
Abstract
MIR34A (microRNA 34a) is a tumor suppressor gene, but how it regulates chemotherapy response and resistance is not completely understood. Here, we show that the microRNA MIR34A-dependent high mobility group box 1 (HMGB1) downregulation inhibits autophagy and enhances chemotherapy-induced apoptosis in the retinoblastoma cell. HMGB1 is a multifaceted protein with a key role in autophagy, a self-degradative, homeostatic process with a context-specific role in cancer. MIR34A inhibits HMGB1 expression through a direct MIR34A-binding site within the HMGB1 3′ untranslated region. MIR34A inhibition of HMGB1 leads to a decrease in autophagy under starvation conditions or chemotherapy treatment. Inhibition of autophagy promotes oxidative injury and DNA damage and increases subsequent CASP3 activity, CASP3 cleavage, and PARP1 [poly (ADP-ribose) polymerase 1] cleavage, which are important to the apoptotic process. Finally, upregulation of MIR34A, knockdown of HMGB1, or inhibition of autophagy (e.g., knockdown of ATG5 and BECN1) restores chemosensitivity and enhances tumor cell death in the retinoblastoma cell. These data provide new insights into the mechanisms governing the regulation of HMGB1 expression by microRNA and their possible contribution to autophagy and drug resistance.
Keywords: :
Introduction
microRNAs (miRNAs) are a class of post-transcriptional regulators.Citation1 They are short, ~22 nucleotide RNA sequences that bind to complementary sequences in the 3′ untranslated region (3′UTR) of multiple target mRNAs (mRNAs). These molecules are expressed in a tissue-specific and developmental stage-specific manner and typically reduce mRNA stability. Alterations in the expression of several miRNAs are often present in human cancers and mediate processes in tumorigenesis and cancer therapy response and resistance.Citation2,Citation3
The retinoblastoma is the most common intraocular malignant tumor in infants and children.Citation4 Understanding the molecular mechanism(s) underlying retinoblastoma pathogenesis is crucial for designing novel targeted therapies to avoid enucleation of the eyeball to prevent metastasis. Recent evidence has indicated that MIR34A functions as a tumor suppressor in retinoblastoma and is a potential therapeutic target.Citation5,Citation6 However, the mechanisms underlying this antitumor effect are not well characterized. MIR34A belongs to one of several evolutionarily conserved families of miRNAs, namely MIR34, and was originally identified as a TP53-targeted microRNA.Citation7 MIR34A is expressed in almost every tissue except lung tissue, whereas MIR34B and MIR34C are mainly expressed in lung tissue but scarcely expressed in other tissues.Citation8 MIR34A has been implicated in multiple cancer types, including retinoblastoma, and its expression is a prognostic parameter.Citation9-Citation13 Numerous studies demonstrate that MIR34A is critically involved in regulating diverse tumor cellular processes such as apoptosis and the cell cycle.Citation7,Citation14-Citation17 Identifying functionally important mRNA targets of MIR34A is essential to unraveling its biological function and is helpful for cancer drug development.Citation18-Citation20
HMGB1 (high mobility group box 1) belongs to a family of highly conserved proteins that contain HMG box domains. HMGB1 is a ubiquitous nuclear protein that regulates and facilitates various DNA-related activities such as transcription, replication, recombination, and repair. In addition to its functions in the nucleus, HMGB1 proteins play a significant extracellular role in inflammation and immunity.Citation21,Citation22 HMGB1 has been proposed as a target for cancer therapy because impaired HMGB1 is associated with the hallmarks of cancer.Citation23 Blocking HMGB1 release and activity reduces tumor incidence, invasion, and metastasis.Citation24 Recent studies suggest that autophagy may be important in the regulation of cancer development and progression and in determining the response of tumor cells to anticancer therapy.Citation25 HMGB1 is an important autophagic regulator.Citation26-Citation28 HMGB1-mediated autophagy inhibits apoptosis and promotes drug resistance in several tumor types, including leukemia, colorectal cancer, pancreatic cancer, and osteosarcoma.Citation29-Citation34 However, little is known about the mechanism of HMGB1 upregulation and its role in retinoblastoma.
In this study, we examined the impact of MIR34A on HMGB1 expression and its molecular actions in the regulation of chemosensitivity in the retinoblastoma cell. Our study suggests that MIR34A targets HMGB1 mRNA, leading to translational repression of HMGB1. MIR34A is a potent inhibitor of autophagy, which promotes chemotherapeutic agent-induced DNA damage and apoptosis in a reactive oxygen species (ROS)-dependent manner. These findings provide the first evidence that the MIR34A-HMGB1 pathway regulates autophagy and apoptosis, as well as the chemotherapy response.
Results
HMGB1 is a target of MIR34A in human retinoblastoma cells
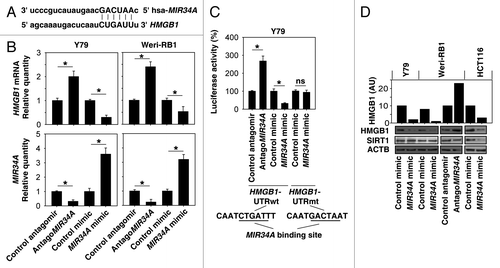
Based on the miRNA database, HMGB1 is a predicted MIR34A target in humans (). A previous study has demonstrated that MIR34A was expressed in human retinoblastoma cells.Citation6 Thus, we determined the effect of MIR34A on the expression of HMGB1 in human retinoblastoma cell lines (Y79 and Weri-RB1). To this end, we used a MIR34A mimic and antagoMIR34A to modulate cellular levels of MIR34A (). The mRNA levels of HMGB1 decreased following MIR34A mimic treatment, whereas antagoMIR34A increased HMGB1 expression (). The 3′ UTR reporter activity of HMGB1 was further assessed by luciferase assays. MIR34A mimic inhibited HMGB1 luciferase activities, whereas antagoMIR34A increased HMGB1 luciferase activities (“HMGB1-UTRwt”) in Y79 cells (). In contrast, MIR34A mimic had no effect on the expression of the luciferase reporter regulated by HMGB1-UTR with mutated MIR34A binding sites (“HMGB1-UTRmt”) (). Consistently, the protein levels of HMGB1 decreased following MIR34A mimic treatment, whereas antagoMIR34A increased HMGB1 protein expression in Y79 and human HCT116 colon cancer cells (). SIRT1 (sirtuin 1) is an NAD-dependent deacetylase that regulates apoptosis and autophagy during stress.Citation17,Citation35 Recent data suggests that SIRT1 may function as a MIR34A-targeted gene in several cancer cells, including HCT116 cells ().Citation17 However, MIR34A did not regulate SIRT1 expression in Y79 and Weri-RB1 cells. These findings suggest that HMGB1 is the target of MIR34A, and MIR34A regulates gene expression in a tissue-specific manner.
