Abstract
Autophagy resembles a recycling process in which proteins, organelles, or regions of the cytoplasm are enveloped and degraded. We have found that two of the central autophagy proteins, MAP1LC3 (microtubule-associated protein 1 light chain 3, also described as LC3) and UVRAG (UV radiation resistance associated/UV radiation associated gene), complex with PGRMC1/S2R (progesterone receptor membrane component 1, also known as sigma-2 receptor). PGRMC1 is a cytochrome that is induced in cancer and is essential for tumor formation, invasion, and metastasis. Autophagy contributes to the turnover of long-lived and/or ubiquitinated proteins and the clearance of damaged organelles, and we have shown that PGRMC1 promotes both processes. Inhibition of PGRMC1 by RNAi or small molecule inhibitors causes autophagy substrates to increase and aberrant mitochondria to accumulate. We propose that this disruption of autophagy upon PGRMC1 inhibition increases AMPK activation, elevating the levels of TSC1 (tuberous sclerosis complex) and TSC2 and inactivating MTOR and RPS6KB/p70S6K, causing cleaved MAP1LC3B levels to increase. Thus, PGRMC1 binds to key components of the autophagy machinery and is required for the degradative activity of autophagy.
Keywords: :
Introduction
Autophagy resembles a recycling system in which cells traffic or envelop aged and misfolded proteins, organelles, or regions of the cytoplasm.Citation1 When autophagy is activated, autophagosomes engulf cargo, which can include entire regions of the cytoplasm, and fuse with other microsomes, including endosomes and lysosomes.Citation1 Autophagy is also essential for the clearance of polyubiquitinated protein aggregates and mitochondria.Citation2,Citation3 During autophagosome elongation and expansion, MAP1LC3 is cleaved and lipidated,Citation4,Citation5 forming LC3-II, and inserted into the membranes of the autophagosome. The entire autophagy process is regulated by kinasesCitation1,Citation2 and multiprotein complexes, and Behrends, et al. have used protein interaction screening to globally characterize complexes controlling autophagy. LC3 and related proteins were central to numerous complexes.Citation6 Another key complex includes BECN1 and UVRAG, which participate in vesicle trafficking and fusion via binding to vesicle sorting proteins.Citation7-Citation9 We demonstrate here that LC3 and UVRAG associate with progesterone receptor membrane component 1 (PGRMC1), also known as the sigma-2 receptor.
PGRMC1 has been studied for decades in various tissues as an intracellular drug binding site that may contribute to signaling and homeostasis.Citation10 PGRMC1 is a microsomal protein related to cytochrome b5.Citation11-Citation16 PGRMC1 expression is elevated in a number of tumor typesCitation13 and is associated with overall survival in lung cancerCitation17 and tumor stage in ovarian cancer.Citation18 Many other studies have linked PGRMC1 to proliferation and tumor progression using PGRMC1 ligands.Citation19-Citation21 PGRMC1 was originally identified as a putative hormone receptorCitation14,Citation16 but it remains unclear whether PGRMC1 directly binds hormones, whereas multiple groups have confirmed that recombinant PGRMC1 binds heme.Citation12,Citation22-Citation24
In normal tissues, PGRMC1 increases lipid synthesis via P450 proteins,Citation23,Citation25-Citation27 while in tumor cells, it has a profound effect on cell signaling.Citation28-Citation30 Specifically, PGRMC1 associates with EGFR in lung and breast cancer cells, where it elevates plasma membrane levels of the receptor.Citation31 In cancer cell lines, we have shown that PGRMC1 increases proliferation,Citation29 invasion,Citation29 tumor growth,Citation29,Citation32 and metastatic colonization.Citation29 Others have reported that PGRMC1 promotes tumor formation and suppresses apoptosis in ovarian cancer.Citation18,Citation33,Citation34 Various PGRMC1 ligands have also been used to initiate cancer cell death and are associated with a lysosomal cell death pathway.Citation21,Citation35-Citation41
In the present study, we demonstrated that PGRMC1 coprecipitates with LC3B-II and UVRAG. Inhibition of PGRMC1 decreases autophagic flux and arrests the degradation of the autophagy substrate SQSTM1 causing aberrant mitochondria to accumulate. We propose that arrested autophagy stimulates further proautophagy signaling, and we observed increased AMPK phosphorylation, TSC1 and TSC2 levels and decreased phosphorylation of MTOR and RPS6KB. We also observed increased LC3B-II levels and morphological changes characteristic of autophagy, likely due to changes in proautophagy signaling. The link between PGRMC1 and autophagy suggests a key role in metabolism that can be targeted with PGRMC1 ligands.
Results
PGRMC1 increases cleaved LC3B without SQSTM1 degradation
Recently, proteomic protein interaction screens have identified PGRMC1 as a binding partner for numerous proteins in autophagy,Citation6 including members of the MAP1LC3 family. To assess the role of PGRMC1 in autophagy, we used a pulse-chase autophagic flux assay with 3H-leucine to measure the breakdown of long-lived proteins by autophagy. As expected, autophagy levels were modest in the presence of serum and were strongly induced by serum starvation (). A PGRMC1 inhibitor, AG-205,Citation10,Citation29,Citation42 reduced 3H-leu release to a significant extent (P = 0.02, t-test) 24 h after a “chase” with unlabeled leucine (, solid line, triangles). This effect occurred in the absence of serum, when autophagy was elevated (, solid line, squares), and AG-205 treatment was not significantly different from treatment with bafilomycin A1 (, dashed lines, triangles, and diamonds, P = 0.6, t-test). Even though AG-205 inhibited autophagic flux, it induced a shift in LC3B localization to a punctate pattern (). The number of LC3B puncta per cell increased from 3.7 ± 1.2 in the vehicle-treated cells to 45 ± 9.5 in the AG-205-treated cells, which was significant (P = 0.002, t-test). A similar change was observed with GFP-LC3B in control and AG-205-treated cells (P = 0.0007).
Figure 1. A PGRMC1 ligand inhibits autophagic flux and increases MAP1LC3B-II. (A) In an autophagic flux assay, serum-fed (dashed lines) or serum-starved (solid lines) A549 cells were vehicle-treated (squares) or treated with 20 μM AG-205 (triangles) or 100 nM bafilomycin A1 (diamonds) for 16 h. Label was then “chased” for 4 h and samples were collected thereafter. Each point is derived from three independent samples, and the entire experiment is representative of four independent assays. The error bars indicate standard deviation. (B) LC3B localized by immunofluorescence to cytoplasmic puncta after AG-205 treatment for 24 h (lower panel). Scale bar: 20 μm. (C) Cells were treated with a vehicle control (lanes 1, 3, and 5) or with the PGRMC1 ligand AG-205 (lanes 2, 4, and 6, 20 μM). Cells were grown in 10% serum (lanes 1–2) or 0% serum without glutamine or glucose (lanes 3 to 6), and lysates were analyzed by western blot for LC3B (upper panels), SQSTM1 (middle panels), and GAPDH (lower panels). In lanes 5 and 6, cells were treated with 100 nM bafilomycin A1. With AG-205 treatment, LC3B-II increased, but there was no change in the levels of SQSTM1, a primary substrate of autophagy. The statistics and quantification for western is shown in Figure S1. (D) A549 cells were treated with 20 μM AG-205 (PGRMC1 ligand) for 0 to 72 h, resulting in increased LC3B-II. (E and F) NB-7 neuroblastoma cells were treated with 0, 50, 15, or 5 μM AG-205 (E) or haloperidol (F) and analyzed as for (C). (G) Electron microscopy showing A549 cells treated with vehicle control in 10% serum (top panel), vehicle control in 0% serum (middle panel), and AG-205 in 10% serum (lower panel). Scale bar: 10 μm. Overall, the results indicate that in multiple cell lines, PGRMC1 ligands initiate autophagy (indicated by LC3 cleavage) that is arrested (indicated by failure to degrade SQSTM1 and decrease starvation-induced increase amino acid release). Throughout, the results shown are representative of at least three independent experiments.
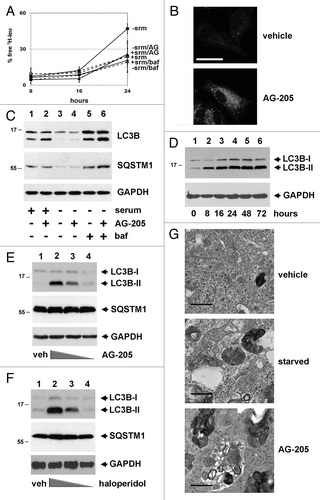
Consistent with the change in LC3B localization, AG-205 induced cleaved, modified LC3B-II levels rapidly in a time- and dose-dependent manner (). However, the levels of SQSTM1 were unchanged (, middle panel, lanes 1 and 2). Densitometry and statistics for western blots are shown in Figure S1. As a positive control, serum deprivation decreased SQSTM1 (a key substrate of autophagy) levels by 24 h (, second panel, lanes 3 and 4). The addition of bafilomycin A1 reversed LC3B-II and SQSTM1 degradation (, lanes 5 and 6). utilized A549 human lung cancer cells, and the same induction by AG-205 of LC3B-II with unchanged SQSTM1 was found in NB7 neuroblastoma cells () and NCI-H226 squamous lung cancer cells (Fig. S2A). The PGRMC1 ligand haloperidol also increased LC3B-II in NB-7 cells (). Like the expression analyses and flux assay, AG-205 inhibited the colocalization of LC3 and LAMP1 by immunofluorescence (Fig. S2B), similar to bafilomycin A1.
