | = | ATP-binding cassette, sub-family D (ALD), member 3 |
| ACOX | = | acyl-CoA oxidase |
| CAT | = | catalase |
| FAO | = | fatty acid oxidation |
| H2O2 | = | hydrogen peroxide |
| LPS | = | lipopolysaccharide |
| PEX14 | = | peroxisomal biogenesis factor 14 |
| PPARA | = | peroxisome proliferator-activated receptor alpha |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.