Figure 1.MIR34A targets HMGB1. (A) Schema representing the functional interaction between MIR34A and the seed sequence (bold) in the 3′UTR of HMGB1 as predicted by miRanda-mirSVR (http://www.microrna.org/microrna/home.do). (B) Y79 and Weri-RB1 cells were transfected with antagomir or mimic of MIR34A or nontarget antagomir or mimic control (100 nM) for 72 h, and the level of MIR34A was assayed by TaqMan qRT-PCR. In parallel, the mRNA level of HMGB1 was assayed by qRT-PCR (n = 3, *P < 0.05). Data represent relative level, with control set as 1. (C) Luciferase assay of Y79 cells cotransfected with reporter constructs containing HMGB1 3′ UTRs with (HMGB1-UTRwt) or without (HMGB1-UTRmt) MIR34a-binding sites and mimic of MIR34A or scrambled control miRNA for 72 h (n = 3, *P < 0.05; ns, nonsignificant). Data represent relative level, with control set as 100%. (D) Y79, Weri-RB1 and HCT116 cells were transfected with indicated miRNAs or antagomir RNAs for 72 h, and the level of HMGB1 and SIRT1 protein expression was assayed by western blot. AU = arbitrary unit.
MIR34A induces apoptosis by modifying HMGB1 expression
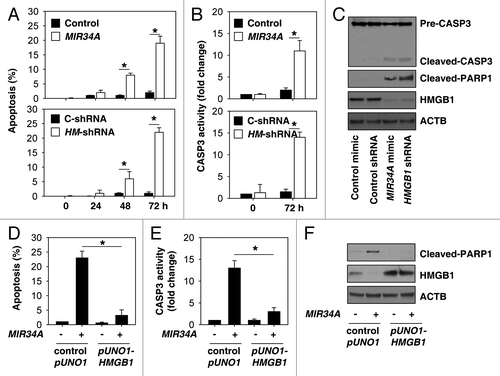
Next, we investigated whether suppression of HMGB1 expression by short hairpin RNA (shRNA) has a similar function compared with treatment with MIR34A mimic. MIR34A can potentially affect several signaling pathways and is implicated in tumor malignancy. Induction of apoptosis has been identified as a major mechanism that contributes to the tumor suppressive function of MIR34A.Citation9 Thus, we determined whether knockdown of HMGB1 by shRNA induces apoptosis in retinoblastoma cells. Similar to MIR34A mimic treatment, suppression of HMGB1 expression promotes apoptosis by flow cytometric analysis of the percentage of cells that are ANXA5/annexin V-positive (). CASP3 (caspase 3) is a critical executioner of apoptosis, as it is either partially or totally responsible for the proteolytic cleavage of many key proteins, such as PARP1.Citation36 Knockdown of HMGB1 or MIR34A mimic increased the activity of CASP3 (), cleaved-CASP3, and cleaved-PARP1 (). These findings suggest that knockdown of HMGB1 or treatment with MIR34A similarly influence the regulation of apoptosis. Furthermore, overexpression of HMGB1 by transfection with full-length HMGB1 cDNA (“pUNO1-HMGB1”) inhibited MIR34A mimic-induced apoptosis (), CASP3 activity (), and the expression of cleaved PARP1 (), suggesting that downregulation of HMGB1 is required for MIR34A mimic-induced apoptosis in retinoblastoma cells.
Figure 2.MIR34A induces apoptosis through suppression of HMGB1 expression. (A and B) Y79 cells were transfected with mimic of MIR34A, non-target mimic control, HMGB1 shRNA (HM-shRNA), or control shRNA (C-shRNA) for 0 to 72 h, and then apoptosis (A) was analyzed by measuring ANXA5-positive cells by flow cytometry and CASP3 activity (B) was measured with the CASP3 Colorimetric Assay Kit (n = 3, *P < 0.05). (C) In parallel, the indicated protein level was assayed by western blot. (D and E) Y79 cells were transfected with mimic of MIR34A, control pUNO1 cDNA, or pUNO1-HMGB1 cDNA for 72 h, and then apoptosis (D) was analyzed by measuring ANXA5-positive cells by flow cytometry and CASP3 activity (E) was measured by CASP3 Colorimetric Assay Kit (n = 3, *P < 0.05). (F) In parallel, the indicated protein level was assayed by western blot.
MIR34A inhibits autophagy by modifying HMGB1 expression
Given that HMGB1 is a target of MIR34A (), we investigated the role of MIR34A in autophagy. The most typical trigger of autophagy is nutrient starvation using Hank's balanced salt solution (HBSS). Starvation increased mRNA expression of HMGB1 at 1 to 2 h, and returned to the baseline levels at 6 h when MIR34A expression was increased in the retinoblastoma cell (). MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3) is now widely used to monitor autophagy because the amount of LC3-II correlates with the number of autophagosomes.Citation37 We found that MIR34A mimic or knockdown of HMGB1 inhibited starvation-induced MAP1LC3-II expression and the number of MAP1LC3 puncta in Y79 and HCT116 cells (). In contrast, treatment with lysosomal protease inhibitors E64d and pepstatin A (E/P) restored MAP1LC3-II expression and MAP1LC3 puncta in MIR34A-transfection cells (). As expected, the levels of polyubiquitin binding protein SQSTM1/p62 (sequestosome 1)Citation38 increased when starvation-induced autophagy was impaired (). An assay measuring the autophagy-mediated degradation of radiolabeled long-lived proteins showed that MIR34A mimic or knockdown of HMGB1 inhibited starvation-induced protein degradation (). Moreover, ultrastructural electron microscopy analysis revealed that MIR34A mimic decreased the number of autophagic vesicles after starvation (). These findings suggest that MIR34A regulates autophagy flux and functions as a potent inhibitor of autophagy.