The results suggested that autophagosome formation is increased by AG-205, while autophagic proteolytic activity is decreased. Electron microscopy revealed few autophagosomes in vehicle-treated serum-replete cells (, upper panel) and large vesicular structures engulfing other cytoplasmic vesicles in serum-starved cells (, middle panel). AG-205 increased the presence of large vesicles with abundant engulfed cargo, consistent with autophagosomes (, lower panel). In a recent paper studying neurons, Park, et al. concluded that haloperidol induced LC3-II levels by blocking autophagosome formation rather than initiating upstream events in autophagic flux.Citation43 Our findings suggest a slightly different interpretation, that PGRMC1 ligands arrest events occurring after autophagosome formation in cancer cells.
We then utilized a stable knockdown of PGRMC1, and found that the release of 3H-leu in serum-starved PGRMC1-knockdown cells decreased significantly compared with control cells at 16 and 24 h (P = 0.02 and 0.03, respectively, ), while there was no significant difference between PGRMC1-knockdown cells and control cells treated with bafilomycin A1 (). The flux assay suggested that autophagy is arrested in PGRMC1-knockdown cells, and we posited that mitochondrial turnover would be inhibited. Mitochondrial staining with MitoTracker Deep Red was increased in PGRMC1-knockdown cells (, top panels). Furthermore, the mitochondrial morphology was aberrant by electron microscopy in PGRMC1-knockdown cells (, lower panels). The Seahorse XF Mito Stress Test analysis confirmed that the basal oxygen consumption rate increased significantly (P = 0.006) in PGRMC1-knockdown cells (), and maximal respiration trended higher (Fig. S3, P = 0.06) consistent with elevated mitochondrial numbers. Surprisingly, there was also no detectable change in lysosomal acidification in PGRMC1-knockdown cells, either by FACS or visual microscopy (Fig. S4), as has been reported in PGRMC1 ligand studies.Citation37
Figure 2.PGRMC1-knockdown disrupts autophagy. (A) Control (solid line, squares, “con”) and PGRMC1-knockdown (solid line, triangles, “shPGR”) A549 cells were analyzed for autophagic flux of 3H-leucine following 24 h of growth in media without serum and a 4 h “chase” period. PGRMC1-knockdown cells had a decrease in free 3H-leu release, which was not significantly different from control cells treated with 100 nM bafilomycin A1 (“baf”). Error bars represent standard deviation of triplicate measurements, and the data shown are representative of triplicate experiments. (B) Elevated MitoTracker Deep Red staining in PGRMC1-knockdown cells (upper right panel) compared with control cells (upper left panel). In the upper panel, the size bar represents 100 μm. Electron microscopy revealed an aberrant mitochondrial morphology in PGRMC1-knockdown cells (lower right panel), in which mitochondria have fewer christae compared with control cells (lower left panel). Scale bar: 1 μm. (C) PGRMC1-knockdown cells have elevated basal mitochondrial activity, (P = 0.006) measured as oxygen consumption rate (OCR, pmole/min/104 cells) but otherwise normal mitochondrial parameters. Error bars represent the standard deviation for five measurements, and the graph is representative of experiments performed in triplicate. Throughout the manuscript, *P ≤ 0.05; **P ≤ 0.01; and ***P ≤ 0.005. (D) LC3B, detected by immunofluorescence, was diffuse in control (top left) and punctate in PGRMC1-knockdown A549 cells (top right). Error bar: 20 μm. In the same cells, electron microscopy revealed increases in crescent-shaped autophagosomes (right, arrowheads) in PGRMC1-knockdown cells. Scale bars: 0.5 μm. (E) A549 control (lanes 1–3) or PGRMC1-knockdown (lanes 4 to 6) cells were maintained for 24 h in media with 10% serum (lanes 1 and 4) or DMEM without glutamine, glucose or serum (lanes 2 to 3 and 5 to 6). In lanes 3 and 6, 100 nM bafilomycin A1 was included. Lysates were analyzed by western blot for LC3B (top), SQSTM1 (middle), and GAPDH (bottom, loading control). LC3B-II increased in PGRMC1-knockdown cells grown in serum, while SQSTM1 levels were unaffected, and the difference in LC3B-II levels was suppressed by inhibiting autophagy. (F) Control (lanes 1–3) and PGRMC1-knockdown (lanes 4 to 6) A549 cells were transfected with a control plasmid (lanes 1 and 4), a plasmid encoding PGRMC1 (pRC40, lanes 2 and 5) or a plasmid encoding a heme-binding-deficient D120G mutant of PGRMC1 (pRC42, lanes 3 and 6). Lysates were analyzed for LC3B (top), SQSTM1 (middle), and GAPDH (bottom). LC3B-II was elevated in PGRMC1-knockdown cells and reversed by PGRMC1 (lane 5), but not the PGRMC1-hbd mutant.
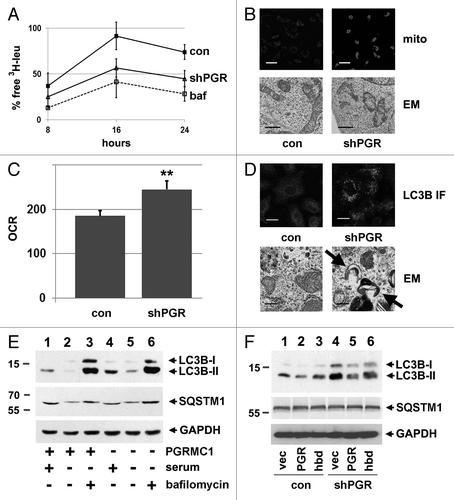
As for AG-205-treated cells, LC3 shifted in localization from a diffuse to a punctate pattern in PGRMC1-knockdown cells (), with a 5.2-fold increase in puncta, which was significant (P = 4 × 10−5, t-test). In addition, electron microscopy revealed an increased number of curved, membranous structures resembling autophagosomes in PGRMC1-knockdown cells (, lower panels). In PGRMC1-knockdown cells, the level of LC3B-II increased compared with control cells (, top panel, lanes 1 and 4) and was statistically significant (P = 0.02, t-test, Fig. S1E). SQSTM1 levels were not significantly affected in PGRMC1-knockdown cells (, second panel, lanes 1 and 4), and the SQSTM1 isoform 1 transcript did not change significantly (0.05 ± 0.01 vs. 0.06 ± 0.1 SQSTM1:actin ratio for control and PGRMC1-knockdown cells respectively). Serum starvation decreased LC3B-II and SQSTM1 levels in control and PGRMC1-knockdown cells (, lanes 2 and 4) through a bafilomycin A1-sensitive process (, lanes 3 and 6).
LC3B-II levels in PGRMC1-knockdown cells were partially complemented by exogenous expression of PGRMC1 (, top panel, lanes 4 and 5). In triplicate measurements in PGRMC1-knockdown cells, LC3B-II levels were lower in cells expressing PGRMC1 compared with a control vector (P = 0.03, t-test), while the PGRMC1 heme-binding-deficient mutant (D120G), did not complement to a significant extent (; Fig. S1F). For experiments using transfected cells, there was an increase in basal levels of LC3-II (, lane 1), which has been reported previously.Citation44 PGRMC1 knockdown with siRNA produced the same effect—increased LC3-II with no change in SQSTM1 levels (Fig. S2C). The results support a model in which PGRMC1 suppresses LC3-II levels but is required for autophagic proteolytic activity.
PGRMC1 coprecipitates with MAP1LC3B-II
Proteomic-based protein interaction screens have predicted PGRMC1 as a binding partner for numerous proteins in autophagy,Citation6 and we found that PGRMC1 precipitates (, top panel) contained abundant LC3B-II in HEK293 cells (, second panel). AG-205 is a PGRMC1 ligand that induces LC3B-II (), and PGRMC1-LC3B-II binding was increased after AG-205 treatment (, second panel, compare lanes 1 and 2), likely because AG-205 increased the abundance of LC3B-II (, lower panel). LC3B-II bound with greater affinity to a PGRMC1D120G heme binding-deficient (hbd) mutant (, second panel, lanes 1 and 2), suggesting that heme antagonizes PGRMC1-LC3B-II coupling.
Figure 3. PGRMC1 associates with LC3B-II. (A) HEK293 human embryonic kidney cells were transfected with plasmid pRC40, encoding HA-tagged PGRMC1 (HA-PGR, lanes 1 and 2), or a control plasmid (lane 3), immunoprecipitated with an anti-HA epitope tag antibody and analyzed by western blot for HA-PGRMC1 (top panel) or LC3B (second panel). In lane 2, the cells were treated with 20 μM AG-205 for 24 h. The lower panels show PGRMC1 and LC3B levels in the lysates. (B) PGRMC1 (lane 1), the heme-binding deficient (hbd) D120G mutant of PGRMC1 (lane 2) or a control plasmid were expressed, precipitated and analyzed as in (A). (C) A549 non-small cell lung cancer cells were transfected with a control plasmid (lane 1) or the PGRMC1 expression plasmid pRC40 and treated with 20 μM AG-205 for 24 h. Lysates (lower panels) were immunoprecipitated with an anti-HA tag antibody and analyzed by western blot for LC3B (top) or HA-PGRMC1 (bottom). The lower band is due to the antibody light chain, which comigrates with PGRMC1. (D) LC3B was immunoprecipitated from A549 cells (lane 2) and probed for LC3B (top) or PGRMC1 (bottom). Lane 1 is an IgG control, and LC3B was weakly detectable. The input lysates are shown in the lower panels.
