
Abstract
Deficiency in autophagy, a lysosome-dependent cell degradation pathway, has been associated with a variety of diseases especially cancer. Recently, the activation of autophagy by hepatitis B virus X (HBx) protein, which is implicated in hepatitis B virus (HBV)-associated hepatocellular carcinoma (HCC), has been identified in hepatic cells. However, the underlying mechanism and the relevance of HBx-activated autophagy to the carcinogenesis caused by HBV remain elusive. Here, by transfection of HBV genomic DNA and HBx in hepatic and hepatoma cells, we showed that HBV- or HBx-induced autophagosome formation was accompanied by unchanged MTOR (mechanistic target of rapamycin) activity and decreased degradation of LC3 and SQSTM1/p62, the typical autophagic cargo proteins. Further functional and morphological analysis indicated that HBx dramatically impaired lysosomal acidification leading to a drop in lysosomal degradative capacity and the accumulation of immature lysosomes possibly through interaction with V-ATPase affecting its lysosome targeting. Moreover, clinical specimen test showed increased SQSTM1 and immature lysosomal hydrolase CTSD (cathepsin D) in human liver tissues with chronic HBV infection and HBV-associated liver cancer. These data suggest that a repressive effect of HBx on lysosomal function is responsible for the inhibition of autophagic degradation, and this may be critical to the development of HBV-associated HCC.
Introduction
Hepatocellular carcinoma is one of the most frequent cancers worldwide, and more than half of the cases are attributable to chronic hepatitis B virus infection.Citation1 Despite the controversy over the role and the underlying molecular mechanism of HBV in HCC formation,Citation2-Citation4 extensive evidence have suggested an important role of hepatitis B virus X protein, a small soluble cytoplasmic protein in the host cells. HBx modulates gene expression or intracellular signal pathway through interaction with either transcriptional machinery or signaling components involved in cell proliferation, apoptosis, and DNA repair, together with those in the immune response.Citation5,Citation6
Macroautophagy (hereafter referred to as autophagy) is a conserved eukaryotic catabolic reaction that sequesters protein aggregates and damaged organelles into double-membrane autophagosomes for lysosomal degradation. The complete autophagic process comprises mainly the formation of autophagosomes, their fusion with lysosomes and cargo degradation in the lysosomes. The formation of autophagosomes involves multiple autophagy-related (ATG) proteins and 2 ubiquitin-like conjugation systems among which the conjugation of microtubule-associated protein 1 light chain 3 (LC3) with phosphatidylethanolamine is crucial to the origination and elongation of phagophore membranesCitation7 and to the autophagic cargo recruitment through LC3 interaction with the cargo receptors.Citation8 As a cytoprotective process, autophagy helps to maintain cell homeostasis in nutrient-rich environments through its constitutive activity, and serves as an alternative energy source for cells under nutrient-poor conditions.Citation9 Deficient autophagy has been associated with a variety of human diseases including aging, metabolic syndrome, neurodegeneration, and especially cancer.Citation10 The inactivation of key autophagy genes, such as Atg4c, Atg5, and Atg7, accelerates spontaneous tumor development in mice,Citation11-Citation13 while frequent monoallelic deletion of BECN1 (VPS30/ATG6 in yeast) has been found in human ovarian, breast, and prostate cancers.Citation14,Citation15 In addition, while tumor suppressor proteins such as PTEN and TP53/p53 positively regulate autophagy,Citation16,Citation17 oncogene products such as BCL2 and AKT-MTOR inhibit it.Citation18,Citation19 With regard to HCC, recently it has been shown that systemic mosaic deletion of Atg5 or liver-specific loss of Atg7 in mouse causes multiple liver tumors, indicating an important suppressive effect of autophagy in liver tumorigenesis.Citation12
Interestingly, 2 recent studies have shown that HBx directly or indirectly promotes autophagy in hepatocytes either by activation of class III phosphatidylinositol 3-kinase (PtdIns3K) or by upregulation of BECN1 expression, sensitizing starvation-induced autophagy.Citation20,Citation21 However, the relevance of HBx-promoted autophagy to HBV-induced carcinogenesis remains elusive, although enhancement of HBV replication or HBV infection by autophagy has been suggested.Citation20 In this study, we investigated the molecular and cellular mechanism of HBV-induced autophagy in hepatocytes by focusing on autophagic flux. We found that HBV significantly inhibited autophagic degradation via HBx, although the number of autophagosomes in the cells was increased. By interfering with the maturation of lysosomes, HBx actually restrained autophagic flux leading to the accumulation of autophagic cargoes such as SQSTM1, which may be linked to HBV-associated HCC.
Results
HBx stimulates autophagosome formation
To date, the effect of HBV on cell autophagy is still ambiguous. To clarify whether HBV infection induces autophagy, we first expressed HBV genomic DNA in human hepatoma Huh7 cells and checked the formation of autophagosomes by staining endogenous LC3. We found that expression of HBV DNA significantly increased intracellular autophagosomes as demonstrated by accumulation of LC3-positive spot-like structures in the cells (). However, expression of the HBVX− DNA, an HBV genomic DNA that is incapable of expressing HBx protein,Citation20 failed to accumulate autophagic puncta ().
Figure 1. HBx induces accumulation of autophagosomes. (A) Huh7 cells were transfected with HBV genomic DNA (HBV) or HBx-negative HBV genomic DNA (HBVX−). At 48 h after transfection, the cells were stained with HBcAg and LC3 antibodies, and were imaged by confocal microscopy. Scale bars: 20 µm. (B) Statistical analysis of the number of LC3-dots per cell in cells with or without expression of HBV or HBVX− in the presence or absence of chloroquine (CQ) or bafilomycin A1 (Baf A1). Data are presented as mean ± SEM, n = 50. (C) L02 and Huh7 cells were transfected with GFP or HBx-GFP. At 48 h after transfection, the cells were immunostained with LC3 antibody and were imaged. Only the images from L02 cells were shown. Scale bars: 20 µm. (D) Statistical analysis of the number of LC3-dots per cell in cells expressing GFP or HBx-GFP. Only GFP- or HBx-GFP-positive cells were counted. Data are presented as mean ± SEM, n = 50. (E) L02 and Huh7 cells were either starved or transfected with GFP or HBx-GFP for 48 h, then the cellular LC3 levels were assessed by western blot. (F) Autophagic vacuoles in cells expressing GFP (control) or HBx-GFP were observed by electron microscopy. The arrow indicates autophagic vacuoles. Scale bars: 0.5 µm. The graph shows a statistical analysis of cytoplasmic occupancy of autophagic vacuoles in the cells. Data are presented as mean ± SEM, n = 20. (G) Autophagic vacuoles were isolated from liver tissues from control individuals and HBV-associated HCC individuals. Then HBx, LC3 and LAMP1 levels were analyzed by western blot. Hom, liver homogenates; AV, autophagic vacuoles. ***P < 0.001.
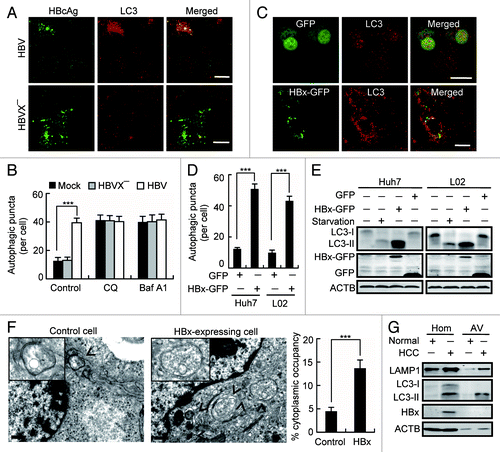
To rule out the possibility that HBVX− expression promoted excessive autophagic degradation which led to the failure in autophaogosome accumulation, we treated the HBV- or HBVX−-expressing cells with lysosome inhibitor bafilomycin A1 (Baf A1) that inactivates the vacuolar-type H+-ATPase (V-ATPase), or chloroquine (CQ) that prevents the acidification of lysosomes. We found that in the absence of Baf A1 or CQ, the number of intracellular autophagic puncta in HBVX−-expressing cells was the same as that in mock-transfected cells, when it was dramatically increased in HBV-expressing cells. Upon Baf A1- or CQ-treatment, the autophagic puncta in mock-transfected cells, HBV-expressing cells and HBVX−-expressing cells arrived at a same level (), suggesting that a promotion of excessive autophagic degradation was not involved in the action of HBVX− expression.