Figure 3.MIR34A inhibits autophagy through modification of HMGB1 expression. (A) Y79 cells were incubated in HBSS for different lengths of time (0 to 6 h); the relative expression levels of HMGB1 mRNA and MIR34A were assessed by qRT-PCR and TaqManqRT-PCR, respectively, with control set as 100%. (B) Y79 and HCT116 were transfected with mimic of MIR34A, mimic control, HMGB1 shRNA, or control shRNA for 72 h, and then treated with HBSS with or without E64D/ pepstatin A (E/P, 10 μg/ml) for 1 h. The levels of MAP1LC3-I/II, SQSTM1, and HMGB1 protein expression were assayed by western blot. (C–E) In parallel, the MAP1LC3 puncta formation (C, shown by red arrow), the long-lived protein degradation and ultrastructural features of autophagic vacuoles (E, shown by red arrow) was assayed as described in Materials and Methods (*P < 0.05). (F and G) Hmgb1+/+ and hmgb1−/− MEFs or SIRT1 knockdown Y79 cells were transfected with antagoMIR34A or nontarget antagomir for 72 h or treated with HBSS in the presence and absence of E64D/pepstatin A (E/P, 10 μg/ml) for 1 h, the levels of MAP1LC3-I/II, SQSTM1, and HMGB1 protein expression were assayed by western blot (F), and MAP1LC3 puncta formation was assayed by confocal microscopic analysis (G). *P < 0.05. (H) Y79 cells were transfected with mimic of MIR34A, control pUNO1 cDNA, or pUNO1-HMGB1 cDNA for 72 h, and then treated with HBSS in the presence and absence of E64D/pepstatin A (E/P, 10 μg/ml) for 1 h; MAP1LC3 puncta formation was assayed by confocal microscopic analysis. *P < 0.05.
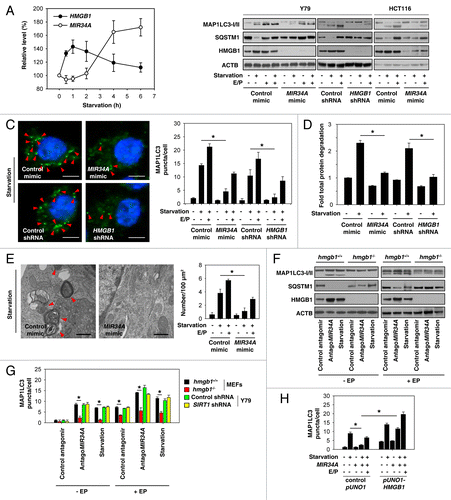
Previous studies have demonstrated that deletion, depletion, or inhibition of HMGB1 in mouse embryonic fibroblasts (MEFs) or tumor cells markedly diminishes autophagy.Citation29-Citation34 Interestingly, antagoMIR34A promoted HMGB1 expression (; ), MAP1LC3-II expression (), and MAP1LC3 puncta () in HMGB1 wild type, but not in HMGB1 knockout MEFs in the presence and absence of E/P, suggesting that upregulation of HMGB1 is required for antagoMIR34A-induced autophagy. However, knockdown of SIRT1 by shRNA did not influence antagoMIR34A- and starvation-induced autophagy in Y79 cells (), suggesting that MIR34A-mediated autophagy is SIRT1-independent in the retinoblastoma cell. Furthermore, overexpression of HMGB1 by transfection with a full-length HMGB1 cDNA (“pUNO1-HMGB1”) restored MIR34A mimic-mediated autophagy inhibition under starvation conditions (), suggesting that upregulation of HMGB1 is required for autophagy under starvation conditions.
Inhibition of autophagy enhances chemotherapeutic efficacy in retinoblastoma cells
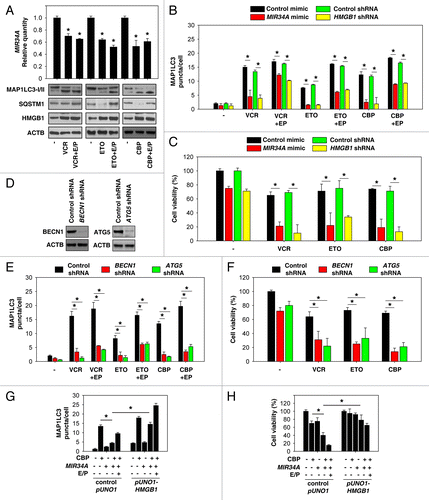
Vincristine (VCR), etoposide (ETO), and carboplatin (CBP) are agents commonly used systemically in retinoblastoma treatment. These anticancer drugs increased MAP1LC3-II and HMGB1 expression () and decreased MIR34A expression () in retinoblastoma cells. Different from ETO and CBP, inhibition of autophagy by the microtubule-destabilizing agent VCR results from an impairment of autolysosome formation because vincristine prevents SQSTM1 degradation, whereas ETO and CBP promote SQSTM1 degradation ().Citation39 Similar to starvation-induced autophagy (), MIR34A mimic and knockdown of HMGB1 significantly decreased VCR, ETO, and CBP-induced autophagy by monitoring MAP1LC3 puncta (), but enhanced chemotherapeutic efficacy in retinoblastoma cells (). BECN1 and ATG5 are critical regulators of autophagy.Citation40 Inhibition of autophagy by knockdown of BECN1 and ATG5 () also increased chemotherapeutic efficacy in retinoblastoma cells (). These findings suggest that enhanced autophagy increases drug resistance in retinoblastoma cells. Furthermore, overexpression of HMGB1 by transfection with a full-length HMGB1 cDNA restored MIR34A mimic-mediated autophagy inhibition () and inhibited chemotherapeutic efficacy after CBP treatment (), suggesting that MIR34A inhibits autophagy and increases cell death induced by chemotherapy in an HMGB1-dependent manner.
Figure 4. Inhibition of autophagy enhances chemotherapeutic efficacy in retinoblastoma cells. (A) Y79 cells were treated with vincristine (VCR, 2.5 nM), etoposide (ETO, 1 µM), and carboplatin (CBP, 1 µM) in the presence and absence of E64D/pepstatin A (E/P, 10 μg/ml) for 48 h, and the levels of MAP1LC3-I/II and HMGB1 were assayed by western blot. In parallel, the level of MIR34A was assayed by TaqMan qRT-PCR (n = 3, *P < 0.05, VS. control group). Data represent relative level, with control set as 1. (B and C) Y79 were transfected with mimic of MIR34A, mimic control, HMGB1 shRNA, or control shRNA for 72 h, and then treated with VCR (2.5 nM), ETO (1 µM), and CBP (1 µM) in the presence and absence of E64D/pepstatin A (E/P, 10 μg/ml) for 48 h. The MAP1LC3 puncta formation (B) was assayed by confocal microscopic analysis and cell viability (C) was assayed by CCK-8 kit (*P < 0.05). (D) Western blot analysis of BECN1 and ATG5 expression in Y79 cells after transfection with indicated shRNA. (E and F) Y79 cells were transfected with BECN1 shRNA, ATG5 shRNA, or control shRNA for 72 h, and then treated with VCR (2.5 nM), ETO (1 µM), and CBP (1 µM) for 48 h. MAP1LC3 puncta formation (E) was assayed by confocal microscopic analysis and cell viability (F) was assayed by CCK-8 kit (*P < 0.05). (G) Y79 cells were transfected with mimic of MIR34A, control pUNO1 cDNA, or pUNO1-HMGB1 cDNA for 72 h, and then treated with carboplatin (CBP, 1 µM) in the presence and absence of E64D/pepstatin A (E/P, 10 μg/ml) for 48 h, and MAP1LC3 puncta formation was assayed by confocal microscopic analysis (*P < 0.05). (H) In parallel, cell viability was assayed by CCK-8 kit (n = 3, *P < 0.05).