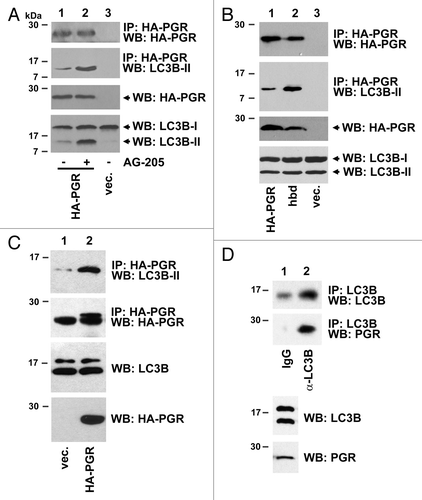
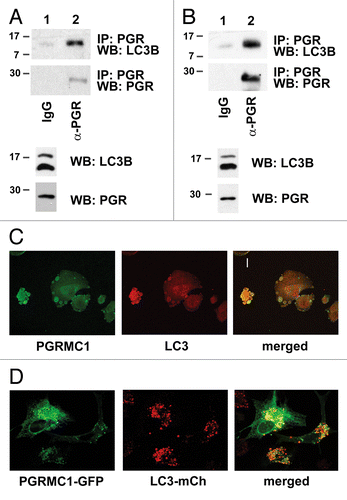
We then tested whether PGRMC1 forms a complex with LC3B-II in cancer cells. In A549 cells transfected with a PGRMC1 expression plasmid, LC3B-II was readily detected in PGRMC1 precipitates (, upper panel). In untransfected A549 cells, PGRMC1 was present in immunoprecipitations of LC3B (). LC3B was also readily detected in PGRMC1 precipitates from untransfected A549 () and HEK293 () cells. Furthermore, PGRMC1 and LC3B colocalized to cytoplasmic vesicles by immunofluorescence () and by coexpression of fluorescent-tagged PGRMC1 and LC3B proteins (). We note that the localization of the endogenous and fluorescent-tagged proteins were slightly different, possibly reflecting the expression of the chimeric proteins, and fluorescent-tagged PGRMC1 was frequently detected in large cytoplasmic vesicles (Fig. S5). We did not detect complete overlap between PGRMC1 and LC3B fluorescence in all cells. Indeed, PGRMC1 did not colocalize with LysoTracker fluorescence, a marker for lysosomes and other highly acidic vesicles (Fig. S5A), but PGRMC1 did have substantial overlap with RAB5, a marker of endosomes (Fig. S5B). The results suggest that PGRMC1 localizes to endosomes and autophagosomes, where it overlaps with LC3B-II.
Figure 4. Endogenous PGRMC1 associates with LC3B. (A) A549 cells were immunoprecipitated with rabbit IgG or an equal amount of anti-PGRMC1 (α-PGR) antibody and analyzed for LC3B (top) or PGRMC1 (bottom). For (A and B), the lysates used in the precipitation were analyzed by western blot (lower panels) as indicated. (B) The same experiment as (A) was performed with lysates from HEK293 cells, where precipitates were analyzed for LC3B (top) or PGRMC1 (bottom), and the input lysates were measured in the two lower panels. (C) PGRMC1 (left) and LC3B (center) were analyzed by immunofluorescence in A549 cells starved for 24 h, and the proteins colocalized (right) in cytoplasmic vesicles. Scale bar: 20 μm. (D) PGRMC1-GFP (left) and LC3B-mCherry (right) were coexpressed in A549 cells and colocalized (right) in cytoplasmic puncta. Scale bar: 20 μm.
PGRMC1 activates MTOR signaling
AMPK acts as a sensor of amino acid pools, which are a product of autophagy, and AMPK promotes autophagy by inhibiting MTOR. MTOR is a PtdIns3K-related protein kinaseCitation45-Citation47 that regulates translation and energy metabolism.Citation48 MTOR is inhibited by the tumor suppressor TSC (tuberous sclerosis), a complex of TSC1 and 2,Citation48-Citation51 and TSC1 is an AMPK substrate.Citation52 To test the model that autophagy signaling is altered in PGRMC1-knockdown cells, we used a proteome profiler antibody array for intracellular kinases and found that phosphorylation of Thr174 in PRKAA1/AMPKa1 and PRKAA2/AMPKa2 was increased in PGRMC1-knockdown cells, while MTOR and RPS6KB phosphorylation decreased (; Fig. S6). Phosphorylation of the MTOR substrate TP53/p53 decreased significantly, but to a lesser extent, and RPS6K phosphorylation was also inhibited (). The experiments were performed in serum-free media to minimize additional effects of EGFR signaling on the AMPK-MTOR pathway.Citation31
Figure 5. PGRMC1 inhibits AMPK signaling. (A) Kinase signaling intermediates in control (gray bars) and PGRMC1-knockdown A549 cells (shPGR, black bars) were screened using proteome profiler antibody array, and significant changes in the MTOR pathway are indicated. Error bars represent standard deviation of four measurements, and the array profiling was performed in duplicate. Cells were serum-starved for 24 h prior to this analysis and for the western blots in (B and C). (B) Decreases in MTOR signaling were verified by western blot analysis, showing decreased MTOR-Ser2448 and RPS6KB-Thr421 phosphorylation in PGRMC1-knockdown cells. (C) Increased AMPK-Thr174 phosphorylation in PGRMC1-knockdown cells was verified by western blot (second panel), which also revealed decreased AMPK levels (top) and increased TSC1 and TSC2 levels (lower panels), which are downstream of AMPK. The samples in (B and C) are identical, so the same loading control is shown for each. (D) Quantification and statistical analysis of the results from (C), measured in duplicate as band intensity relative to the loading control.
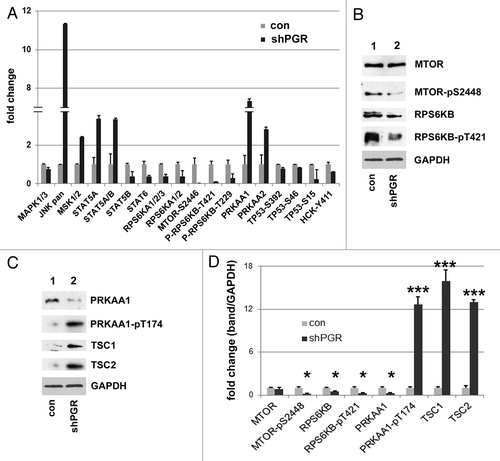
We verified by western blot that MTOR and RPS6KB phosphorylation is inhibited in PGRMC1-knockdown cells (, quantified in ). While AMPK phosphorylation increased, as expected (, second panel), AMPK levels declined (, top panel), but TSC1 and 2 were elevated (, lower panels). We propose that arrested autophagy in PGRMC1-knockdown cells deprives cells of amino acids, activating AMPK and inhibiting MTOR, a major suppressor of autophagy.Citation49 However, we cannot exclude the possibility that pathways other than autophagy, such as EGFR signaling, may contribute to the suppression of MTOR.
PGRMC1-UVRAG complexes alter levels of ubiquitinated proteins
LC3 has numerous binding partners, including SQSTM1 and tubulin,Citation53,Citation54 and we found that in LC3B immunoprecipitates (, top panel), LC3B-SQSTM1 increased in PGRMC1-knockdown cells (, second panel), with a similar increase in TUBA/α-tubulin binding (, lower panel). In contrast, SQSTM1 and TUBA levels changed little in the source lysates (). The primary function of LC3B-bound SQSTM1 is thought to involve ubiquitinated proteins,Citation54 and we found that ubiquitinated proteins were elevated in lysates from PGRMC1-knockdown cells (, lanes 1 and 2). Furthermore, AG-205 treatment increased the levels of ubiquitinated proteins (, lanes 3 and 4). In triplicate experiments, AG-205 treatment increased ubiquitination by 2.7-fold (Fig. S1G), which was significant (P = 0.003, 2-sided t-test).
Figure 6. PGRMC1 promotes the degradation of ubiquitinated proteins. (A) LC3B was immunoprecipitated from control cells (lane 2) or PGRMC1-knockdown cells (shPGR, lane 3). Lane 1 is an identical precipitation reaction of control cell lysates (identical to lane 2) with a control antibody. Precipitates were analyzed for LC3B (top), SQSTM1 (second panel) or TUBA (third panel). (B) The loading controls for the source lysates for (A) revealed little change in SQSTM1 and TUBA levels. (C) Lysates from control (lane 1) or PGRMC1-knockdown cells (lane 2) were probed by western blot for ubiquitinated proteins. In lanes 3 and 4, A549 cells were treated with a vehicle (lane 3) or 20 μM AG-205 (lane 4) for 24 h. (D) Control cells (lanes 1 to 3) or PGRMC1-knockdown cells (lanes 4 to 6) were grown in serum-containing media, treated with vehicle (lanes 1 and 4), 30 μM chloroquine (lanes 2 and 5) or 100 nM bafilomycin A1 (lanes 3 and 6) for 24 h and analyzed for LC3B (top), ubiquitin (middle panel) or TUBA (lower panel). (E) A549 cells were treated with vehicle (lanes 1–3) or 20 μM AG-205 (lanes 4 to 6) and bafilomycin A1 or chloroquine, as described for (D) and analyzed by western blot as for (D).