To clarify the effect of HBx on autophagosome formation, a GFP-tagged HBx was transfected in human hepatic L02 cells and human hepatoma Huh7 cells. Clearly, expression of HBx-GFP caused growth in intracellular autophagic puncta (). Expression of HBx-GFP also dramatically stimulated the conversion of LC3-І to LC3-II in the cells, indicating an increase in membrane-associated LC3 (). Autophagosome induction by HBx was further confirmed by electron microscopy. Clearly, expression of HBx-GFP but not GFP significantly increased intracellular autophagic vacuoles shown as double-membrane vesicles with visible cytoplasm contents (). Finally, to exclude that accumulation of autophagosomes was due to artificial aggregation of HBx caused by overexpression of HBx, since the level of HBx is quite low during HBV infection,Citation22 we examined human HCC tissues for the possible association of HBx with autophagosomes or autolysosomes. Using density-gradient centrifugation, we isolated the autophagic vacuoles from the tissues and checked HBx level in the fraction. In the autophagosomal fraction marked LC3-II and lysosomal-associated membrane protein 1 (LAMP1), no detectable HBx was found, suggesting that HBx is not majorly associated with or included in autophagosomes during HBV infection ().
Taken together, these data are consistent with a previous reportCitation20 and suggest that expression of HBx alone induces the formation of autophagosomes in hepatic cells.
HBx-induced autophagosome formation is MTOR inhibition-independent
MTOR is an important modulator of autophagy by which the upstream signals from deletion of growth factors, reduction in intracellular energy or amino acid availability, are transferred to downstream effectors.Citation23 To investigate whether HBx stimulates autophagosome formation through inhibition of MTOR, the activity of MTOR was checked by measuring the phosphorylation of MTOR itself and its specific substrate RPS6KB/p70S6K. We found that, in contrast to cells treated with rapamycin, an MTOR inhibitor, in which the phosphorylation of both MTOR and RPS6KB was suppressed, the expression of HBx-GFP downregulated neither phosphorylated MTOR nor phosphorylated RPS6KB (), suggesting that HBx stimulates autophagosome formation without changing MTOR activity in these cells.
Figure 2. Characterization of HBx-triggered autophagosome accumulation. (A) Huh7 cells were treated with rapamycin or transfected with GFP or HBx-GFP for 48 h. Phosphorylation of MTOR and RPS6KB was analyzed by western blot using specific anti-phospho-MTOR or anti-phospho-RPS6KB antibodies. (B) Huh7 cells with or without HBx-GFP expression for 48 h were either starved or treated with rapamycin. Then the cells were fixed and stained with LC3 antibody. The number of LC3-dots per cell was quantified and is presented as mean ± SEM, n = 50. **P < 0.01. GFP-expressing cells were used as a control for HBx-GFP-expressing cells. (C) Western blot analysis of LC3 in cells treated as in (B).
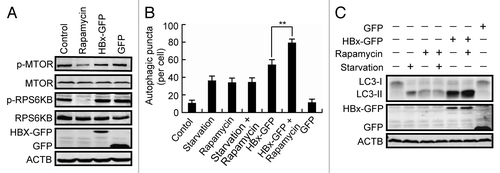
If HBx stimulates autophagosome formation through a pathway independent of MTOR inhibition, a synergistic effect might be expected in cells with both HBx expression and rapamycin treatment. To test this, cells expressing HBx-GFP were treated with or without rapamycin, and the production of LC3-II was measured. We found that while rapamycin treatment failed to further enhance the autophagic puncta and the production of LC3-II in starved cells, addition of rapamycin evidently increased the autophagic puncta number and the LC3-II level in HBx-GFP-expressing cells (), confirming that a pathway independent of MTOR inhibition is involved in the HBx-stimulated production of autophagosomes.
HBx inhibits autophagic degradation
During autophagy, formed autophagosomes need to fuse with lysosomes so that the autophagic cargoes together with the LC3 associated with the inner membrane of the autophagosomes are degraded.Citation24 To determine whether the formation of autophagosomes stimulated by HBx is followed by an increase in autophagic degradation, we first measured the total LC3 level in HBV DNA- or HBx-expressing Huh7 cells. Surprisingly, we found that expression of HBV or HBx-GFP, but not HBVX− or GFP, dramatically raised the LC3 level in the cells (). The increase in LC3 was not due to increased LC3 RNA synthesis, because, like starvation or treatment with Baf A1, HBx did not change the LC3 mRNA level (Fig. S1).
Figure 3. HBx inhibits autophagic degradation. (A) Pseudocolor-coded images showing LC3 intensity in Huh7 cells expression of GFP, HBx-GFP, HBV, or HBVX−. (B) Quantification of the relative mean LC3 intensity per cell in cells expression of GFP, HBx-GFP, HBV, or HBVX−, n = 30. (C) L02 cells stably expressing GFP-LC3 were either starved or treated with 100 nM bafilomycin A1 (Baf A1) for 12 h or transfected with HBx-HA for 48 h. Then the cells were analyzed by western blot using anti-GFP antibody. Note the production of GFP fragment. (D) Huh7 cells transfected with HBV or HBVX− were fixed and stained with HBcAg and SQSTM1 antibodies at 48 h post-transfection. Quantification shown on the right represents the relative fluorescence intensity of SQSTM1 in cells expressing HBV or HBVX− in the presence or absence of 50 µM chloroquine (CQ), n = 30. (E) Huh7 cells with GFP or HBx-GFP expression were treated with 100 nM Baf A1 for 12 h. The cellular SQSTM1 level was analyzed by western blot. Quantification shown on the right represents the relative SQSTM1 levels of the cells normalized to ACTB, and the fold change vs. GFP-expressing control cells was quantified from 3 independent experiments. All the quantitative data are presented as mean ± SEM ***P < 0.001. Scale bars: 20 µm.
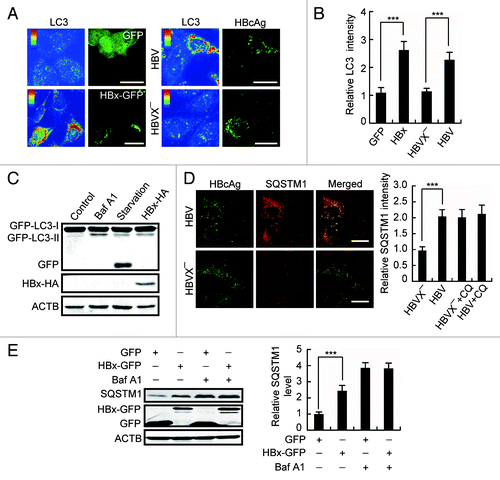
The expression of GFP-tagged LC3 is widely used to study autophagy. In cells expressing GFP-LC3, the detection of free GFP fragments generated by degradation of GFP-LC3 in the autolysosomes is a useful assay for autophagic degradation.Citation24 We found that in GFP-LC3-expressing L02 cells, although starvation, HBx-HA expression and Baf A1 treatment all triggered the production of GFP-LC3-II, only cell starvation clearly produced GFP fragments (). These data suggest that, unlike cell starvation, which enhances lysosome-dependent autophagic degradation, HBx expression rather suppresses lysosomal degradation, mimicking the effect of Baf A1.
The protein SQSTM1 is a selective autophagic receptor and is incorporated into autophagosomes and degraded along with other substrates by lysosomal hydrolyses.Citation24 In search of further evidence that HBx inhibits autophagic degradation, we first observed intracellular SQSTM1 in HBV DNA- or HBVX− DNA-transfected Huh7 cells by immunostaining. We found expression of HBV DNA but not the HBVX− DNA dramatically raised SQSTM1 level in the cells and the cytoplasmic SQSTM1 assembled into aggregates (). In addition, in cell expression of HBV DNA, addition of CQ only slightly furthered the accumulation of SQSTM1 (), suggesting that lysosome-dependent pathway is the major target of HBV. We then transfected the HBx-GFP into Huh7 cells and checked the intracellular level of SQSTM1 by western blot. As expected, like that in Baf A1-treated cells, expression of HBx-GFP significantly accumulated cellular SQSTM1, and the accumulation of SQSTM1 by Baf A1 was not furthered by HBx-GFP expression (), indicating no synergy between Baf A1 and HBx-GFP on SQSTM1 degradation. Results from RT-PCR demonstrated that HBx expression had no effect on SQSTM1 mRNA level (Fig. S2). Taken together, these data suggest an inhibitory effect of HBx on autophagic degradation.