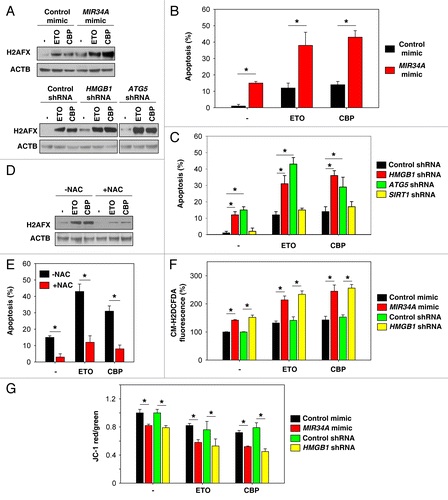
Inhibition of autophagy enhances DNA damage in chemotherapy
The anticancer activity of most chemotherapy drugs relies on the induction of DNA damage in rapidly cycling tumor cells concomitant with the inhibition of cellular DNA repair processes.Citation41 Exposure of Y79 cells to ETO and CBP resulted in the phosphorylation of histone H2A at Ser139 (gamma-H2AFX), a marker for DNA damage (). MIR34A mimic, knockdown of HMGB1, and knockdown of ATG5 increased chemotherapy drug-induced gamma-H2AFX expression, suggesting that inhibition of autophagy enhances DNA damage in chemotherapy (). Apoptosis induced by DNA damage is an important mechanism of tumor suppression, and is also significant in cancer chemotherapy. As expected, MIR34A mimic, knockdown of HMGB1, and knockdown of ATG5 increased chemotherapy-induced apoptosis (). However, knockdown of SIRT1 cannot increase chemotherapy-induced apoptosis (). Moreover, the antioxidant N-Acetyl-L-Cysteine (NAC) attenuated ETO- and CBP-induced DNA damage () and apoptosis () in ATG5 knockdown cells, suggesting that ROS-induced DNA damage is required for induction of apoptosis in autophagy-deficient cells. Mitochondrial membrane potential is an important parameter of mitochondrial function used as an indicator of cell health. There is a tight link between mitochondrial membrane potential and the rate of ROS formation. As expected, MIR34A mimic and knockdown of HMGB1 increased chemotherapy drug-induced ROS production () and decreased mitochondrial membrane potential (). Taken together, these data support the notion that miRNAs regulate autophagy, thereby largely influencing mitochondrial function as well as ROS production.
Figure 5. Inhibition of autophagy enhances DNA damage in chemotherapy. (A) Indicated Y79 cells were treated with etoposide (ETO, 1 µM) and carboplatin (CBP, 1 µM) for 48 h, and the level of gamma-H2AFX was assayed by western blot. (B and C) In parallel, apoptosis was analyzed by measuring ANXA5-positive cells (n = 3, *P < 0.05). (D) ATG5 knockdown Y79 cells were treated with ETO (1 µM) and CBP (1 µM) with or without antioxidant NAC (50 mM) for 48 h, and the level of gamma-H2AFX was assayed by western blot. (E) In parallel, apoptosis was analyzed by measuring ANXA5-positive cells (n = 3, *P < 0.05). (F and G) Indicated Y79 cells were treated with ETO (1 µM) and CBP (1 µM) for 48 h. ROS production (F) was assessed by measuring the fluorescent intensity of CM-H2DCFDA in a fluorescent plate reader, and mitochondrial membrane potential depolarization (G) was measured by JC-1 fluorescence (n = 3, *P < 0.05).
Discussion
Autophagy is a catabolic process involving the degradation of a cell's own components, such as waste or excess proteins and organelles through the lysosomal machinery.Citation42 It has been a fast-growing field of research in recent years, and is controlled by an ever-growing number of regulators, signalers, and miRNAs.Citation40,Citation43-Citation45 In this study, we identified that tumor suppressive miRNA, MIR34A, is a potent inhibitor of autophagy by downregulation of HMGB1, but not SIRT1 in the retinoblastoma cell. MIR34A was shown to target SIRT1 mRNA, leading to translational repression of SIRT1 in several human cancer cells. Recent evidence has demonstrated that activating SIRT1 induces autophagy, and loss of endogenous SIRT1 inhibits autophagy in several cell types. However, we found that the expression of SIRT1 in retinoblastoma cells is not required for antagoMIR34A and starvation-induced autophagy. Indeed, a recent study suggests that network organization of the human autophagy system is complex and each autophagic pathway has different components.Citation46
HMGB1, a conserved nuclear protein that enhances transcription, was recently discovered to be a crucial regulator of autophagy that mediates the stress response.Citation47 HMGB1 plays subcellular localization-dependent roles in the regulation of autophagy. Nuclear HMGB1 regulates small heat-shock protein HSPB1 expression.Citation28 Phosphorylation of HSPB1 is necessary for the regulation of the actin cytoskeleton, which affects the cellular transport required for autophagy and mitophagy in response to mitochondrial injury.Citation28 Cytosolic HMGB1 is a novel BECN1-binding protein that dissociates its inhibitory partner BCL2 to induce autophagy.Citation26 Moreover, ULK1 and TP53 positively and negatively regulate interaction between HMGB1 and BECN1 in cancer cells, respectively.Citation30 Extracellular HMGB1 binds AGER/RAGE (advanced glycosylation end product-specific receptor), but not TLR4, which inhibits MTOR and promotes the formation of BECN1-PIK3C3 core autophagic complexes in cancer cells. Induction of autophagy by both intracellular and extracellular HMGB1 is important for tumor development and a novel target for cancer therapy.Citation23,Citation30,Citation32,Citation48 Our findings suggest that MIR34A inhibits HMGB1 expression and reduces its protein levels, thereby preventing the activation of autophagy.