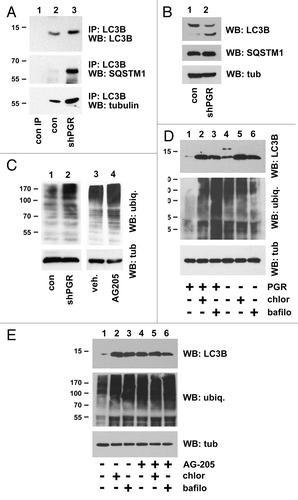
The results suggested that PGRMC1 promoted the degradation of ubiquitinated proteins via autophagy, and if true, addition of autophagy inhibitors should have no additional effect on ubiquitinated protein levels. As expected, control cells treated with bafilomycin A1 and chloroquine had elevated levels of ubiquitinated proteins after 24 h of treatment (, middle panel, lanes 2 and 3; Fig. S1H), but the same proteins did not increase in PGRMC1-knockdown cells after bafilomycin A1 and chloroquine treatment (, middle panel, lanes 5 and 6). Similarly, ubiquitinated proteins did not increase in cells treated with AG-205 combined with bafilomycin A1 or chloroquine (, middle panel, lanes 5 and 6). The results support a model in which PGRMC1 inhibits the degradation of ubiquitinated proteins through bafilomycin A1 and chloroquine-sensitive mechanisms. Under the same conditions, LC3 was similar to ubiquitinated proteins. AG-205 caused LC3B-II to accumulate, and chloroquine and bafilomycin A1 had no additional effect (, upper panel, lanes 5 and 6; Fig. S1J). Surprisingly, bafilomycin A1 and chloroquine treatment increased LC3B-II levels in PGRMC1-knockdown cells (, upper panel, lanes 5 and 6; Fig. S1H), suggesting that the PGRMC1-knockdown may inhibit LC3B-II generation in addition to its degradation. It is possible that the pathways affected by rapid treatment with an inhibitor are slightly different from those of a stable knockdown. We conclude that LC3B-SQSTM1 binding is elevated at the autophagy arrest point in PGRMC1-knockdown cells, while the degradation of ubiquitinated proteins is inhibited. However, we cannot exclude the possibility that PGRMC1 also inhibits the ubiquitination machinery.
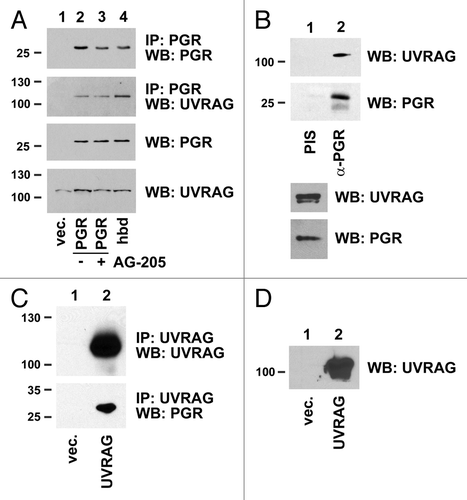
The traffic of ubiquitinated proteins requires the fusion of autophagic vesicles with lysosomal vesicles, and one of the key proteins in this process, UVRAG, is a predicted PGRMC1 binding partner.Citation6 We confirmed the PGRMC1-UVRAG interaction in HEK293 cells expressing PGRMC1 (, top two panels). In contrast to LC3B-II, AG-205 treatment did not affect PGRMC1-UVRAG binding (, second panel, lanes 2 and 3), although UVRAG bound with greater affinity to the PGRMC1D120G heme-binding-deficient (hbd) mutant (, second panel, lanes 2 and 4). UVRAG coprecipitated with PGRMC1 in A549 lung cancer cells (), where preimmune serum for the PGRMC1 antibody served as a negative control. PGRMC1 was also present in UVRAG immunoprecipitates (), and fluorescent forms of the two proteins colocalized in punctate sites A549 lung cancer cells (Fig. S5C).
Figure 7. PGRMC1 associates with UVRAG. (A) HEK293 cells were transfected with a control plasmid (lane 1), pRC40, encoding PGRMC1 (PGR, lanes 2 and 3), or a plasmid encoding heme-binding deficient PGRMC1 (hbd, lane 4). In lane 3, cells were treated with 20 μM AG-205 for 24 h. PGRMC1 was immunoprecipitated and analyzed by western blot for PGRMC1 (top panel) or UVRAG (second panel). The lower panels show PGRMC1 and UVRAG levels, respectively, in the lysates. (B) PGRMC1 (lane 2) was immunoprecipitated from UVRAG-transfected A549 cells and probed for UVRAG (top panel) or PGRMC1 (lower panel). Lane 1 shows an identical precipitation reaction with preimmune serum from the same animal (PIS). The offset panel shows UVRAG expression in the lysate. (C) UVRAG fused to a flag epitope tag was expressed in A549 cells, immunoprecipitated and analyzed by western blot for UVRAG (top panel) or PGRMC1 (lower panel). (D) The input loading control is shown for (C).
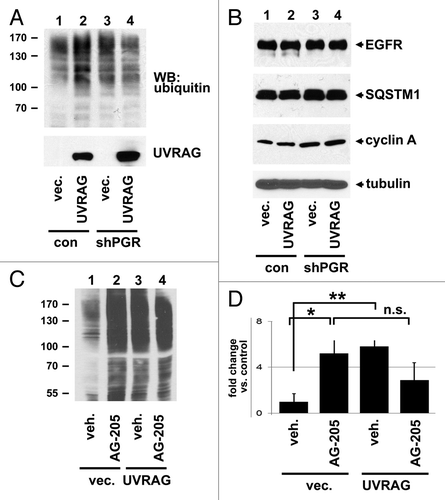
While LC3B-SQSTM1 binding corresponded with elevated levels of ubiquitinated proteins, UVRAG has been implicated in the turnover of ubiquitinated EGFR.Citation8,Citation9 In contrast, UVRAG overexpression in A549 cells caused an increase in the pool of ubiquitinated proteins (; Fig. S1L) but had no effect on EGFR levels (). CCNA1/Cyclin A1, which is degraded via ubiquitination, was also not affected by UVRAG expression (), suggesting that the effect of UVRAG is specific to certain classes of proteins. Levels of SQSTM1 were unaffected, suggesting that UVRAG did not dramatically induce autophagy in this setting (). As for , PGRMC1-knockdown increased the pool of ubiquitinated proteins (, lanes 1 and 3), but the ability of UVRAG to increase ubiquitinated protein levels was lost in PGRMC1-knockdown cells (, lane 4). Similarly, A549 cells treated with AG-205 or expressing UVRAG increased protein ubiquitination (, lanes 2 and 3, P = 0.009 and 0.01, respectively), but the combined treatment was not additive and not significantly different from either treatment alone (, lanes 2 and 4, P = 0.12, t-test). The results (plotted in ) are consistent with a model in which UVRAG suppresses the degradation of ubiquitinated proteins, an effect antagonized by PGRMC1.
Figure 8. PGRMC1 counteracts UVRAG in regulating ubiquitinated protein levels. (A) Control (lanes 1 and 2) and PGRMC1-knockdown A549 cells (shPGR, lanes 3 and 4) were transfected with a control plasmid (lanes 1 and 3) or a UVRAG expression vector (lanes 2 and 4), maintained in serum, and analyzed by western blot for ubiquitin (top) or UVRAG (bottom). (B) The lysates from (A) were analyzed by western blot for EGFR, SQSTM1, CCNA1/cyclin A1, and TUBA. (C) A549 cells were transfected with a control plasmid (lanes 1 and 2) or a UVRAG expression vector (lanes 3 and 4) and treated with either vehicle (lanes 1 and 3) or the PGRMC1 ligand AG205 at 20 μM (lanes 2 and 4). Samples were then analyzed for ubiquitin. (D) Quantification of triplicate experiments described in (C), where the error bars represent standard deviation of triplicate measurements. The experiment was performed in quadruplicate. The results suggest that PGRMC1 associates with UVRAG and that UVRAG requires PGRMC1 to increase levels of ubiquitinated proteins.
Discussion
PGRMC1 has been implicated in multiple steps in growth and survival, including proliferation, apoptosis resistance, lysosomal maintenance, receptor signaling, anchorage-independent growth, invasion, tumor formation, metastasis, and angiogenesis. PGRMC1 is attractive as a therapeutic target because it has numerous ligands, including investigational compounds and FDA-approved drugs with an extensive clinical history. Many of these compounds have antiproliferative activity, and some induce nonapoptotic cell death through a lysosomal degradation pathway. PGRMC1 has been implicated in autophagy previously through ligand-binding studies, but until recently, the identity of the sigma-2 receptor was unknown. In the present study, we provide a mechanistic link between PGRMC1 and key events in autophagy and ubiquitinated protein degradation.