HBx does not affect autophagosome-lysosome fusion
To investigate the mechanism underlying the inhibition of autophagic degradation caused by HBx, we first analyzed the effect of HBx on the autolysosome formation. RAB7A, a small GTPase localized to late endosome and lysosome, is implicated in the fusion of autophagosome with lysosome.Citation25 Colocalization of endogenous LC3 with RAB7A-GFP was checked in cells coexpressing HBx-Cherry and RAB7A-GFP. While LC3 poorly colocalized with RAB7A-GFP because of a dominant cytosolic distribution of LC3 in control cells, starvation significantly promoted the colocalization (), demonstrating increased autolysosome formation. Expression of HBx-Cherry but not Cherry resulted in a colocalization coefficient of LC3 and RAB7A-GFP almost equal to that of cell starvation (), indicating a similar formation of autolysosomes.
Figure 4. HBx does not affect autophagosome-lysosome fusion. (A) L02 cells expressing RAB7A-GFP were either starved or cotransfected with HBx-Cherry or Cherry. Then the cells were fixed, stained with anti-LC3 antibody and imaged by confocal microscopy. (B) Statistical analysis of the colocalization coefficient of RAB7A-GFP and LC3. The colocalization coefficient was represented as percentage of punctate signals of LC3 that were positive for RAB7A-GFP. (C) Colocalization of endogenous LC3 and LAMP1 in starved L02 cells with or without RAB7AT22N-GFP expression, or in HBx-GFP-expressing L02 cells. RAB7AT22N-GFP-expressing cells were used as a negative control. (D) Statistical analysis of the colocalization coefficient of LC3 and LAMP1. The colocalization coefficient was represented as percentage of punctate signals of LC3 that were positive for LAMP1. (E) Huh7 cells expression of Cherry-LC3 were either starved or cotransfected with HBV genome DNA for 48 h. Then the cells were fixed, stained with HBcAg and LAMP1 antibodies, and were imaged. (F) Statistical analysis of the colocalization coefficient of Cherry-LC3 and LAMP1. The colocalization coefficient was represented as percentage of punctate signals of Cherry-LC3 that were positive for LAMP1. Quantifications were performed using Velocity software, and the values represent mean ± SEM of 30 cells. **P < 0.01. Scale bars: 5 µm.
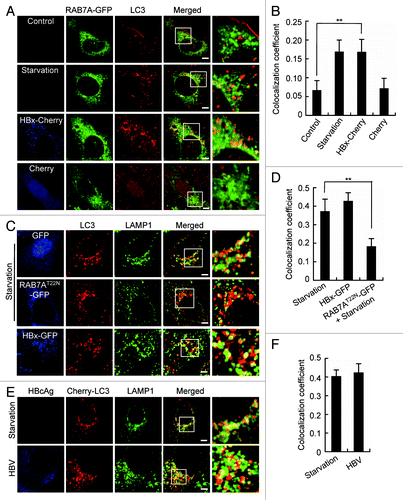
The autophagosome-lysosome fusion was further studied by observing the colocalization of LC3 with LAMP1. Starvation led to an overlap of LC3 with LAMP1, and the colocalization coefficient was markedly higher than that of LC3 with RAB7A (0.39 vs. 0.17), reflecting a better localization of LAMP1 on lysosomes than RAB7A (). Overexpression of a dominant-negative mutant of RAB7A (RAB7A T22N) clearly reduced the cell starvation-induced colocalization of LC3 with LAMP1, confirming a mediating function of RAB7A in the process. Expression of HBx-GFP led to a degree of LC3-LAMP1 colocalization similar to that caused by cell starvation (). Finally, the colocalization of LC3 and LAMP1 was checked in HBV-expressing Huh7 cells. Like that in starved cells, HBV expression led to a similar colocalization coefficient of Cherry-LC3 and LAMP1 (), indicating an unharmed autolysosome formation. Taken together, these data suggest that the inhibition of autophagic degradation by HBx is not due to impaired autophagosome-lysosome fusion.
HBx impairs lysosomal degradative capacity
Because lysosomes are the final destination for autophagic cargo degradation, we then focused on dissecting the effect of HBx on lysosomal function. First, we assessed the lysosomal degradation of the epidermal growth factor receptor (EGFR) in Huh7 cells. Endogenous EGFR localized primarily to the cell surface before EGF treatment. Following EGF treatment, EGFR was internalized and clustered into puncta, and the normalized EGFR mean intensity reached the highest level at 1 h after the treatment both in GFP- and HBx-GFP-expressing cells (). However, at 2 h and 3 h after EGF treatment, when a significant portion of the internalized EGFR was degraded in nontransfected and GFP-expressing cells, the EGFR puncta were retained in HBx-GFP-expressing cells (), indicating weakened lysosomal proteolysis. This result was confirmed by data from western blot demonstrating a lower reduction of total cellular EGFR in HBx-expressing cells (). Both in GFP- and HBx-GFP expressing cells, EGFR was significantly retained by Baf A1, confirming a specific degradation of EGFR in lysosomes ().
Figure 5. HBx impairs lysosomal degradative capacity. (A) Huh7 cells expressing GFP or HBx-GFP were incubated with 100 ng/ml of EGF for 15 min, then the EGF was washed out. At indicated time points after EGF stimulation, the cells were fixed and immunostained with EGFR antibody. Note the accumulation and subsequent degradation of EGFR puncta. (B) Quantification of the mean EGFR intensity per cell in cells expression of GFP or HBx-GFP. The values were normalized against the intensity in cells 1 h after EGF treatment. Data are presented as mean ± SEM, n = 30. **P < 0.01. (C) Western blot analysis of EGFR at indicated time points after EGF incubation in GFP or HBx-GFP expression cells with or without 100 nM bafilomycin A1 (Baf A1) treatment for 12 h. (D) Huh7 cells were cotransfected with Cherry-LC3 and GFP or Cherry-LC3 and HBx-GFP. At 48 h after transfection, the GFP-expressing cells were starved to accumulate autophagosomes. Then 10 mM of 3-methyladenine (3-MA) was added to the GFP- or HBx-GFP-expressing cells to block the formation of new autophagosomes and the degradation of formed Cherry-LC3-positive autophagosomes was observed at indicated time points. (E) Huh7 cells expressing GFP or HBx-GFP were treated as in (D). LC3 levels in the cells were assessed by western blot. Scale bars: 20 µm.
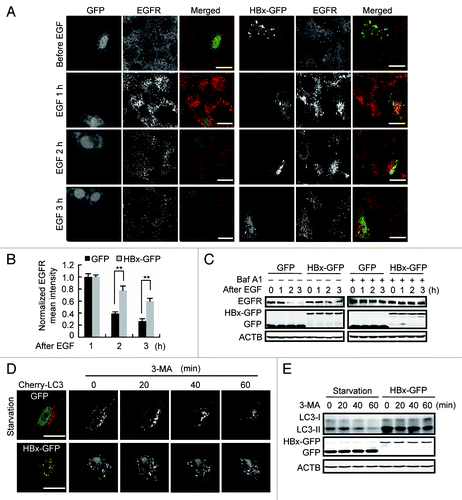
To further investigate lysosomal activity, we designed a pulse-chase assay to check the degradation of formed autophagosomes. We first confirmed that the conversion of LC3-I to LC3-II by HBx-GFP expression was suppressed by 3-methyladenine (3-MA), a specific inhibitor of class III PtdIns3K (Fig. S3), indicating HBx-induced accumulation of autophagosomes is sensitive to PtdIns3K inhibition. Huh7 cells expressing Cherry-LC3 were either transfected with HBx-GFP or starved to form autophagosomes. Then the cells were treated with 3-MA to prevent the synthesis of new autophagosomes, and the degradation of formed autophagosomes was tracked by time-lapse confocal microscopy. We found, in contrast to starved GFP-expressing cells in which the formed autophagosomes were gradually degraded, HBx-GFP-expressing cells retained almost all the formed autophagosomes (). The degradation of endogenous LC3 was observed in the same way by western blot, and the result demonstrated that in comparison to starved GFP-expressing cells, in cells expressing HBx-GFP, LC3 degradation was markedly inhibited (). Taken together, these results suggest that HBx impairs the degradative capacity of lysosomes.