Current cancer therapies, including chemotherapy, radiotherapy, and immunotherapy, are known to induce a tight interplay between autophagy and apoptosis.Citation49 However, autophagy plays the dual role of either promoting cell survival or promoting cell death (“autophagic cell death”) in response to these cancer treatments, depending on the cellular context and the nature of the treatment.Citation25 Indeed, accumulating evidence challenges the concept of autophagic cell death in mammalian cells.Citation50 Inhibition of autophagy results in a shift from an autophagic morphology to nonautophagic cell death, including apoptosis and necrosis.Citation50 In many cases, autophagic cell death is cell death with autophagy, but not cell death by autophagy.Citation51 We demonstrated that MIR34A-mediated downregulation of HMGB1 and inhibition of autophagy can sensitize retinoblastoma cells to chemotherapy drug-mediated apoptosis. Of note, the molecular mechanism of drug-mediated autophagy in cancer cells is different. We showed that VCR blocks autolysosome formation, whereas ETO and CBP inhibit autophagosome formation. Therefore, mechanisms of the observed anticancer effects with MIR34A mimic and knockdown of HMGB1 could vary in different chemotherapies. These data support the notion that miRNAs regulate autophagy-related genes and autophagy regulators, thereby largely influencing the efficacy of chemotherapy.
The production of ROS by mitochondria is important because it underlies oxidative damage and cell death in many diseases.Citation52 Autophagy has been implicated in the removal of damaged mitochondria (termed mitophagy) and is an important mechanism for mitochondrial quality control.Citation53 Autophagy deficiency in cells and yeast leads to mitochondrial injury and ROS production.Citation54,Citation55 A previous study showed that HMGB1-mediated autophagy and mitophagy is important for mitochondrial quality control.Citation28 Knockout or knockdown of HMGB1 in fibroblasts and cancer cells significantly inhibits mitochondrial energy production and increases ROS production.Citation28 PTEN-induced putative kinase-1 (PINK1) is a kinase of the outer mitochondrial membrane and PARK2/PARKIN is a gene implicated in autosomal recessive juvenile Parkinsonism. The PINK1-PARK2 pathway has been implicated in mitophagy and mitochondrial dysfunction in humans,Citation56 and is required for HMGB1-mediated mitophagy and mitochondrial quality control.Citation28 We found that loss of the MIR34A-HMGB1 pathway inhibits autophagy and results in increased ROS production during chemotherapy in retinoblastoma cells. Excess ROS may induce oxidative DNA damage and chromosomal aberrations. Defects in DNA damage-induced apoptosis contribute to tumorigenesis and to the resistance of cancer cells to a variety of therapeutic agents. Thus, the level of MIR34A expression may affect the outcome of either autophagy or apoptosis.
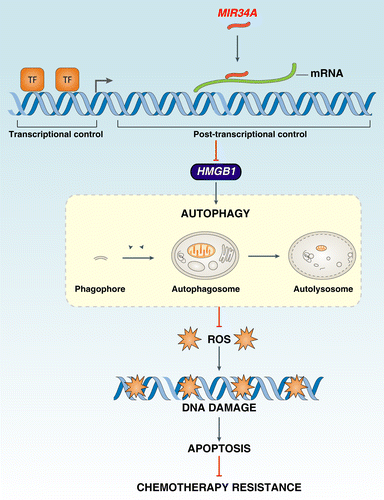
Taken together, this work suggests that HMGB1 may be one of several distinct targets of MIR34A that contribute to its ability to regulate cell survival and death by regulation of autophagy and apoptosis (). MIR34A inhibits HMGB1 expression through a MIR34A-binding site within the 3′UTR of HMGB1 in the retinoblastoma cell. Accumulating data suggest that HMGB1 is an important autophagy regulator during cell stress. MIR34A inhibition of HMGB1 leads to a significant decrease in autophagy. Chemotherapy is one of the main methods of cancer treatment and is known to induce autophagy in cancer cells. Our findings further suggest that MIR34A-HMGB1 pathway-mediated autophagy increases chemotherapy-induced apoptotic resistance by modification of the ROS-mediated DNA damage response. Thus, targeting the MIR34A-HMGB1 pathway in cancer research will provide new opportunities for drug development.
Figure 6.MIR34A regulates autophagy and apoptosis by targeting HMGB1. HMGB1 is a direct transcriptional target of MIR34A. MIR34A inhibition of HMGB1 leads to a decrease in autophagy that promotes oxidative injury, DNA damage, and subsequent apoptosis during chemotherapy.
Materials and Methods
Reagents
The antibodies to CASP3 (9665), cleaved-PARP1 (5625), and SIRT1 (9475) were obtained from Cell Signaling Technology. The antibodies to HMGB1 (ab18256) and ACTB/actin (ab3280) were obtained from Abcam. The antibody to MAP1LC3 (L8918) was obtained from Sigma. The antibody to SQSTM1 (sc-28359) came from Santa Cruz Biotechnology. Hoechst 33342 (H1399) was obtained from Invitrogen. All other anticancer and chemical reagents not listed were obtained from Sigma.
Cell culture
The human retinoblastoma cell lines Y79 (HTB-18), Weri-Rb1 (HTB-169), and HCT116 (CCL-247) were obtained from American Type Culture Collection and cultured in RPMI-1640 (11875-093) or McCoy’s 5a (Life Technologies, 16600-082) plus 10% heat-inactivated fetal bovine serum and 1% penicillin-streptomycin (Invitrogen, 15140) in a humidified incubator with 5% CO2 and 95% air. Hmgb1+/+ and hmgb1−/− immortalized MEFs were a kind gift from Dr Marco E Bianchi (San Raffaele Institute, Italy).
Transfection and RNAi
Short hairpin RNA (shRNA) against HMGB1 (SHCLNV-NM_002128), BECN1 (SHCLNV-NM_003766), ATG5 (SHCLNV-NM_004849), and SIRT1 (SHCLNV-NM_012238) were obtained from Sigma. The mimic and antagomir against MIR34A (C-301145-01; IH-301145-02) were obtained from Dharmacon. The pUNO1-HMGB1 plasmid (puno1-hhmgb1-s) was obtained from InvivoGen. HMGB1 3′UTR Lenti-reporter-Luciferase Vector (MT-h09960) was obtained from ABM. This HMGB1 3′UTR-containing vector (pLenti-UTR-Luc) was used as a template to create mutated MIR34A binding sites (HMGB1-UTRmt) by using the GeneArt® Site-Directed Mutagenesis PLUS System (Invitrogen, A14604) according to the manufacturer’s instructions. These shRNAs, miRNAs, and cDNA were transfected into cells by lentiviral delivery systems (Sigma, SHP001) or Lipofectamine 2000 reagent (Invitrogen, 11668) according to the manufacturer’s instructions.Citation57
Cell viability assay
Cells were plated at a density of 4 × 104 cells/well in 96-well plates in 100 μl medium. After treatment, cell viability was evaluated using the Cell Counting Kit-8 (Dojindo Laboratories, CK04-01) according to the manufacturer’s instructions.