We have found that PGRMC1 binds to MAP1LC3-II and is essential for the degradative activity of autophagy. Several lines of evidence support this conclusion. First, autophagic flux is inhibited by either a PGRMC1 inhibitor or PGRMC1-knockdown ( and ). Second, PGRMC1 decreased the pools of aberrant mitochondria, suggesting a role in mitophagy. Indeed, PGRMC1 is evolutionarily conserved with yeast, and mutants lacking DAP1, the yeast PGRMC1 homologue have decreased mitochondrial function.Citation25 As a third line of evidence supporting a role of PGRMC1 in autophagy, PGRMC1-knockdown cells had increased levels of ubiquitinated proteins, a substrate of autophagy. Ubiquitinated proteins increased markedly when autophagy was inhibited by bafilomycin A1 and chloroquine (), and this increase was nonadditive with PGRMC1 inhibition, suggesting a common mechanism. PGRMC1 has been implicated in autophagy and lysosomal function previously,Citation37 and the present results suggest a new mechanism in this pathway.
It is somewhat paradoxical that PGRMC1 is required for autophagy even though MAP1LC3-II levels increase. MAP1LC3 (or simply LC3) is an ubiquitin-like ortholog of yeast Atg8,Citation53 which is required for autophagy.Citation55 We propose that loss of PGRMC1 function disrupts autophagy, depriving the cells of nutrients and stimulating the initiating steps of autophagy. Indeed, PGRMC1-knockdown decreases the phosphorylation of MTOR, an autophagy suppressor,Citation48,Citation56-Citation58 and increases the phosphorylation of AMPK (), a proautophagy kinase,Citation59-Citation61 likely increasing the levels of TSC1 and TSC2 (). The latter is a GTPase-activating protein for RHEB, a GTPase that activates MTOR,Citation50,Citation62-Citation64 which in turn suppresses the initiation of autophagy. When MTOR is inhibited in PGRMC1-knockdown cells, autophagy is initiated, and increased production of the specific marker MAP1LC3B-II is the observed result ( and ). The initiating signal linking PGRMC1 inhibition to AMPK activation is unclear but could include decreased energy production from aberrant mitochondria or suppressed metabolite levels due to arrested autophagy. JNK kinases are autophagy regulators that may contribute to the suppression of autophagy by PGRMC1. JNK1 plays a key role in regulating BECN1 expressionCitation65 and BCL2 phosphorylation,Citation66 promoting autophagy, and PGRMC1-knockdown dramatically increased pan-JNK phosphorylation. The mechanism linking PGRMC1 and JNK is under investigation.
We propose that, upon PGRMC1 inhibition, the proteolytic activity of autophagy is suppressed. In support of this model, combined treatment with AG-205 and bafilomycin A1 or chloroquine does not result in a further increase in LC3-II (), suggesting that AG-205 arrests LC3-II degradation. Furthermore, the levels of ubiquitinated proteins did not increase in AG-205-treated cells or PGRMC1-knockdown cells after bafilomycin A1 or chloroquine treatment (). Indeed, we have previously found that PGRMC1-knockdown cells have diminished activation of MMP9 and CTSD/cathepsin D,Citation67 suggesting that some lysosome proteolytic events are promoted by PGRMC1. Notably, lysosomal acidification was unaffected in PGRMC1-knockdown cells (Fig. S4), suggesting that other lysosomal events are activated by PGRMC1. One candidate is the fusion of autophagosomes with lysosomes, which was impaired in PGRMC1-knockdown cells. Mutants lacking DAP1, the yeast PGRMC1 homologue have defects in vacuole structure,Citation27 and vacuoles are the yeast equivalent of lysosomes. The dap1Δ fragmented vacuole phenotype is similar to the vps16Δ vacuolar phenotype,Citation68 suggesting a defect in lysosomal fusion in mutants of the yeast PGRMC1 ortholog. Notably, mammalian VPS16 is a UVRAG-associated protein that localizes to autophagosomes, although its role in autophagy is unclear.Citation9 A caveat to this model is that LC3-II levels are elevated by bafilomycin A1 and chloroquine in PGRMC1-knockdown cells (), suggesting that PGRMC1-knockdown cells are not defective for LC3-II degradation. It may be that the temporal differences between the two types of inhibition (rapid for drug treatment vs. prolonged for a stable knockdown) are responsible for the discrepancy. An additional mechanism is that PGRMC1 antagonizes LC3-SQSTM1 binding, where SQSTM1 is a receptor for ubiquitinated cargo.Citation54,Citation69 However, SQSTM1 docks ubiquitinated proteins to LC3-positive puncta, and does not directly increase their degradation per se.Citation54
UVRAG increased the levels of ubiquitinated proteins (), and we note that these experiments were performed in serum-rich media, which suppresses basal autophagy. In earlier experiments, HCT116 or HeLa cells have been starved and stimulated with ligand, whereupon UVRAG decreases levels of EGFR, an ubiquitinated protein.Citation8,Citation9 In A549 lung cancer cells fed with serum, UVRAG did not decrease EGFR levels (), and EGFR ubiquitination was minimal (data not shown). The ability of UVRAG to increase ubiquitinated protein levels required PGRMC1 (), and UVRAG bound PGRMC1, suggesting that the UVRAG-PGRMC1 complex inhibits the function of PGRMC1. Indeed, UVRAG bound preferentially to a heme-binding-deficient mutant of PGRMC1 (). However, the targets of PGRMC1 in the degradation of ubiquitinated proteins are the subject of ongoing investigation.
One of the features that makes PGRMC1 compelling as a therapeutic target is that numerous ligands have been developed for the protein, including novel compounds that act alone or as delivery platformsCitation29,Citation36,Citation40,Citation41 and FDA-approved drugs with extensive clinical histories. Indeed, a PGRMC1 ligand has been linked to the lysosomal degradation process,Citation36,Citation37,Citation70 and the current findings suggest a mechanism through which PGRMC1 increases cell survival. We propose that PGRMC1 ligands like AG-205 and haloperidol arrest autophagy, at least in part by altering interactions with key autophagy proteins, disrupting protein, and organelle turnover. PGRMC1 has been implicated in a variety of disease states in which autophagy plays a key role, including neurodegeneration,Citation71,Citation72 cardiovascular disease,Citation73 and cancer, and the findings suggest a new mechanism for PGRMC1-based therapeutics in these diseases.
Materials and Methods
Cell lines, treatments, and transfections
A549 human non-small lung cancer cells, H226 squamous cell lung cancer cells, and HEK293 human embryonic kidney cells were purchased from ATCC, and their identity was verified by Genetica, Inc. Cells were cultured in DMEM containing antibiotics and 10% fetal bovine serum. Where indicated, cells were maintained in DMEM lacking glucose or L-glutamine (Sigma, D5030). Cells were treated with AG-205 (TimTec, Inc. ST050150), haloperidol (Sigma, H1512), LysoTracker Red (Invitrogen, 1020133), bafilomycin A1 (Cayman Chemical Co., 11038), and chloroquine (Pfaltz and Bauer, Inc., C19070) at the indicated concentrations. Plasmids encoding LC3-GFP and LC3-mCherry were from Dr Haining Zhu (University of Kentucky) and the plasmids encoding UVRAG-flag and UVRAG-GFP were from Addgene, Inc., while the plasmid for PGRMC1-HA (pRC40) and PGRMC1-hbd-HA (pRC42) have been described.Citation29 The conditions for siRNA knockdown of PGRMC1 have been described previously.Citation24,Citation31 All transfections were performed using the Transgin (APS Bio, 10102) or Effectgene (Qiagen, 301425) reagents according to the manufacturer’s instructions, and cells were transferred to Martek glass bottom microwell dishes by trypsinization for imaging.
Lysosomal and mitochondrial staining and immunofluorescence
LysoTracker Red fluorescence was measured in triplicate by incubating trypsinized cells in 75 nM dye for 15 min prior to fluorescence-activated cell sorting using the iCyt sy3200 analyzer and Flow-Jo software (Becton-Dickinson) at the University of Kentucky Flow Cytometry Core Facility. For mitochondrial staining, cells were seeded onto coverslips in 6-well plates. After 24 h, mitochondria were labeled by incubating cells with prewarmed DMEM without serum containing 100 nM MitoTracker Deep Red FM probe (Invitrogen, M22426) for 15 min at 37 °C. Cells were immediately observed under a fluorescence microscope at 644 nm excitation and 665 nm emission. In all cases, images were captured using a Leica DM IRBE inverted microscope at the University of Kentucky Imaging Core Facility. For all experiments, a 63× objective was used. Images were captured at room temperature in culture media with green fluorescent protein (GFP), mCherry, LysoTracker Red or MitoTracker Deep Red as fluorochromes.
For immunofluorescence, cells were fixed in 3:1 methanol:acetone at 4 °C for 45 min, as recommended for the LC3B antibody, and then washed, blocked with 10% normal goat serum and probed with rabbit anti-LC3B (Cell Signaling, D11) and, where indicated, goat anti-PGRMC1 (Novus, NB100-2841). All other procedures for staining followed the manufacturer’s recommendations. For puncta scoring with LC3B staining, LC3B puncta were counted in three separate images containing at least 20 cells and averaged.