HBx impairs lysosomal acidification and accumulates immature lysosomes
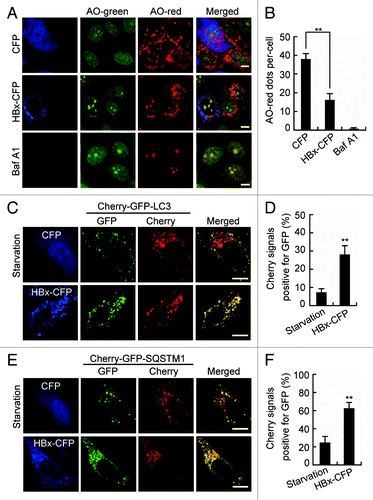
Since acidification is required for the maturation and activation of most lysosomal enzymes, the maintenance of acidity is a hallmark of functionally mature lysosomes. To further determine the action of HBx on lysosomes, we checked the acidification of lysosomes in HBx-expressing cells by assessing their retention of acridine orange (AO). The nonprotonated monomeric form of AO emits green fluorescence in the cytosol. But, when the dye enters acidic lysosomes, the protonated form becomes trapped in aggregates that fluoresces bright red.Citation26 We found, compared with cells expression of CFP, the cytoplasmic AO-red dots dramatically decreased in HBx-CFP-expressing cells (), indicating a reduced number of acidified compartments. No AO-red dots emerged in the cytoplasm of Baf A1-treated cells confirmed the representation of lysosomes by the compartments (). Lysosomal acidification was further examined by expression of the devised fusion protein Cherry-GFP-LC3.Citation27 Under nonlysosomal and neutral pH condition, both GFP and Cherry fluoresce. However, the low pH in the lumen of the lysosome quenches the GFP signal, but not the Cherry signal. Using this approach, we found that in starved cells, most of the puncta lost the GFP signal and retained the Cherry signal (). But in cells expressing HBx-CFP, quenching of the GFP was significantly diminished as indicated by the retention of both the Cherry and GFP signals (). A similar phenomenon was also detected in cells expressing Cherry-GFP-SQSTM1 (). Because we demonstrated already that HBx had no effect on autophagosome-lysosome fusion, these results suggest an impairment of lysosomal acidification by HBx.
Figure 6. HBx inhibits lysosomal acidification. (A) Live Huh7 cells expressing CFP or HBx-CFP were labeled with 5 µM acridine orange (AO). AO was imaged through a 515/530-nm band-pass filter (green) or a 580-nm long-pass filter (red). Note the decrease of cytoplasmic AO-red dots in HBx-CFP-expressing cells. Cells treated with 100 nM bafilomycin A1 (Baf A1) for 24 h were used as a positive control showing the complete disappearance of AO-red dots from the cytoplasm. (B) Statistical analysis of the number of AO-red dots per cell in cells expressing CFP or HBx-CFP, or treated with Baf A1, n = 30. (C andE) Live Huh7 cells expressing Cherry-GFP-LC3 (C) or Cherry-GFP-SQSTM1 (E) were starved or cotransfected with HBx-CFP for 48 h. Then the cells were imaged by confocal microscopy. (D and F) Statistical analysis of the percentage of punctate Cherry signals that were positive for GFP in (C and E), n = 30. All the quantitative data are presented as mean ± SEM **P < 0.01. Scale bars: 10 µm.
The maturation of lysosomes requires integration of the endocytic pathway, which involves the gradual remodeling of endosomal intermediates to form early endosomes and then late endosomes and lysosomes.Citation28 To investigate the effect of HBx on lysosomal maturation, we first assessed the early endosomes. Expression of HBx led to no change in the subcellular distribution and expression of EEA1 (early endosome antigen 1), a marker for early endosomes (). However, the number of LAMP1-positive puncta in HBx-expressing cells was significantly increased () accompanied by elevation in LAMP1 and LAMP2 protein levels (), suggesting a stabilization of LAMPs and accumulation of immature lysosomes by HBx expression. We further investigated the maturation of the lysosomal hydrolase CTSD. CTSD is synthesized in the endoplasmic reticulum as a 54-kDa inactive glycosylated propeptide. After transport to endosomes, it is cleaved into a 44-kDa intermediate form, which is then cleaved in the acidic environment of mature lysosomes to form the mature enzyme comprising noncovalently associated 31-kDa and 14-kDa polypeptides.Citation29 Any change in the mature forms reflects altered late endocytic trafficking or perturbed lysosomal maturation. We found that expression of HBx-GFP clearly reduced the 31-kDa and increased the 44-kDa CTSD (), indicating a suppression of the conversion from the intermediate to the mature form. The activation of CTSD was also assessed by incubating cells with Bodipy-FL-pepstatin A, which binds selectively to mature active CTSD.Citation30 Compared with that in nontransfected or Cherry-transfected cells in which Bodipy-FL-pepstatin A stained spot-like structures representing lysosomes with mature CTSD, Bodipy-FL-pepstatin A labeling was nearly completely abolished in HBx-Cherry-expressing cells (). Because the LAMP1-positive puncta () and the total CTSD () were not decreased in the cells, these results further confirmed an accumulation of immature lysosomes in HBx-expressing cells.
Figure 7. HBx inhibits the maturation of lysosomes. (A and B) Huh7 cells expressing GFP or HBx-GFP for 48 h were analyzed by either immunostaining (A) or western blot (B) using a specific EEA1 antibody. (C) Huh7 cells expressing GFP or HBx-GFP for 48 h were stained with LAMP1 antibody and imaged by confocal microscopy. Quantitation shown on the right represents the number of LAMP1 dots per cell in cells with GFP or HBx-GFP expression, n = 30. (D) Western blot analysis of LAMP1 and LAMP2 levels in Huh7 cells expressing GFP or HBx-GFP for 48 h, quantitation shown on the right represents the relative LAMP1/2 protein levels normalized to ACTB and the data were from 3 independent experiments. (E) Western blot analysis of CTSD in GFP- or HBx-GFP-transfected Huh7 cells. Note the increase in Inter-CTSD and decrease in Mature-CTSD. Quantification shown on the right represents the relative CTSD protein levels normalized to ACTB and the data were from 3 independent experiments. Pre-CTSD, precursor cathepsin D; Inter-CTSD, intermediate cathepsin D; Mature-CTSD, mature form of cathepsin D; *, nonspecific bands. (F) Huh7 cells expressing Cherry or HBx-Cherry for 48 h were loaded with 1 mM Bodipy-FL-pepstatin A for 1 h. After 3 washes with PBS, the cells were visualized by confocal microscopy. Quantification shown on the right represents the number of Bodipy-FL-pepstatin A dots per cell in cells with Cherry or HBx-Cherry expression, n = 30. (G) Huh7 cells were transfected with HA-tagged V-ATPase V1D and GFP or HA-tagged V-ATPase V1D and HBx-GFP for 48 h. Then the cells were fixed, stained with HA and LAMP1 antibodies, and were imaged. (H) Huh7 cells expressing GFP or HBx-GFP were immunoprecipitated with anti-GFP antibody, and the immunocomplexes were analyzed by western blot with an anti-V-ATPase V1D antibody. All the quantitative data are presented as mean ± SEM ***P < 0.001. Scale bars: 10 µm.
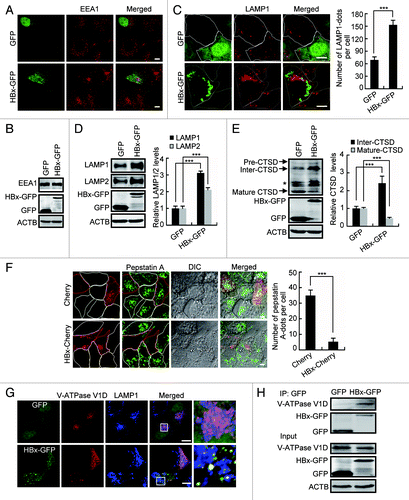
V-ATPase is an ATP-driven proton pump that imports protons into lysosomal lumen responsible for the acidification of the compartment. Trying to understand the basis for the acidification defect and subsequent lysosome immaturation in cells expression of HBx, we investigated the V-ATPase V1D subunit as a marker of proton pump function in lysosomes. We first investigated the subcellular localization of V-ATPase in cells transfected with HBx-GFP and HA-tagged V-ATPase V1D subunit. In Huh7 cells and HepG2 cells, V-ATPase V1D colocalized strongly with LAMP1-positive compartments. This colocalization was clearly disrupted in Huh7 cells expressing HBx-GFP, and in HepG2.2.15 cells, a HBV-replicating cell line carrying HBV DNA (; Fig. S4), suggesting a failure in the targeting of the V-ATPase V1D subunit to lysosomes by HBx. A colocalization of HBx-GFP with V-ATPase V1D was also observed () prompting us to look for their possible interaction. We examined whether HBx and V-ATPase V1D underwent coimmunoprecipitation. In Huh7 cells expressing HBx-GFP, immunoprecipitation of HBx-GFP using GFP antibody resulted in coimmunoprecipitation of endogenous V-ATPase V1D (), indicating an interaction between HBx and V-ATPase V1D.