Luciferase reporter assay
Cells were lysed in passive lysing buffer and then analyzed for firefly and Renilla luciferase activities using the commercial Dual-Luciferase reporter assay system (Promega, E1910) according to the manufacturer’s instructions; firefly luciferase activity was normalized to the Renilla luciferase activity.
Western blotting
Protein quantity from cell extracts was determined using the Bradford method. Protein was separated by electrophoresis in 4–12% Criterion XT Bis-Tris gels (Bio-Rad, 345-0124) and was transferred to nitrocellulose membrane, blocked with 5% milk in Tris-buffered saline with Tween 20, and probed with the indicated primary antibodies. After incubation with peroxidase-conjugated secondary antibodies, bands were visualized with enhanced chemiluminescence (Pierce, 34079). The relative band intensities were quantified using Gel-pro Analyzer® software (Media Cybernetics).
RNA isolation and qRT-PCR
Total RNA was extracted by using Trizol (Invitrogen, 15596-026) according to the manufacturer’s instructions and reverse transcribed for quantification using the TaqMan microRNA Reverse Transcription Kit (Applied Biosystems, 4366596) according to the manufacturer’s instructions. Mature miRNAs were quantified using 2-step TaqManqRT-PCR with the TaqMan microRNA kit. miRNA expression level was normalized using RNU66rRNA as an internal control as previously described.Citation58 qRT-PCR for HMGB1 with specific primers (PPH00999F) from SABiosciences was performed using a RTCitation2 First Strand Kit (SABiosciences, 330401) according to the manufacturer’s instructions. Fold changes of mRNA levels in target gene relative to the POLR2A/RNA polymerase II [polymerase (RNA) II (DNA directed) polypeptide A, 220 kDa] control were calculated by relative quantification analysis.
Autophagy assays
The number of MAP1LC3 puncta was determined by quantifying the number of positively stained cells from 100 randomly chosen cells by Image-Pro Plus 5.1 software (Media Cybernetics) as previously described.Citation26 Autophagic flux assays were performed by western blotting for MAP1LC3-I/II and SQSTM1 in the presence or absence of lysosomal protease inhibitors or by long-lived protein degradation assays as previously described.Citation57,Citation59 Transmission electron microscopy assessment of autophagic vacuoles was performed as previously described.Citation28
Apoptosis assays
Apoptosis in cells was assessed using the ANXA5-FITC Apoptosis Detection Kit (BD PharMingen, 556570) by flow cytometric analysis. CASP3 activity assays were performed using the CASP3 Colorimetric Assay Kit from Calbiochem (235419-1KIT) according to the manufacturer’s instructions. Cleaved PARP1 and cleaved CASP3 were measured by western blot analysis. Mitochondrial membrane potential depolarization was measured by flow cytometry using the fluorescent cationic dye JC-1, (5,5′,6,6′-tetrachloro-1,1′3,3′-tetraethylbenzamidazolocarbocyanin iodide; Molecular Probes, M34152). JC-1 dye exhibits potential-dependent accumulation in mitochondria, indicated by a fluorescence emission shift from green to red. Consequently, mitochondrial depolarization was assessed by a decrease in the red/green fluorescence intensity ratio.
Measurement of intracellular ROS
Cells were seeded in 96-well plates and cultured in the presence of a given stimuli for the indicated time. ROS were assessed with cell-permeable dye CM-H2DCFDA (Invitrogen, C6827) as described previously.Citation26 Analysis of signal intensity was performed using a fluorescent plate reader.
Statistical analysis
One-way ANOVA was used for comparison among the different groups. When the ANOVA was significant, post hoc testing of differences between groups was performed using an LSD test. A P value < 0.05 was considered significant.
| Abbreviations: | ||
| AGER | = | advanced glycosylation end product-specific receptor |
| CBP | = | carboplatin |
| E/P | = | E64d and pepstatin A |
| ETO | = | etoposide |
| HBSS | = | Hank’s balanced salt solution |
| HMGB1 | = | high mobility group box 1 |
| H2AFX | = | H2A histone family, member X |
| gamma-H2AFX | = | phosphorylation of histone H2AFX at Ser139 |
| MAP1LC3 | = | microtubule-associated protein 1 light chain 3 |
| MEFs | = | mouse embryonic fibroblasts |
| miRNAs | = | microRNAs |
| MIR34A | = | microRNA 34a |
| mRNAs | = | messenger RNAs |
| NAC | = | N-acetyl-L-cysteine |
| PARP1 | = | poly (ADP-ribose) polymerase 1 |
| PINK1 | = | PTEN-induced putative kinase 1 |
| ROS | = | reactive oxygen species |
| SQSTM1 | = | sequestosome 1 |
| shRNA | = | short hairpin RNA |
| UTR | = | untranslated region |
| VCR | = | vincristine |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Christine Heiner (Departments of Surgery and Anesthesiology, University of Pittsburgh) for her critical reading of the manuscript. This work was supported by grants from The National Natural Sciences Foundation of China (30973234 and 31171328 to LC; 81270616 to YY) and a grant from the US. National Institutes of Health (NIH) (R01CA160417 to DT).