Labeling with 3H-leucine
For protein degradation assays using 3H-leucine, cells were cultured in 8-well plates and labeled for 16 to 18 h with 1 μCi L-3H-leucine (Perkin Elmer, 689483). Cells were then washed with PBS containing a 10-fold excess of cold leucine 3 times for 5 min/wash to remove unincorporated 3H-leucine. Cells were then incubated in fresh medium containing cold leucine for 4 h to account for short-lived protein degradation. The media was then replaced with fresh media, which was itself removed at various time points and precipitated with 10% trichloroacetic acid. The supernatant was counted in Biosafe II scintillation fluid for 1 min on a Beckman LS6500 scintillation counter.
RNA analysis
Total RNA was isolated using the TRIzol reagent (Invitrogen, 15596026), and cDNA was synthesized from 2 μg of RNA using Superscript II (Invitrogen, 18064014) and random hexamers, as described previously.Citation26 Quantitative PCR was performed in triplicate using the SYBR Green I system (Bio-Rad, 13172) and a Stratagene MX3000P PCR system with MXPro software. The relative gene expression values were calculated as 2(Ct-SQSTM1 – Ct-ACTB) value was determined after normalization to actin. The primer sequences targeting SQSTM1 isoform 1 were SQSTM1i1+978F- 5′-AAGCCGGGTG GGAATGTTG and SQSTM1i1+1190R- 5′-GCTTGGCCCT TCGGATTCT.
Western blots, immunoprecipitations, and antibody arrays
The antibodies used were anti-RPS6KB (Genscript, Ab-421), anti-RPS6KB-pThr421 (R&D Systems, AB8965), anti-AMPK (R&D Systems, AF2509), anti-AMPK-pThr174 (R&D Systems, AF2509), anti-flag (Sigma, F3165), anti-GAPDH (Millipore, MAB374), anti-HA, anti-LC3A (Genscript, A00971), anti-LAMP1 (Santa Cruz Biotechnology, H4A3), anti-LC3B (Cell Signaling, D11), anti-MTOR (Cell Signaling), anti-MTOR-pSer2448 (Cell Signaling), anti-SQSTM1 (Cell Signaling), rabbit anti-PGRMC1,Citation29 goat anti-PGRMC1 (Novus), anti-TSC1 (Cell Signaling, 28A7), anti-TSC2 (Cell Signaling), anti-TUBA/tubulin (Cell Signaling, 11H10), anti-ubiquitin (Santa Cruz Biotechnology, P4D1), and anti-UVRAG (Cell Signaling, 5320). Protein lysis and immunoprecipitation conditions have been described.Citation31 Briefly, cells were lysed in 1% NP-40 Buffer (1% NP-40, 20 mM Tris, pH 7.4, 150 mM NaCl, 5 mM EDTA with the Halt protease inhibitor cocktail [Thermo, ML16387]). Lysed cells were incubated on ice for 15 min and then centrifuged for 15 min at 13,000 × g at 4 °C. Protein concentrations were determined by bicinchroninic acid assay.
HA immunoprecipitations of transfected PGRMC1 utilized anti-HA μMACS Microbeads (Myltenyi Biotec, Inc., 5110927339), substituting 1× PBS/0.5% NP-40 for wash buffer 1 and 1× PBS for wash buffer 2. The proteome profiler human phospho-kinase antibody array was from R&D Systems and was used according to their instructions with lysates from serum-starved cells. Spots were visualized using the WestPico chemiluminescent substrate and quantified by densitometry.
Electron microscopy
Cells were seeded into 6-well plates and grown in DMEM containing 10% FBS until confluent. Cells were rinsed in 0.1 M cacodylate and fixed with 3.0% glutaraldehyde in 0.1 cacodylate buffer, pH 7.4 at 4 °C for 45 min. Wells were rinsed 4 times in 0.1 M cacodylate with 5% sucrose for 5 min each. Cells were osmicated in 1% OsO4 for 45 min at 4 °C, dehydrated in a graded series of ethanols 50 to 100%, infiltrated with eponate 12 resin, and placed in a 60 °C oven for 48 h. Sectioning was performed with on a Reichert Ultracut E microtome and mounted on copper 300 mesh grids before staining with uranyl acetate and lead citrate. Grids were examined in a Philips Tecnai 12 Bioitwin transmission electron microscope and images were captured with a Gatan 4 K × 4 K digital camera.
Metabolic analyses
The XF96 Extracellular Flux analyzer (Seahorse Biosciences) is a 96-well instrument that measures metabolite uptake and release. Each XF96-assay well contains a disposable sensor cartridge, embedded with 96 pairs of fluorescent biosensors (oxygen and pH), coupled to fiber-optic wave guides. The wave guides deliver light at various excitation wavelengths (oxygen = 532 nm, pH = 470 nm) and transmits a fluorescent signal (oxygen = 650 nm, pH = 530 nm) to a set of photodetectors. Oxygen consumption rate (OCR) was expressed in pMoles/min in control and PGRMC1-knockdown cell lines. Cells were cultured for 2 h in DMEM media for the basal measurements, then were treated with oligomycin (1 μg/ml), FCCP (300 nM) and rotenone (10 μM), and the OCR was measured. The data shown represent triplicate repeats of at least three independent measurements and were analyzed for significance by Student t-test.
Statistical methods
All data are expressed as mean ± standard deviation and analyzed using a Student t-test to assess the significance between groups. All measurements were considered significant if the P-value ≤ 0.05 (*), ≤ 0.01 (**), and ≤ 0.005 (***).
| Abbreviations: | ||
| AMPK | = | AMP-dependent protein kinase |
| BCL2 | = | B-cell lymphoma 2 |
| BECN1 | = | Beclin 1, autophagy related |
| CCNA1 | = | cyclin A1 |
| CTSD | = | cathepsin D |
| EGFR | = | epidermal growth factor receptor |
| FACS | = | fluorescence-activated cell sorting |
| GAPDH | = | glyceraldehyde 3-phosphate dehydrogenase |
| GFP | = | green fluorescent protein |
| JNK | = | c-JUN N-terminal kinase |
| LAMP1 | = | lysosomal-associated membrane protein 1 |
| MAP1LC3 | = | microtubule-associated protein 1 light chain 3 |
| MMP9 | = | matrix metallopeptidase 9 (gelatinase B, 92 kDa gelatinase, 92 kDa type IV collagenase) |
| MTOR | = | mechanistic target of rapamycin |
| PGRMC1 | = | progesterone-associated membrane component 1 |
| RPS6KB/p70S6K | = | ribosomal protein S6 kinase, 70 kDa, polypeptide/p70 S6 kinase |
| RNAi | = | RNA interference |
| SQSTM1 | = | sequestosome 1 |
| TSC | = | tuberous sclerosis complex |
| TUBA | = | tubulin A |
| UVRAG | = | UV radiation-resistance associated |
| VPS16 | = | vacuolar protein sorting 16 homolog (S. cerevisiae) |
Additional material
Download Zip (645.5 KB)Acknowledgments
The authors thank Mary Gail Engle and Jim Begley for their expertise with confocal and electron microscopy and Matthew Thacker for expert technical assistance. This work supported by grants from the Kentucky Lung Cancer Research Program, the Bonnie Addario Lung Cancer Foundation and the Kentucky Science and Education Fund (KSEF-2064-RDE-013).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: http://www.landesbioscience.com/journals/autophagy/article/25889
Related Research Data
References
- Kimmelman AC. The dynamic nature of autophagy in cancer. Genes Dev 2011; 25:1999 - 2010; http://dx.doi.org/10.1101/gad.17558811; PMID: 21979913
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27 - 42; http://dx.doi.org/10.1016/j.cell.2007.12.018; PMID: 18191218
- Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol Cell 2009; 34:259 - 69; http://dx.doi.org/10.1016/j.molcel.2009.04.026; PMID: 19450525
- Kirisako T, Ichimura Y, Okada H, Kabeya Y, Mizushima N, Yoshimori T, Ohsumi M, Takao T, Noda T, Ohsumi Y. The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J Cell Biol 2000; 151:263 - 76; http://dx.doi.org/10.1083/jcb.151.2.263; PMID: 11038174
- Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, Mizushima N, Tanida I, Kominami E, Ohsumi M, et al. A ubiquitin-like system mediates protein lipidation. Nature 2000; 408:488 - 92; http://dx.doi.org/10.1038/35044114; PMID: 11100732
- Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature 2010; 466:68 - 76; http://dx.doi.org/10.1038/nature09204; PMID: 20562859
- Funderburk SF, Wang QJ, Yue Z. The Beclin 1-VPS34 complex--at the crossroads of autophagy and beyond. Trends Cell Biol 2010; 20:355 - 62; http://dx.doi.org/10.1016/j.tcb.2010.03.002; PMID: 20356743