HBV infection increases autophagosomes and immature lysosomes in human liver tissues
To search for the possible link between HBx-caused inhibition in autophagic degradation and the development of HBV-associated HCC, we used immunostaining and western blot to assess the autophagosomes and autophagic degradation in liver tumor and nontumor tissues in subject with HBV-associated HCC, and in samples from normal control individuals. Compared with the control group, both the nontumor and tumor samples demonstrated increased autophagosomes as indicated by accumulation of LC3-positive puncta () and increased LC3 protein level () (5 of the 7 sample pairs). When SQSTM1 protein was increased in nontumor and tumor samples, the tumor samples showed an even higher SQSTM1 level than that in nontumor tissues (5 of the 7 sample pairs, ).
Figure 8. Lysosomal function assessed in human tissues. (A andB) Representative immunostaining (A) and western blot (B) analysis of LC3 in normal liver tissue from control individuals, liver tumor, and nontumor tissue from HBV-associated HCC individuals. Note the accumulation of LC3-puncta and LC3 in the tumor and nontumor samples. (C andD) Immunostaining (C) and western blot (D) analysis of SQSTM1 in the human tissues. (E) Immunostaining of LAMP1 in the human tissues. (F) Western blot analysis of LAMP1 and CTSD in the tissues. Note the reduction of the mature form and accumulation of the intermediate form. Scale bars: 10 µm.
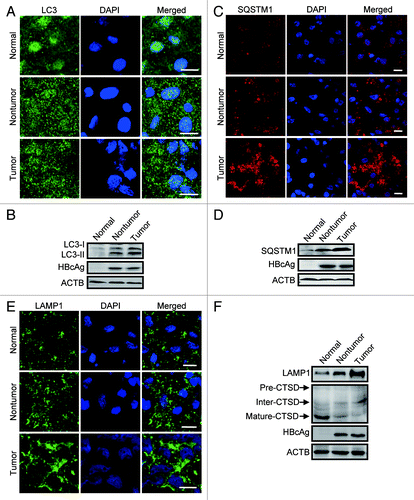
We also checked the lysosomal function in the human tissue samples. We found that the expression of LAMP1 in HCC samples, including the tumor and nontumor tissues, was dramatically increased in contrast to the normal group (5 of the 7 sample pairs, ). In addition, in these samples, the mature form of CTSD reduced significantly accompanied by accumulation of its intermediate form (5 of the 7 sample pairs, ). These data suggest that inhibition of autophagic degradation by impairing lysosomal function is involved in the HBV-associated HCC.
Discussion
Given that HBV and autophagy are closely associated with hepatocarcinogenesis, we investigated the role of HBV in hepatocellular autophagy and characterized the underlying mechanisms. Our results showed that through the HBx protein, HBV suppressed rather than stimulated autophagy in hepatocytes and hepatoma cells. Furthermore, our data suggested that inhibition of lysosomal maturation was involved in the process.
Despite the fact that the molecular mechanism underlying the role of autophagy in carcinogenesis remains far from clear, so far accumulating evidence suggests that defects in autophagy are associated with the development of carcinoma. Nevertheless, with regard to the close association of HBV with HCC, it has recently been reported that HBx itself either stimulates autophagy or sensitizes starvation-induced autophagy.Citation20,Citation21 Although enhancement of HBV replication has been interpreted as a result of HBx-induced autophagy,Citation20 this cannot explain the involvement of HBx in the transformation of hepatic cells and the generation of HCC.Citation31 In fact, as a lysosome-dependent pathway, one of the final outcomes and hallmarks of enhanced autophagy is the increase in cargo degradation. However, previous studies either missed or failed to detect this.Citation20,Citation21 Here, we demonstrated that HBx impaired lysosomal maturation. Our data suggest that the inactivation of lysosomes caused by HBx is one of the fundamental events responsible for the accumulation of autophagosomes and autophagic cargo proteins, including LC3 and SQSTM1. Also, our data support a recent suggestion that accumulation of SQSTM1 by either loss in essential autophagy genes or inhibition in lysosomal function may contribute to the development of HCC. Because it has been shown that assembly of excess SQSTM1 causes DNA damageCitation32 and stimulates a protumorigenic inflammatory response.Citation33,Citation34 In addition, persistent activation of nuclear factor NFE2L2/Nrf2 by accumulated SQSTM1 has been shown to connect autophagy-deficiency with liver tumors.Citation35
We showed that the expression of HBx increased intracellular autophagosomes without other costimulators. This is inconsistent with the report of Tang et al., in which HBx fails to stimulate the formation of autophagosome but sensitizes starvation-induced autophagosomes by upregulating BECN1 expression.Citation21 On the contrary, our data partly support the results of Sir et al., showing that expression of HBx by itself is sufficient to induce autophagosomes and is essential for this action of HBV.Citation20 However, our results indicated an additional mechanism underlying the increase in intracellular autophagosomes triggered by HBx. In addition to stimulating the early signaling to enhance the formation of autophagosomes,Citation20 our data suggest at least an involvement of an inhibitory effect of HBx on the late stage of autophagy. This conclusion derives from 3 lines of evidence. First, HBx had a synergistic effect on starvation- or rapamycin-induced production of LC3-II, mimicking the action of Baf A1; second, HBx arrested the degradation of LC3 and SQSTM1, the selective autophagy substrates; finally, HBx prevented the maturation of lysosomes, leading to lysosomal dysfunction. Using this strategy, HBV possibly intends to achieve 2 purposes. One is to enhance HBV DNA replication by accumulating autophagosomes in host cells. Multiple studies have indicated that induction of autophagosomes contributes to virus production and inhibition of autophagosome formation suppresses virus replication,Citation20,Citation36-Citation38 suggesting that these viruses can develop mechanisms to utilize autophagy machinery as proviral host factors favoring viral replication. Accumulation of autophagosomes by inhibiting the maturation of autophagosomes has also been observed with some viruses to achieve the same effect.Citation39,Citation40 Increased autophagosomes are of importance for HBV viral envelopment,Citation37 and contribute to the effective production of HCV particles with little effect on the intracellular production of HCV mRNA and HCV-related proteins,Citation41 supporting a speculation that autophagosome-derived membranes could be used as a source of membranes for viral envelopment. Another possibility for HBV to inhibit lysosomal activity may be that the virus uses it as a survival mechanism to escape direct destruction and/or attenuating antigen presentation by suppressing the generation of antigenic peptides.Citation42,Citation43
Cytoplasmic HBx influences intracellular signal transduction by interacting directly with signaling components. With regard to the possible mechanism for the impediment in lysosomal maturation caused by HBx, in this study, we found very little localization of HBx to either endosomes or lysosomes, implying that a direct interaction between HBx and the organelles is unlikely. In addition, in HBx-expressing cells, autophagosomes fused normally with lysosomes, ruling out a possible defect of lysosomes received from autophagosomes. Furthermore, accumulation of LAMPs and intermediate CTSD in late endosomes suggests unobstructed transport of integral lysosomal membrane proteins and soluble lysosomal hydrolases to the endosomes. Based on the lowered acidity of lysosome with a deficiency in CTSD maturation, a potential target of HBx could be V-ATPase. It has been suggested that lysosomal targeting of such a multiprotein complex exhibits an additional level of complexity compared with the LAMPs, and possibly through a direct mannose-6-phosphate receptor-independent trans-Golgi-network-to-lysosome transport pathway.Citation28,Citation44 In fact, a dislocation of V-ATPase has been observed in HCV replicon cells and considered to be involved in the impairment of the autolysosomal acidification in the cells.Citation43 In our study, we detected coimmunoprecipitation of HBx with the V1D subunit of V-ATPase accompanied with a disruption in lysosomal targeting of V-ATPase. Although further study is required, our data suggest that HBx-caused defect of lysosomal acidification may involve HBx-V-ATPase interactions and subsequent impairment in V-ATPase transport to lysosomes.