References
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004; 116:281 - 97; http://dx.doi.org/10.1016/S0092-8674(04)00045-5; PMID: 14744438
- Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer 2006; 6:857 - 66; http://dx.doi.org/10.1038/nrc1997; PMID: 17060945
- Garzon R, Calin GA, Croce CM. MicroRNAs in Cancer. Annu Rev Med 2009; 60:167 - 79; http://dx.doi.org/10.1146/annurev.med.59.053006.104707; PMID: 19630570
- Dimaras H, Kimani K, Dimba EA, Gronsdahl P, White A, Chan HS, Gallie BL. Retinoblastoma. Lancet 2012; 379:1436 - 46; http://dx.doi.org/10.1016/S0140-6736(11)61137-9; PMID: 22414599
- Jo DH, Kim JH, Park WY, Kim KW, Yu YS, Kim JH. Differential profiles of microRNAs in retinoblastoma cell lines of different proliferation and adherence patterns. J Pediatr Hematol Oncol 2011; 33:529 - 33; http://dx.doi.org/10.1097/MPH.0b013e318228280a; PMID: 21941147
- Dalgard CL, Gonzalez M, deNiro JE, O’Brien JM. Differential microRNA-34a expression and tumor suppressor function in retinoblastoma cells. Invest Ophthalmol Vis Sci 2009; 50:4542 - 51; http://dx.doi.org/10.1167/iovs.09-3520; PMID: 19443717
- He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, Xue W, Zender L, Magnus J, Ridzon D, et al. A microRNA component of the p53 tumour suppressor network. Nature 2007; 447:1130 - 4; http://dx.doi.org/10.1038/nature05939; PMID: 17554337
- Bommer GT, Gerin I, Feng Y, Kaczorowski AJ, Kuick R, Love RE, Zhai Y, Giordano TJ, Qin ZS, Moore BB, et al. p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr Biol 2007; 17:1298 - 307; http://dx.doi.org/10.1016/j.cub.2007.06.068; PMID: 17656095
- Hermeking H. The miR-34 family in cancer and apoptosis. Cell Death Differ 2010; 17:193 - 9; http://dx.doi.org/10.1038/cdd.2009.56; PMID: 19461653
- Yin D, Ogawa S, Kawamata N, Leiter A, Ham M, Li D, Doan NB, Said JW, Black KL, Phillip Koeffler H. miR-34a functions as a tumor suppressor modulating EGFR in glioblastoma multiforme. Oncogene 2013; 32:1155 - 63; http://dx.doi.org/10.1038/onc.2012.132; PMID: 22580610
- Mudduluru G, Ceppi P, Kumarswamy R, Scagliotti GV, Papotti M, Allgayer H. Regulation of Axl receptor tyrosine kinase expression by miR-34a and miR-199a/b in solid cancer. Oncogene 2011; 30:2888 - 99; http://dx.doi.org/10.1038/onc.2011.13; PMID: 21317930
- Sotillo E, Laver T, Mellert H, Schelter JM, Cleary MA, McMahon S, Thomas-Tikhonenko A. Myc overexpression brings out unexpected antiapoptotic effects of miR-34a. Oncogene 2011; 30:2587 - 94; http://dx.doi.org/10.1038/onc.2010.634; PMID: 21297663
- Wei JS, Song YK, Durinck S, Chen QR, Cheuk AT, Tsang P, Zhang Q, Thiele CJ, Slack A, Shohet J, et al. The MYCN oncogene is a direct target of miR-34a. Oncogene 2008; 27:5204 - 13; http://dx.doi.org/10.1038/onc.2008.154; PMID: 18504438
- Raver-Shapira N, Marciano E, Meiri E, Spector Y, Rosenfeld N, Moskovits N, Bentwich Z, Oren M. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell 2007; 26:731 - 43; http://dx.doi.org/10.1016/j.molcel.2007.05.017; PMID: 17540598
- Chang TC, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH, Feldmann G, Yamakuchi M, Ferlito M, Lowenstein CJ, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell 2007; 26:745 - 52; http://dx.doi.org/10.1016/j.molcel.2007.05.010; PMID: 17540599
- Tarasov V, Jung P, Verdoodt B, Lodygin D, Epanchintsev A, Menssen A, Meister G, Hermeking H. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle 2007; 6:1586 - 93; http://dx.doi.org/10.4161/cc.6.13.4436; PMID: 17554199
- Yamakuchi M, Ferlito M, Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci U S A 2008; 105:13421 - 6; http://dx.doi.org/10.1073/pnas.0801613105; PMID: 18755897
- Liu C, Kelnar K, Liu B, Chen X, Calhoun-Davis T, Li H, Patrawala L, Yan H, Jeter C, Honorio S, et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat Med 2011; 17:211 - 5; http://dx.doi.org/10.1038/nm.2284; PMID: 21240262
- Genovese G, Ergun A, Shukla SA, Campos B, Hanna J, Ghosh P, Quayle SN, Rai K, Colla S, Ying H, et al. microRNA regulatory network inference identifies miR-34a as a novel regulator of TGF-β signaling in glioblastoma. Cancer Discov 2012; 2:736 - 49; http://dx.doi.org/10.1158/2159-8290.CD-12-0111; PMID: 22750848
- Li Y, Guessous F, Zhang Y, Dipierro C, Kefas B, Johnson E, Marcinkiewicz L, Jiang J, Yang Y, Schmittgen TD, et al. MicroRNA-34a inhibits glioblastoma growth by targeting multiple oncogenes. Cancer Res 2009; 69:7569 - 76; http://dx.doi.org/10.1158/0008-5472.CAN-09-0529; PMID: 19773441
- Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol 2005; 5:331 - 42; http://dx.doi.org/10.1038/nri1594; PMID: 15803152
- Harris HE, Andersson U, Pisetsky DS. HMGB1: a multifunctional alarmin driving autoimmune and inflammatory disease. Nat Rev Rheumatol 2012; 8:195 - 202; http://dx.doi.org/10.1038/nrrheum.2011.222; PMID: 22293756
- Tang D, Kang R, Zeh HJ 3rd, Lotze MT. High-mobility group box 1 and cancer. Biochim Biophys Acta 2010; 1799:131 - 40; http://dx.doi.org/10.1016/j.bbagrm.2009.11.014; PMID: 20123075
- Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol 2010; 28:367 - 88; http://dx.doi.org/10.1146/annurev.immunol.021908.132603; PMID: 20192808
- White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer 2012; 12:401 - 10; http://dx.doi.org/10.1038/nrc3262; PMID: 22534666
- Tang D, Kang R, Livesey KM, Cheh CW, Farkas A, Loughran P, Hoppe G, Bianchi ME, Tracey KJ, Zeh HJ 3rd, et al. Endogenous HMGB1 regulates autophagy. J Cell Biol 2010; 190:881 - 92; http://dx.doi.org/10.1083/jcb.200911078; PMID: 20819940
- Tang D, Kang R, Cheh CW, Livesey KM, Liang X, Schapiro NE, Benschop R, Sparvero LJ, Amoscato AA, Tracey KJ, et al. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene 2010; 29:5299 - 310; http://dx.doi.org/10.1038/onc.2010.261; PMID: 20622903