- Thoresen SB, Pedersen NM, Liestøl K, Stenmark H. A phosphatidylinositol 3-kinase class III sub-complex containing VPS15, VPS34, Beclin 1, UVRAG and BIF-1 regulates cytokinesis and degradative endocytic traffic. Exp Cell Res 2010; 316:3368 - 78; http://dx.doi.org/10.1016/j.yexcr.2010.07.008; PMID: 20643123
- Liang C, Lee JS, Inn KS, Gack MU, Li Q, Roberts EA, Vergne I, Deretic V, Feng P, Akazawa C, et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol 2008; 10:776 - 87; http://dx.doi.org/10.1038/ncb1740; PMID: 18552835
- Xu J, Zeng C, Chu W, Pan F, Rothfuss JM, Zhang F, Tu Z, Zhou D, Zeng D, Vangveravong S, et al. Identification of the PGRMC1 protein complex as the putative sigma-2 receptor binding site. Nat Commun 2011; 2:380; http://dx.doi.org/10.1038/ncomms1386; PMID: 21730960
- Mifsud W, Bateman A. Membrane-bound progesterone receptors contain a cytochrome b5-like ligand-binding domain. Genome Biol 2002; 3:H0068; http://dx.doi.org/10.1186/gb-2002-3-12-research0068; PMID: 12537557
- Min L, Strushkevich NV, Harnastai IN, Iwamoto H, Gilep AA, Takemori H, Usanov SA, Nonaka Y, Hori H, Vinson GP, et al. Molecular identification of adrenal inner zone antigen as a heme-binding protein. FEBS J 2005; 272:5832 - 43; http://dx.doi.org/10.1111/j.1742-4658.2005.04977.x; PMID: 16279947
- Rohe HJ, Ahmed IS, Twist KE, Craven RJ. PGRMC1 (progesterone receptor membrane component 1): a targetable protein with multiple functions in steroid signaling, P450 activation and drug binding. Pharmacol Ther 2009; 121:14 - 9; http://dx.doi.org/10.1016/j.pharmthera.2008.09.006; PMID: 18992768
- Falkenstein E, Meyer C, Eisen C, Scriba PC, Wehling M. Full-length cDNA sequence of a progesterone membrane-binding protein from porcine vascular smooth muscle cells. Biochem Biophys Res Commun 1996; 229:86 - 9; http://dx.doi.org/10.1006/bbrc.1996.1761; PMID: 8954087
- Selmin O, Lucier GW, Clark GC, Tritscher AM, Vanden Heuvel JP, Gastel JA, Walker NJ, Sutter TR, Bell DA. Isolation and characterization of a novel gene induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin in rat liver. Carcinogenesis 1996; 17:2609 - 15; http://dx.doi.org/10.1093/carcin/17.12.2609; PMID: 9006096
- Gerdes D, Wehling M, Leube B, Falkenstein E. Cloning and tissue expression of two putative steroid membrane receptors. Biol Chem 1998; 379:907 - 11; PMID: 9705155
- Mir SU, Ahmed IS, Arnold S, Craven RJ. Elevated progesterone receptor membrane component 1/sigma-2 receptor levels in lung tumors and plasma from lung cancer patients. Int J Cancer 2012; 131:E1 - 9; http://dx.doi.org/10.1002/ijc.26432; PMID: 21918976
- Peluso JJ, Liu X, Saunders MM, Claffey KP, Phoenix K. Regulation of ovarian cancer cell viability and sensitivity to cisplatin by progesterone receptor membrane component-1. J Clin Endocrinol Metab 2008; 93:1592 - 9; http://dx.doi.org/10.1210/jc.2007-2771; PMID: 18319313
- Hou C, Tu Z, Mach R, Kung HF, Kung MP. Characterization of a novel iodinated sigma-2 receptor ligand as a cell proliferation marker. Nucl Med Biol 2006; 33:203 - 9; http://dx.doi.org/10.1016/j.nucmedbio.2005.10.001; PMID: 16546674
- Wheeler KT, Wang LM, Wallen CA, Childers SR, Cline JM, Keng PC, Mach RH. Sigma-2 receptors as a biomarker of proliferation in solid tumours. Br J Cancer 2000; 82:1223 - 32; http://dx.doi.org/10.1054/bjoc.1999.1067; PMID: 10735510
- Hornick JR, Xu J, Vangveravong S, Tu Z, Mitchem JB, Spitzer D, Goedegebuure P, Mach RH, Hawkins WG. The novel sigma-2 receptor ligand SW43 stabilizes pancreas cancer progression in combination with gemcitabine. Mol Cancer 2010; 9:298; http://dx.doi.org/10.1186/1476-4598-9-298; PMID: 21092190
- Thompson AM, Reddi AR, Shi X, Goldbeck RA, Moenne-Loccoz P, Gibney BR, et al. Measurement of the Heme Affinity for Yeast Dap1p, and Its Importance in Cellular Function. Biochem 2007.
- Hughes AL, Powell DW, Bard M, Eckstein J, Barbuch R, Link AJ, Espenshade PJ. Dap1/PGRMC1 binds and regulates cytochrome P450 enzymes. Cell Metab 2007; 5:143 - 9; http://dx.doi.org/10.1016/j.cmet.2006.12.009; PMID: 17276356
- Crudden G, Chitti RE, Craven RJ. Hpr6 (heme-1 domain protein) regulates the susceptibility of cancer cells to chemotherapeutic drugs. J Pharmacol Exp Ther 2006; 316:448 - 55; http://dx.doi.org/10.1124/jpet.105.094631; PMID: 16234411
- Hand RA, Jia N, Bard M, Craven RJ. Saccharomyces cerevisiae Dap1p, a novel DNA damage response protein related to the mammalian membrane-associated progesterone receptor. Eukaryot Cell 2003; 2:306 - 17; http://dx.doi.org/10.1128/EC.2.2.306-317.2003; PMID: 12684380
- Mallory JC, Crudden G, Johnson BL, Mo C, Pierson CA, Bard M, Craven RJ. Dap1p, a heme-binding protein that regulates the cytochrome P450 protein Erg11p/Cyp51p in Saccharomyces cerevisiae. Mol Cell Biol 2005; 25:1669 - 79; http://dx.doi.org/10.1128/MCB.25.5.1669-1679.2005; PMID: 15713626
- Craven RJ, Mallory JC, Hand RA. Regulation of iron homeostasis mediated by the heme-binding protein Dap1 (damage resistance protein 1) via the P450 protein Erg11/Cyp51. J Biol Chem 2007; 282:36543 - 51; http://dx.doi.org/10.1074/jbc.M706770200; PMID: 17954932
- Neubauer H, Clare SE, Wozny W, Schwall GP, Poznanovic S, Stegmann W, Vogel U, Sotlar K, Wallwiener D, Kurek R, et al. Breast cancer proteomics reveals correlation between estrogen receptor status and differential phosphorylation of PGRMC1. Breast Cancer Res 2008; 10:R85; http://dx.doi.org/10.1186/bcr2155; PMID: 18922159
- Ahmed IS, Rohe HJ, Twist KE, Mattingly MN, Craven RJ. Progesterone receptor membrane component 1 (Pgrmc1): a heme-1 domain protein that promotes tumorigenesis and is inhibited by a small molecule. J Pharmacol Exp Ther 2010; 333:564 - 73; http://dx.doi.org/10.1124/jpet.109.164210; PMID: 20164297
- Hand RA, Craven RJ. Hpr6.6 protein mediates cell death from oxidative damage in MCF-7 human breast cancer cells. J Cell Biochem 2003; 90:534 - 47; http://dx.doi.org/10.1002/jcb.10648; PMID: 14523988
- Ahmed IS, Rohe HJ, Twist KE, Craven RJ. Pgrmc1 (progesterone receptor membrane component 1) associates with epidermal growth factor receptor and regulates erlotinib sensitivity. J Biol Chem 2010; 285:24775 - 82; http://dx.doi.org/10.1074/jbc.M110.134585; PMID: 20538600
- Peluso JJ, Liu X, Gawkowska A, Lodde V, Wu CA. Progesterone inhibits apoptosis in part by PGRMC1-regulated gene expression. Mol Cell Endocrinol 2010; 320:153 - 61; http://dx.doi.org/10.1016/j.mce.2010.02.005; PMID: 20144686
- Peluso JJ, Pappalardo A, Losel R, Wehling M. Progesterone membrane receptor component 1 expression in the immature rat ovary and its role in mediating progesterone’s antiapoptotic action. Endocrinology 2006; 147:3133 - 40; http://dx.doi.org/10.1210/en.2006-0114; PMID: 16513825
- Peluso JJ, Romak J, Liu X. Progesterone receptor membrane component-1 (PGRMC1) is the mediator of progesterone’s antiapoptotic action in spontaneously immortalized granulosa cells as revealed by PGRMC1 small interfering ribonucleic acid treatment and functional analysis of PGRMC1 mutations. Endocrinology 2008; 149:534 - 43; http://dx.doi.org/10.1210/en.2007-1050; PMID: 17991724
- Kashiwagi H, McDunn JE, Simon PO Jr., Goedegebuure PS, Vangveravong S, Chang K, Hotchkiss RS, Mach RH, Hawkins WG. Sigma-2 receptor ligands potentiate conventional chemotherapies and improve survival in models of pancreatic adenocarcinoma. J Transl Med 2009; 7:24; http://dx.doi.org/10.1186/1479-5876-7-24; PMID: 19323815
- Ostenfeld MS, Fehrenbacher N, Høyer-Hansen M, Thomsen C, Farkas T, Jäättelä M. Effective tumor cell death by sigma-2 receptor ligand siramesine involves lysosomal leakage and oxidative stress. Cancer Res 2005; 65:8975 - 83; http://dx.doi.org/10.1158/0008-5472.CAN-05-0269; PMID: 16204071