We found that HBx impaired lysosome maturation by inhibiting lysosomal acidification without disturbing autophagosome-lysosome fusion. This is consistent with a previous report,Citation45 suggesting that acidification of lysosome is not a prerequisite for autolysosome formation. Accumulating data have suggested that autophagosome-lysosome fusion requires the presence of particular molecular modulators on the membranes of both compartments,Citation46,Citation47 although currently we know little about the molecular mechanism underlying the process. It seems that, as in the case of LAMPs, the transport of SNAREs to endosomes was not blocked by HBx. Our data also suggest that activation of soluble lysosomal hydrolases and possibly the V-ATPase in lysosomal membranes are not involved in autophagosome-lysosome fusion.
In summary, we showed that the degradative endocytic pathway is a new target for HBV. Through HBx protein, dysfunction of lysosomes by HBV resulted in not only an increase of intracellular autophagosomes but also the accumulation of selective autophagic cargoes that may be linked to HBV-associated HCC.
Materials and Methods
Cell culture and transfection
Huh7 cells were grown in DMEM, L02 cells were grown in RPMI-1640, with 10% FBS at 37 °C under 5% CO2. Transient transfection was performed using FuGENE (Roche, 04-709-713-001) for cell imaging and Lipofectamine 2000 (Invitrogen, 11668-019) for western blotting, according to the manufacturer’s instructions.
Plasmids, reagents, and antibodies
Plasmids with the 1.3mer HBV genomic DNA (pUC19 HBV), the HBx-negative 1.3mer HBV genomic DNA (pUC19 HBVX−) and HBx-HA were gifts from Jing-hsiung Ou (University of Southern California). The RAB7A-GFP and RAB7AT22N-GFP were gifts from Eeva-Liisa Eskelinen (University of Kiel). The Cherry-GFP-SQSTM1 and Cherry-GFP-LC3 were provided by Terje Johansen (University of Tromsø). HA-V-ATPase V1D was a gift from Xiaodong Shu (Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Science). The HBx-GFP and Cherry-LC3 plasmids were described previously.Citation6,Citation48 The HBx-CFP and HBx-Cherry plasmids were constructed by replacing GFP of HBx-GFP with CFP and Cherry. Rapamycin (Sigma, R8781), bafilomycin A1 (Baf A1) (Sigma, B1793), chloroquine (CQ) (Sigma, C6628) and 3-methyladenine (3-MA) (Sigma, M9281) were from Sigma. EGF (PeproTech, AF-100-15) was from PeproTech. Bodipy-FL-pepstatin A (Invitrogen, P12271) was from Invitrogen. The following antibodies were used: anti-LC3 (Sigma, L7643), anti-ACTB/β-actin (Sigma, A5316), anti-RPS6KB/p70S6 (Cell Signaling, 9202), anti-phospho-RPS6KB/p70S6 (Cell Signaling, 9205), anti-MTOR (Cell Signaling, 2972), anti-phospho-MTOR (Cell Signaling, 2971), anti-SQSTM1 (Santa Cruz, sc28359), anti-HA (Santa Cruz, sc7392), anti-LAMP1 (Santa Cruz, sc20011), anti-V-ATPase V1D (Santa Cruz, sc166218), anti-CTSD (Santa Cruz, sc6487), anti-GFP (Santa Cruz, sc9996), anti-EGFR (BD Biosciences, 610016) and anti-EEA1 (BD Biosciences, 610456); anti-LAMP2 (Abcam, ab25631), anti-HBcAg (Abcam, ab8637), anti-HBx (Abcam, ab39716), donkey anti-rabbit IRDye800CW (LI-COR Biosciences, 926-32213) and donkey anti-mouse IRDye680 (LI-COR Biosciences, 926-32222).
Human tissues
Frozen fresh liver or liver cancer tissues were anonymously taken from the First Affiliated Hospital, Zhejiang University School of Medicine. Seven pairs of tumor liver tissues and their peripheral nontumor tissues after surgical resection were collected from HCC patients with chronic HBV infection. Seven normal liver tissues after surgical resection were collected from patients with liver hemangiomas. The diagnoses were based on clinical laboratory examination. All the human tissues were taken with written informed consent and with the approval of the Medical Ethical Committee of Zhejiang University School of Medicine (No. 1-009).
Autophagy induction by cell starvation or rapamycin treatment
Cells were washed 3 times with prewarmed PBS and then incubated in starvation medium (1% BSA, 140 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, 5 mM glucose and 20 mM HEPES, pH 7.4) at 37 °C for 1.5 h. For rapamycin treatment, cells were cultured with 100 nM rapamycin for 24 h.
Analysis of lysosomal acidification
Cells grown in LabTek chambers were cultured with 5 μg/ml acridine orange for 15 min at 37 °C. After washing with PBS to remove excess AO, the cells were visualized by confocal microscopy. For the Cherry-GFP-tagged LC3 and SQSTM1 assay, cells grown in LabTek chambers were cotransfected with HBx-CFP and Cherry-GFP-LC3 or HBx-CFP and Cherry-GFP-SQSTM1. At 48 h after transfection, the cells were visualized by live cell imaging. The percentage of punctate Cherry signals that were positive for GFP was analyzed using Volocity 5.4.2 (PerkinElmer).
Bodipy-FL-Pepstatin A labeling
Cells grown in LabTek chambers were transfected with Cherry or HBx-Cherry. At 48 h after transfection, the cells were incubated with 1 mM Bodipy-FL-pepstatin A for 1 h at 37 °C. After washing with PBS to remove excess Bodipy-FL-pepstatin A, the cells were visualized by confocal microscopy.
Analysis of EGFR degradation
Cells were transfected with HBx-GFP or GFP for 36 h. After culture in serum-free DMEM for 12 h, the cells were incubated on ice in serum-free DMEM containing 100 ng/ml of EGF for 15 min followed by culturing in EGF-free medium at 37 °C. At determined time points, the cells were either lysed and analyzed by western blot or were fixed in 4% formaldehyde, stained with anti-EGFR antibody and imaged by confocal microscopy.
Western blot and immunoprecipitation
Western blot was performed as described previously.Citation48 Briefly, the proteins from lysed cells or human tissues were denatured and loaded on sodium dodecyl sulfate polyacrylamide gels. Afterwards, the proteins were transferred to PVDF membranes, blocked in TBS-T (150 mM NaCl, 10 mM TRIS-HCl pH 7.5, and 0.1% Tween 20) containing 5% (w/v) bovine serum albumin, and incubated with the corresponding primary and secondary antibodies. The specific bands were analyzed by the western blot infrared imaging system (LI-COR Biosciences). The protein levels were quantified by using NIH Image program (Developed at the National Institutes of Health and available on line at rsb.info.nih.gov/nih-image/). For immunoprecipitation, cells were lysed with Triton X-100 lysis buffer containing protease inhibitors. After centrifugation, the supernatants were incubated with antibody overnight and then Protein A agarose for 2 h at 4 °C. Immunocomplexes were washed and analyzed by western blot.
Immunostaining and confocal microscopy
For immunostaining, cells or frozen tissue sections were fixed in 4% formaldehyde. After washing twice with PBS, they were incubated in PBS/FBS (PBS, pH 7.4, containing 10% FBS) to block nonspecific sites of antibody adsorption. Then, the cells or tissues sections were incubated with appropriate primary and secondary antibodies in 0.1% saponin as indicated in the figure legends. Images were captured on a Zeiss LSM510 Meta laser scanning confocal microscope (Carl Zeiss). For quantification of fluorescence intensities, nonsaturated images were taken with 40× 1.3 NA objective and a full-open pinhole, whereas a 63× 1.4 NA objective and a pinhole diameter equivalent to 1 to 3 Airy units were used for nonquantitative imaging. For quantification of intracellular puncta, a pinhole diameter equivalent to 5 to 6 Airy units was used. Live cell imaging was performed in LabTek chambers (Nalge Nunc International) maintained at 37 °C with 5% CO2.