- Tang D, Kang R, Livesey KM, Kroemer G, Billiar TR, Van Houten B, Zeh HJ 3rd, Lotze MT. High-mobility group box 1 is essential for mitochondrial quality control. Cell Metab 2011; 13:701 - 11; http://dx.doi.org/10.1016/j.cmet.2011.04.008; PMID: 21641551
- Livesey KM, Kang R, Vernon P, Buchser W, Loughran P, Watkins SC, Zhang L, Manfredi JJ, Zeh HJ 3rd, Li L, et al. p53/HMGB1 complexes regulate autophagy and apoptosis. Cancer Res 2012; 72:1996 - 2005; http://dx.doi.org/10.1158/0008-5472.CAN-11-2291; PMID: 22345153
- Huang J, Ni J, Liu K, Yu Y, Xie M, Kang R, Vernon P, Cao L, Tang D. HMGB1 promotes drug resistance in osteosarcoma. Cancer Res 2012; 72:230 - 8; http://dx.doi.org/10.1158/0008-5472.CAN-11-2001; PMID: 22102692
- Liu L, Yang M, Kang R, Wang Z, Zhao Y, Yu Y, Xie M, Yin X, Livesey KM, Loze MT, et al. DAMP-mediated autophagy contributes to drug resistance. Autophagy 2011; 7:112 - 4; http://dx.doi.org/10.4161/auto.7.1.14005; PMID: 21068541
- Liu L, Yang M, Kang R, Wang Z, Zhao Y, Yu Y, Xie M, Yin X, Livesey KM, Lotze MT, et al. HMGB1-induced autophagy promotes chemotherapy resistance in leukemia cells. Leukemia 2011; 25:23 - 31; http://dx.doi.org/10.1038/leu.2010.225; PMID: 20927132
- Yang L, Yu Y, Kang R, Yang M, Xie M, Wang Z, Tang D, Zhao M, Liu L, Zhang H, et al. Up-regulated autophagy by endogenous high mobility group box-1 promotes chemoresistance in leukemia cells. Leuk Lymphoma 2012; 53:315 - 22; http://dx.doi.org/10.3109/10428194.2011.616962; PMID: 21864037
- Kang R, Zhang Q, Zeh HJ 3rd, Lotze MT, Tang D. HMGB1 in cancer: good, bad, or both?. Clin Cancer Res 2013; 19:4046 - 57; http://dx.doi.org/10.1158/1078-0432.CCR-13-0495; PMID: 23723299
- Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, Tsokos M, Alt FW, Finkel T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A 2008; 105:3374 - 9; http://dx.doi.org/10.1073/pnas.0712145105; PMID: 18296641
- Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature 1995; 376:37 - 43; http://dx.doi.org/10.1038/376037a0; PMID: 7596430
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19:5720 - 8; http://dx.doi.org/10.1093/emboj/19.21.5720; PMID: 11060023
- Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005; 171:603 - 14; http://dx.doi.org/10.1083/jcb.200507002; PMID: 16286508
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445 - 544; http://dx.doi.org/10.4161/auto.19496; PMID: 22966490
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009; 43:67 - 93; http://dx.doi.org/10.1146/annurev-genet-102808-114910; PMID: 19653858
- Bouwman P, Jonkers J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat Rev Cancer 2012; 12:587 - 98; http://dx.doi.org/10.1038/nrc3342; PMID: 22918414
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069 - 75; http://dx.doi.org/10.1038/nature06639; PMID: 18305538
- Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell 2010; 40:280 - 93; http://dx.doi.org/10.1016/j.molcel.2010.09.023; PMID: 20965422
- Frankel LB, Lund AH. MicroRNA regulation of autophagy. Carcinogenesis 2012; 33:2018 - 25; http://dx.doi.org/10.1093/carcin/bgs266; PMID: 22902544
- Xu J, Wang Y, Tan X, Jing H. MicroRNAs in autophagy and their emerging roles in crosstalk with apoptosis. Autophagy 2012; 8:873 - 82; http://dx.doi.org/10.4161/auto.19629; PMID: 22441107
- Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature 2010; 466:68 - 76; http://dx.doi.org/10.1038/nature09204; PMID: 20562859
- Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev 2012; 249:158 - 75; http://dx.doi.org/10.1111/j.1600-065X.2012.01146.x; PMID: 22889221
- Amaravadi RK, Lippincott-Schwartz J, Yin XM, Weiss WA, Takebe N, Timmer W, DiPaola RS, Lotze MT, White E. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res 2011; 17:654 - 66; http://dx.doi.org/10.1158/1078-0432.CCR-10-2634; PMID: 21325294
- Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 2007; 8:741 - 52; http://dx.doi.org/10.1038/nrm2239; PMID: 17717517
- Shen S, Kepp O, Michaud M, Martins I, Minoux H, Métivier D, Maiuri MC, Kroemer RT, Kroemer G. Association and dissociation of autophagy, apoptosis and necrosis by systematic chemical study. Oncogene 2011; 30:4544 - 56; http://dx.doi.org/10.1038/onc.2011.168; PMID: 21577201
- Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol 2008; 9:1004 - 10; http://dx.doi.org/10.1038/nrm2529; PMID: 18971948
- Murphy MP. How mitochondria produce reactive oxygen species. Biochem J 2009; 417:1 - 13; http://dx.doi.org/10.1042/BJ20081386; PMID: 19061483
- Ashrafi G, Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ 2013; 20:31 - 42; http://dx.doi.org/10.1038/cdd.2012.81; PMID: 22743996
- Kurihara Y, Kanki T, Aoki Y, Hirota Y, Saigusa T, Uchiumi T, Kang D. Mitophagy plays an essential role in reducing mitochondrial production of reactive oxygen species and mutation of mitochondrial DNA by maintaining mitochondrial quantity and quality in yeast. J Biol Chem 2012; 287:3265 - 72; http://dx.doi.org/10.1074/jbc.M111.280156; PMID: 22157017
- Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011; 469:221 - 5; http://dx.doi.org/10.1038/nature09663; PMID: 21124315
- Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 2010; 12:119 - 31; http://dx.doi.org/10.1038/ncb2012; PMID: 20098416
- Yu Y, Yang L, Zhao M, Zhu S, Kang R, Vernon P, Tang D, Cao L. Targeting microRNA-30a-mediated autophagy enhances imatinib activity against human chronic myeloid leukemia cells. Leukemia 2012; 26:1752 - 60; http://dx.doi.org/10.1038/leu.2012.65; PMID: 22395361
- Peltier HJ, Latham GJ. Normalization of microRNA expression levels in quantitative RT-PCR assays: identification of suitable reference RNA targets in normal and cancerous human solid tissues. RNA 2008; 14:844 - 52; http://dx.doi.org/10.1261/rna.939908; PMID: 18375788
- Bauvy C, Meijer AJ, Codogno P. Assaying of autophagic protein degradation. Methods Enzymol 2009; 452:47 - 61; PMID: 19200875