- Ostenfeld MS, Høyer-Hansen M, Bastholm L, Fehrenbacher N, Olsen OD, Groth-Pedersen L, Puustinen P, Kirkegaard-Sørensen T, Nylandsted J, Farkas T, et al. Anti-cancer agent siramesine is a lysosomotropic detergent that induces cytoprotective autophagosome accumulation. Autophagy 2008; 4:487 - 99; PMID: 18305408
- Groth-Pedersen L, Ostenfeld MS, Høyer-Hansen M, Nylandsted J, Jäättelä M. Vincristine induces dramatic lysosomal changes and sensitizes cancer cells to lysosome-destabilizing siramesine. Cancer Res 2007; 67:2217 - 25; http://dx.doi.org/10.1158/0008-5472.CAN-06-3520; PMID: 17332352
- Brent PJ, Pang GT. Sigma binding site ligands inhibit cell proliferation in mammary and colon carcinoma cell lines and melanoma cells in culture. Eur J Pharmacol 1995; 278:151 - 60; http://dx.doi.org/10.1016/0014-2999(95)00115-2; PMID: 7671999
- Crawford KW, Bowen WD. Sigma-2 receptor agonists activate a novel apoptotic pathway and potentiate antineoplastic drugs in breast tumor cell lines. Cancer Res 2002; 62:313 - 22; PMID: 11782394
- Spitzer D, Simon PO Jr., Kashiwagi H, Xu J, Zeng C, Vangveravong S, Zhou D, Chang K, McDunn JE, Hornick JR, et al. Use of multifunctional sigma-2 receptor ligand conjugates to trigger cancer-selective cell death signaling. Cancer Res 2012; 72:201 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-11-1354; PMID: 22065721
- Yoshitani N, Satou K, Saito K, Suzuki S, Hatanaka H, Seki M, Shinozaki K, Hirota H, Yokoyama S. A structure-based strategy for discovery of small ligands binding to functionally unknown proteins: combination of in silico screening and surface plasmon resonance measurements. Proteomics 2005; 5:1472 - 80; http://dx.doi.org/10.1002/pmic.200401032; PMID: 15798990
- Park J, Chung S, An H, Kim J, Seo J, Kim DH, Yoon SY. Haloperidol and clozapine block formation of autophagolysosomes in rat primary neurons. Neuroscience 2012; 209:64 - 73; http://dx.doi.org/10.1016/j.neuroscience.2012.02.035; PMID: 22390943
- Mo RH, Zaro JL, Ou JH, Shen WC. Effects of Lipofectamine 2000/siRNA complexes on autophagy in hepatoma cells. Mol Biotechnol 2012; 51:1 - 8; http://dx.doi.org/10.1007/s12033-011-9422-6; PMID: 21660602
- Heitman J, Movva NR, Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 1991; 253:905 - 9; http://dx.doi.org/10.1126/science.1715094; PMID: 1715094
- Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P, Snyder SH. RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell 1994; 78:35 - 43; http://dx.doi.org/10.1016/0092-8674(94)90570-3; PMID: 7518356
- Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, Lane WS, Schreiber SL. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 1994; 369:756 - 8; http://dx.doi.org/10.1038/369756a0; PMID: 8008069
- Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 2011; 12:21 - 35; http://dx.doi.org/10.1038/nrm3025; PMID: 21157483
- van Veelen W, Korsse SE, van de Laar L, Peppelenbosch MP. The long and winding road to rational treatment of cancer associated with LKB1/AMPK/TSC/mTORC1 signaling. Oncogene 2011; 30:2289 - 303; http://dx.doi.org/10.1038/onc.2010.630; PMID: 21258412
- Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr Biol 2003; 13:1259 - 68; http://dx.doi.org/10.1016/S0960-9822(03)00506-2; PMID: 12906785
- Tee AR, Fingar DC, Manning BD, Kwiatkowski DJ, Cantley LC, Blenis J. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc Natl Acad Sci U S A 2002; 99:13571 - 6; http://dx.doi.org/10.1073/pnas.202476899; PMID: 12271141
- Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003; 115:577 - 90; http://dx.doi.org/10.1016/S0092-8674(03)00929-2; PMID: 14651849
- Mann SS, Hammarback JA. Molecular characterization of light chain 3. A microtubule binding subunit of MAP1A and MAP1B. J Biol Chem 1994; 269:11492 - 7; PMID: 7908909
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131 - 45; http://dx.doi.org/10.1074/jbc.M702824200; PMID: 17580304
- Tsukada M, Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett 1993; 333:169 - 74; http://dx.doi.org/10.1016/0014-5793(93)80398-E; PMID: 8224160
- Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem 1998; 273:3963 - 6; http://dx.doi.org/10.1074/jbc.273.7.3963; PMID: 9461583
- Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, Gray NS. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem 2009; 284:8023 - 32; http://dx.doi.org/10.1074/jbc.M900301200; PMID: 19150980
- Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol 2000; 150:1507 - 13; http://dx.doi.org/10.1083/jcb.150.6.1507; PMID: 10995454
- Alers S, Löffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol 2012; 32:2 - 11; http://dx.doi.org/10.1128/MCB.06159-11; PMID: 22025673
- Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol 2007; 8:774 - 85; http://dx.doi.org/10.1038/nrm2249; PMID: 17712357
- Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol 2011; 13:1016 - 23; http://dx.doi.org/10.1038/ncb2329; PMID: 21892142
- Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev 2003; 17:1829 - 34; http://dx.doi.org/10.1101/gad.1110003; PMID: 12869586
- Garami A, Zwartkruis FJ, Nobukuni T, Joaquin M, Roccio M, Stocker H, Kozma SC, Hafen E, Bos JL, Thomas G. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell 2003; 11:1457 - 66; http://dx.doi.org/10.1016/S1097-2765(03)00220-X; PMID: 12820960
- Zhang Y, Gao X, Saucedo LJ, Ru B, Edgar BA, Pan D. Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat Cell Biol 2003; 5:578 - 81; http://dx.doi.org/10.1038/ncb999; PMID: 12771962
- Li DD, Wang LL, Deng R, Tang J, Shen Y, Guo JF, Wang Y, Xia LP, Feng GK, Liu QQ, et al. The pivotal role of c-Jun NH2-terminal kinase-mediated Beclin 1 expression during anticancer agents-induced autophagy in cancer cells. Oncogene 2009; 28:886 - 98; http://dx.doi.org/10.1038/onc.2008.441; PMID: 19060920
- Lorin S, Borges A, Ribeiro Dos Santos L, Souquère S, Pierron G, Ryan KM, Codogno P, Djavaheri-Mergny M. c-Jun NH2-terminal kinase activation is essential for DRAM-dependent induction of autophagy and apoptosis in 2-methoxyestradiol-treated Ewing sarcoma cells. Cancer Res 2009; 69:6924 - 31; http://dx.doi.org/10.1158/0008-5472.CAN-09-1270; PMID: 19706754
- Mir SU, Jin L, Craven RJ. Neutrophil gelatinase-associated lipocalin (NGAL) expression is dependent on the tumor-associated sigma-2 receptor S2RPgrmc1. J Biol Chem 2012; 287:14494 - 501; http://dx.doi.org/10.1074/jbc.M111.324921; PMID: 22418433
- Sambade M, Alba M, Smardon AM, West RW, Kane PM. A genomic screen for yeast vacuolar membrane ATPase mutants. Genetics 2005; 170:1539 - 51; http://dx.doi.org/10.1534/genetics.105.042812; PMID: 15937126
- Vadlamudi RK, Joung I, Strominger JL, Shin J. p62, a phosphotyrosine-independent ligand of the SH2 domain of p56lck, belongs to a new class of ubiquitin-binding proteins. J Biol Chem 1996; 271:20235 - 7; http://dx.doi.org/10.1074/jbc.271.34.20235; PMID: 8702753
- Hornick JR, Vangveravong S, Spitzer D, Abate C, Berardi F, Goedegebuure P, Mach RH, Hawkins WG. Lysosomal membrane permeabilization is an early event in Sigma-2 receptor ligand mediated cell death in pancreatic cancer. J Exp Clin Cancer Res 2012; 31:41; http://dx.doi.org/10.1186/1756-9966-31-41; PMID: 22551149
- Wei Z, Mousseau DD, Dai Y, Cao X, Li XM. Haloperidol induces apoptosis via the sigma2 receptor system and Bcl-XS. Pharmacogenomics J 2006; 6:279 - 88; PMID: 16462815
- Bowen WD. Sigma receptors: recent advances and new clinical potentials. Pharm Acta Helv 2000; 74:211 - 8; http://dx.doi.org/10.1016/S0031-6865(99)00034-5; PMID: 10812960
- Monassier L, Manoury B, Bellocq C, Weissenburger J, Greney H, Zimmermann D, Ehrhardt JD, Jaillon P, Baró I, Bousquet P. sigma(2)-receptor ligand-mediated inhibition of inwardly rectifying K(+) channels in the heart. J Pharmacol Exp Ther 2007; 322:341 - 50; http://dx.doi.org/10.1124/jpet.107.122044; PMID: 17460149