For quantification of the number of autophagosomes (diameters 0.3–1.0 μM) and AO-red dots (diameters 0.1–1.0 μM), a total of 50 cells were recorded and analyzed using the Axiovision Automatic measurement program on the Zeiss LSM510 Meta laser scanning confocal microscope. Quantification of LC3 and EGFR fluorescence intensity and analysis of protein colocalization coefficients in 30 cells were performed using Volocity 5.4.2 (PerkinElmer).
Electron microscopy
Electron microscopy was performed as described previously.Citation49 Briefly, cells were fixed in 2.5% glutaraldehyde in 0.2 M HEPES (pH 7.4), and post-fixed in aqueous 1% OsO4 followed by 2% uranyl acetate. After ethanol and propylene oxide dehydration and embedding in polybed 812 resin (Polysciences, 025950-1), ultrathin (80 nm) sections were poststained with 2% uranyl acetate followed by 0.3% lead citrate. Sections were imaged using a TECNAI 10 transmission electron microscope (FEI Company) at 80 kV. For autophagic vacuole quantification, 20 micrographs, primary magnification ×15,000, were taken with systematic random sampling from each sample. The cytoplasmic volume fraction of autophagic vacuoles was estimated using MetaMorph (Universal Imaging Corp).
Subcellular fractionation
Purified autophagosomes were prepared by a procedure described previously.Citation50 Briefly, liver tissue was minced and homogenized by a Dounce homogenizer in homogenization buffer (HB; 0.25 M sucrose, 1 mM EDTA, 20 mM HEPES, pH 7.4). The homogenate were centrifuged at 2,000 g for 2 min to obtain a nuclear pellet and a supernatant fraction. The nuclear pellet fraction was washed twice in HB and the total supernatant fractions were purified on a discontinuous Nycodenz gradient. The Nycodenz gradient was prepared by carefully layering stepwise 7 ml of 22.5% and 17 ml of 9.5% Nycodenz solution in centrifuge tubes. The Nycodenz gradient was centrifuged at 141,000 g for 1 h. The interface band, which contained the autophagosomes, was diluted with 5 ml of HB and layered on top of a discontinuous gradient of 21 ml of 33% Percoll in HB on top of 7 ml of 22.5% Nycodenz in HB, and centrifuged at 72,000 g for 30 min. The autophagosomes banded at the lower interface. Percoll was removed from the autophagosome fraction by mixing 5 ml of the fraction with 3.5 ml of isotonic 60% (w/v) iodixanol in water, overlaying with 1.5 ml of 30% iodixanol and a top layer of 2.5 ml of HB, and centrifuging at 71,000 g for 30 min. The autophagosomes banded at the iodixanol/HB interface.
Statistical analysis
All the statistical data were presented as mean ± SEM. Statistical significance of the differences was determined using Student t test. Differences were considered significant at values of P < 0.05.
| Abbreviations: | ||
| 3-MA | = | 3-methyladenine |
| ATG | = | autophagy-related |
| AO | = | acridine orange |
| Baf A1 | = | bafilomycin A1 |
| BCL2 | = | B-cell CLL/lymphoma 2 |
| BECN1 | = | Beclin 1 |
| CQ | = | chloroquine |
| CTSD | = | cathepsin D |
| EEA1 | = | early endosome antigen 1 |
| EGF | = | epidermal growth factor |
| EGFR | = | epidermal growth factor receptor |
| GFP | = | green fluorescent protein |
| HBV | = | hepatitis B virus |
| HBx | = | hepatitis B virus X protein |
| HCC | = | hepatocellular carcinoma |
| LAMP1/2 | = | lysosomal-associated membrane protein 1/2 |
| MAP1LC3 (LC3) | = | microtubule-associated protein 1 light chain 3 beta |
| MTOR | = | mechanistic target of rapamycin |
| NFE2L2/Nrf2 | = | nuclear factor, erythroid 2-like 2 |
| PtdIns3K | = | phosphatidylinositol 3-kinase |
| RPS6KB/p70S6K | = | ribosomal protein S6 kinase, 70 kDa |
| SQSTM1 | = | sequestosome 1 |
| TP53/p53 | = | tumor protein p53 |
| V-ATPase | = | vacuolar-type H+-ATPase |
Additional material
Download Zip (240.2 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Drs. Jing-hsiung Ou, Eeva-Liisa Eskelinen, Terje Johansen and Xiaodong Shu for sharing plasmids, Dr. IC Bruce for correction of the manuscript. This work was supported by National Basic Research Program of China (2011CB910100 and 2013CB910200), National Natural Science Foundation of China (31171288 and 31271431), and a Qianjiang Talents project of the Technology Office of Zhejiang Province (2010R10059). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- Yang JD, Roberts LR. Hepatocellular carcinoma: A global view. Nat Rev Gastroenterol Hepatol 2010; 7:448 - 58; http://dx.doi.org/10.1038/nrgastro.2010.100; PMID: 20628345
- Robinson WS. Molecular events in the pathogenesis of hepadnavirus-associated hepatocellular carcinoma. Annu Rev Med 1994; 45:297 - 323; http://dx.doi.org/10.1146/annurev.med.45.1.297; PMID: 8198385
- Nakamoto Y, Guidotti LG, Kuhlen CV, Fowler P, Chisari FV. Immune pathogenesis of hepatocellular carcinoma. J Exp Med 1998; 188:341 - 50; http://dx.doi.org/10.1084/jem.188.2.341; PMID: 9670046
- Zheng Y, Chen WL, Louie SG, Yen TS, Ou JH. Hepatitis B virus promotes hepatocarcinogenesis in transgenic mice. Hepatology 2007; 45:16 - 21; http://dx.doi.org/10.1002/hep.21445; PMID: 17187428
- Zhang X, Zhang H, Ye L. Effects of hepatitis B virus X protein on the development of liver cancer. J Lab Clin Med 2006; 147:58 - 66; http://dx.doi.org/10.1016/j.lab.2005.10.003; PMID: 16459163
- Xiang WQ, Feng WF, Ke W, Sun Z, Chen Z, Liu W. Hepatitis B virus X protein stimulates IL-6 expression in hepatocytes via a MyD88-dependent pathway. J Hepatol 2011; 54:26 - 33; http://dx.doi.org/10.1016/j.jhep.2010.08.006; PMID: 20937539
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19:5720 - 8; http://dx.doi.org/10.1093/emboj/19.21.5720; PMID: 11060023
- Kraft C, Peter M, Hofmann K. Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol 2010; 12:836 - 41; http://dx.doi.org/10.1038/ncb0910-836; PMID: 20811356
- Lum JJ, DeBerardinis RJ, Thompson CB. Autophagy in metazoans: cell survival in the land of plenty. Nat Rev Mol Cell Biol 2005; 6:439 - 48; http://dx.doi.org/10.1038/nrm1660; PMID: 15928708
- Rubinsztein DC, Mariño G, Kroemer G. Autophagy and aging. Cell 2011; 146:682 - 95; http://dx.doi.org/10.1016/j.cell.2011.07.030; PMID: 21884931
- Mariño G, Salvador-Montoliu N, Fueyo A, Knecht E, Mizushima N, López-Otín C. Tissue-specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin-3. J Biol Chem 2007; 282:18573 - 83; http://dx.doi.org/10.1074/jbc.M701194200; PMID: 17442669
- Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, Mizushima N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev 2011; 25:795 - 800; http://dx.doi.org/10.1101/gad.2016211; PMID: 21498569
- Inami Y, Waguri S, Sakamoto A, Kouno T, Nakada K, Hino O, Watanabe S, Ando J, Iwadate M, Yamamoto M, et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol 2011; 193:275 - 84; http://dx.doi.org/10.1083/jcb.201102031; PMID: 21482715
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999; 402:672 - 6; http://dx.doi.org/10.1038/45257; PMID: 10604474
- Aita VM, Liang XH, Murty VV, Pincus DL, Yu W, Cayanis E, Kalachikov S, Gilliam TC, Levine B. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics 1999; 59:59 - 65; http://dx.doi.org/10.1006/geno.1999.5851; PMID: 10395800
- Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer 2006; 6:184 - 92; http://dx.doi.org/10.1038/nrc1819; PMID: 16453012
- Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A 2005; 102:8204 - 9; http://dx.doi.org/10.1073/pnas.0502857102; PMID: 15928081
- Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005; 122:927 - 39; http://dx.doi.org/10.1016/j.cell.2005.07.002; PMID: 16179260
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27 - 42; http://dx.doi.org/10.1016/j.cell.2007.12.018; PMID: 18191218
- Sir D, Tian Y, Chen WL, Ann DK, Yen TS, Ou JH. The early autophagic pathway is activated by hepatitis B virus and required for viral DNA replication. Proc Natl Acad Sci U S A 2010; 107:4383 - 8; http://dx.doi.org/10.1073/pnas.0911373107; PMID: 20142477
- Tang H, Da L, Mao Y, Li Y, Li D, Xu Z, Li F, Wang Y, Tiollais P, Li T, et al. Hepatitis B virus X protein sensitizes cells to starvation-induced autophagy via up-regulation of beclin 1 expression. Hepatology 2009; 49:60 - 71; http://dx.doi.org/10.1002/hep.22581; PMID: 19065679
- Su Q, Schröder CH, Hofmann WJ, Otto G, Pichlmayr R, Bannasch P. Expression of hepatitis B virus X protein in HBV-infected human livers and hepatocellular carcinomas. Hepatology 1998; 27:1109 - 20; http://dx.doi.org/10.1002/hep.510270428; PMID: 9537452
- Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol 2010; 12:814 - 22; http://dx.doi.org/10.1038/ncb0910-814; PMID: 20811353
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313 - 26; http://dx.doi.org/10.1016/j.cell.2010.01.028; PMID: 20144757
- Gutierrez MG, Munafó DB, Berón W, Colombo MI. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J Cell Sci 2004; 117:2687 - 97; http://dx.doi.org/10.1242/jcs.01114; PMID: 15138286
- Kirkegaard T, Roth AG, Petersen NH, Mahalka AK, Olsen OD, Moilanen I, Zylicz A, Knudsen J, Sandhoff K, Arenz C, et al. Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature 2010; 463:549 - 53; http://dx.doi.org/10.1038/nature08710; PMID: 20111001
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131 - 45; http://dx.doi.org/10.1074/jbc.M702824200; PMID: 17580304
- Saftig P, Klumperman J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol 2009; 10:623 - 35; http://dx.doi.org/10.1038/nrm2745; PMID: 19672277
- Cai Q, Lu L, Tian JH, Zhu YB, Qiao H, Sheng ZH. Snapin-regulated late endosomal transport is critical for efficient autophagy-lysosomal function in neurons. Neuron 2010; 68:73 - 86; http://dx.doi.org/10.1016/j.neuron.2010.09.022; PMID: 20920792
- Chen CS, Chen WN, Zhou M, Arttamangkul S, Haugland RP. Probing the cathepsin D using a BODIPY FL-pepstatin A: applications in fluorescence polarization and microscopy. J Biochem Biophys Methods 2000; 42:137 - 51; http://dx.doi.org/10.1016/S0165-022X(00)00048-8; PMID: 10737220
- Kim CM, Koike K, Saito I, Miyamura T, Jay G. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature 1991; 351:317 - 20; http://dx.doi.org/10.1038/351317a0; PMID: 2034275
- Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009; 137:1062 - 75; http://dx.doi.org/10.1016/j.cell.2009.03.048; PMID: 19524509
- Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, Moscat J. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell 2008; 13:343 - 54; http://dx.doi.org/10.1016/j.ccr.2008.02.001; PMID: 18394557
- Ling J, Kang Y, Zhao R, Xia Q, Lee DF, Chang Z, Li J, Peng B, Fleming JB, Wang H, et al. KrasG12D-induced IKK2/β/NF-κB activation by IL-1α and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell 2012; 21:105 - 20; http://dx.doi.org/10.1016/j.ccr.2011.12.006; PMID: 22264792
- Hayes JD, McMahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci 2009; 34:176 - 88; http://dx.doi.org/10.1016/j.tibs.2008.12.008; PMID: 19321346
- Dreux M, Gastaminza P, Wieland SF, Chisari FV. The autophagy machinery is required to initiate hepatitis C virus replication. Proc Natl Acad Sci U S A 2009; 106:14046 - 51; http://dx.doi.org/10.1073/pnas.0907344106; PMID: 19666601
- Li J, Liu Y, Wang Z, Liu K, Wang Y, Liu J, Ding H, Yuan Z. Subversion of cellular autophagy machinery by hepatitis B virus for viral envelopment. J Virol 2011; 85:6319 - 33; http://dx.doi.org/10.1128/JVI.02627-10; PMID: 21507968
- Tian Y, Sir D, Kuo CF, Ann DK, Ou JH. Autophagy required for hepatitis B virus replication in transgenic mice. J Virol 2011; 85:13453 - 6; http://dx.doi.org/10.1128/JVI.06064-11; PMID: 21957292
- Gannagé M, Dormann D, Albrecht R, Dengjel J, Torossi T, Rämer PC, Lee M, Strowig T, Arrey F, Conenello G, et al. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 2009; 6:367 - 80; http://dx.doi.org/10.1016/j.chom.2009.09.005; PMID: 19837376
- Berkova Z, Crawford SE, Trugnan G, Yoshimori T, Morris AP, Estes MK. Rotavirus NSP4 induces a novel vesicular compartment regulated by calcium and associated with viroplasms. J Virol 2006; 80:6061 - 71; http://dx.doi.org/10.1128/JVI.02167-05; PMID: 16731945
- Tanida I, Fukasawa M, Ueno T, Kominami E, Wakita T, Hanada K. Knockdown of autophagy-related gene decreases the production of infectious hepatitis C virus particles. Autophagy 2009; 5:937 - 45; http://dx.doi.org/10.4161/auto.5.7.9243; PMID: 19625776
- Tardif KD, Siddiqui A. Cell surface expression of major histocompatibility complex class I molecules is reduced in hepatitis C virus subgenomic replicon-expressing cells. J Virol 2003; 77:11644 - 50; http://dx.doi.org/10.1128/JVI.77.21.11644-11650.2003; PMID: 14557650
- Taguwa S, Kambara H, Fujita N, Noda T, Yoshimori T, Koike K, Moriishi K, Matsuura Y. Dysfunction of autophagy participates in vacuole formation and cell death in cells replicating hepatitis C virus. J Virol 2011; 85:13185 - 94; http://dx.doi.org/10.1128/JVI.06099-11; PMID: 21994453
- Vergarajauregui S, Puertollano R. Two di-leucine motifs regulate trafficking of mucolipin-1 to lysosomes. Traffic 2006; 7:337 - 53; http://dx.doi.org/10.1111/j.1600-0854.2006.00387.x; PMID: 16497227
- Renna M, Schaffner C, Brown K, Shang S, Tamayo MH, Hegyi K, Grimsey NJ, Cusens D, Coulter S, Cooper J, et al. Azithromycin blocks autophagy and may predispose cystic fibrosis patients to mycobacterial infection. J Clin Invest 2011; 121:3554 - 63; http://dx.doi.org/10.1172/JCI46095; PMID: 21804191
- Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012; 151:1256 - 69; http://dx.doi.org/10.1016/j.cell.2012.11.001; PMID: 23217709
- Fader CM, Sánchez DG, Mestre MB, Colombo MI. TI-VAMP/VAMP7 and VAMP3/cellubrevin: two v-SNARE proteins involved in specific steps of the autophagy/multivesicular body pathways. Biochim Biophys Acta 2009; 1793:1901 - 16; http://dx.doi.org/10.1016/j.bbamcr.2009.09.011; PMID: 19781582
- Guo Y, Chang C, Huang R, Liu B, Bao L, Liu W. AP1 is essential for generation of autophagosomes from the trans-Golgi network. J Cell Sci 2012; 125:1706 - 15; http://dx.doi.org/10.1242/jcs.093203; PMID: 22328508
- Ylä-Anttila P, Vihinen H, Jokitalo E, Eskelinen EL. Monitoring autophagy by electron microscopy in Mammalian cells. Methods Enzymol 2009; 452:143 - 64; PMID: 19200881
- Strømhaug PE, Berg TO, Fengsrud M, Seglen PO. Purification and characterization of autophagosomes from rat hepatocytes. Biochem J 1998; 335:217 - 24; PMID: 9761717