
Abstract
Oxidative stress has been implicated in both normal aging and various neurodegenerative disorders and it may be a major cause of neuronal death. Chaperone-mediated autophagy (CMA) targets selective cytoplasmic proteins for degradation by lysosomes and protects neurons against various extracellular stimuli including oxidative stress. MEF2A (myocyte enhancer factor 2A), a key transcription factor, protects primary neurons from oxidative stress-induced cell damage. However, the precise mechanisms of how the protein stability and the transcriptional activity of MEF2A are regulated under oxidative stress remain unknown. In this study, we report that MEF2A is physiologically degraded through the CMA pathway. In pathological conditions, mild oxidative stress (200 μM H2O2) enhances the degradation of MEF2A as well as its activity, whereas excessive oxidative stress (> 400 μM H2O2) disrupts its degradation process and leads to the accumulation of nonfunctional MEF2A. Under excessive oxidative stress, an N-terminal HDAC4 (histone deacetylase 4) cleavage product (HDAC4-NT), is significantly induced by lysosomal serine proteases released from ruptured lysosomes in a PRKACA (protein kinase, cAMP-dependent, catalytic, α)-independent manner. The production of HDAC4-NT, as a MEF2 repressor, may account for the reduced DNA-binding and transcriptional activity of MEF2A. Our work provides reliable evidence for the first time that MEF2A is targeted to lysosomes for CMA degradation; oxidative stress-induced lysosome destabilization leads to the disruption of MEF2A degradation as well as the dysregulation of its function. These findings may shed light on the underlying mechanisms of pathogenic processes of neuronal damage in various neurodegenerative-related diseases.
Introduction
The 4 MEF2 proteins, MEF2A to MEF2D, are members of the MADS (MCM1, Agamous, Deficiens and SRF) family of transcription factors.Citation1 The MEF2 isoforms share a highly homologous N-terminal region (over 95% similarity), including an N-terminal 56-amino-acid MADS domain and an adjacent 29-amino-acid MEF2 domain, which together mediate DNA binding and dimerization.Citation2 The sequences outside these 2 domains contain a divergent large C-terminal transcriptional activation domain, as well as other regulatory minidomains, including a nuclear localization sequence and multiple phosphorylation motifs.Citation3 The structural organization of MEF2 isoforms allows them to receive and respond to multiple signals from the extracellular environment.
MEF2 isoforms have initially been identified in cells of muscle lineage and verified to be important regulators of gene expression during the development of skeletal, cardiac, and smooth muscle.Citation4 In these tissues, MEF2 interact with myogenic basic helix-loop-helix transcription factors such as MYOD1 (myogenic differentiation 1) to activate myogenesis.Citation5 However, MEF2 transcripts and proteins are not restricted to muscle but are expressed at different levels in nonmuscle tissues, particularly in brain.Citation6 All MEF2 isoforms are expressed at variable levels in distinct, but overlapping regions of brain during embryogenesis, as well as in adults.Citation7 MEF2 proteins play an important role in the regulation of neuronal proliferation, differentiation, survival, and synapse development.Citation8 Many neurotoxins, such as excitotoxic insults, 1-methyl-4-phenyl-1, 2, 5, 6-tetrahydropyridine (MPTP), 6-hydroxydopamine, β-amyloid peptide (Aβ), 3-nitropropionic acid, and oxidative stress could regulate the transcriptional activity of MEF2.Citation9 Inhibition of MEF2 by stress and toxic signals contributes to neuronal death, while enhancing MEF2 activity rescues cultured primary neurons from death in vitro.Citation10 Therefore, dysregulation of MEF2 activity has been linked to many neurodegenerative diseases, including Alzheimer and Parkinson diseases.Citation11,Citation12
In response to diverse physiological or pathological stimuli, the regulation of MEF2 function is quite complex. Through MEF2 phosphorylation,Citation13 acetylation,Citation14 sumoylation,Citation15 or interaction with other cofactors such as HDAC4,Citation16 many key steps of MEF2 regulation can be tightly modulated, including MEF2 mRNA alternative splicing, translation, transactivation domain activity, DNA binding, subcellular localization, and protein stability.
Among the above-mentioned steps, the regulation of MEF2 protein stability is particularly important to neuronal cell survival. It is well known that the rate of protein synthesis vs. degradation controls protein stability. Two major pathways accomplish protein and organelle clearance: the ubiquitin-proteasome system degrades specific short-lived proteins, whereas the lysosomal (autophagy) pathway is involved in the bulk degradation of long-lived cytosolic proteins and organelles.Citation17 Autophagy takes place in mammalian cells mainly through 3 different mechanisms, namely macroautophagy, microautophagy, and chaperone-mediated autophagy.Citation18 In 2 of these mechanisms, macroautophagy and microautophagy, the substrates are engulfed or sequestered in bulk, whereas in CMA, the substrates are selectively transported across the lysosomal membrane on a one-by-one basis.Citation19 During CMA, protein substrates containing peptide regions similar to Lys-Phe-Glu-Arg-Gln (KFERQ) are targeted to lysosomes through the interaction with a cytosolic chaperone, HSPA8/HSC70. The targeted substrate-chaperone complex docks at lysosomes through interaction with the cytosolic tail of LAMP2A (lysosomal-associated membrane protein 2A). After docking, the substrate protein unfolds and crosses the lysosomal membrane through a multimeric translocation complex with the coordinated action of chaperones located at both sides of the membrane. After translocation, substrate proteins are rapidly degraded to single amino acids by an abundant array of lysosomal hydrolases. These amino acids are recycled for synthesis of new proteins or serve as an energy source.
According to the criterion that putative CMA substrates have a KFERQ-like motif in their sequences,Citation20 it was estimated that 30% of cytosolic proteins are candidates for CMA.Citation21 However, only about 25 proteins have been classified as bona fide CMA substrates thus far and more proteins are pending further validation.Citation22 Recently, it was found that the degradation of MEF2D, 1 of the 4 isoforms of MEF2, was mediated by CMA under basal conditions. Disruption of this process by both wild-type or mutant SNCA/α-synuclein leads to the accumulation of nonfunctional MEF2D, and it may underlie the pathogenic process in Parkinson disease.Citation23 As stated above, although the C-terminal amino acid sequences of the 4 MEF2 isoforms differ considerably, they share a highly homologous N-terminal region, which contains the motifs required for lysosome targeting. This raises the interesting possibility that other MEF2 isoforms may also be regulated by CMA.
Accumulating evidence indicates that oxidative stress which disturbs the autophagy-lysosomal degradation pathway is a major cause of cellular injuries in a variety of human diseases including neurodegenerative disorders. MEF2A and its isoforms play an important role in the survival of several types of neurons. However, the precise mechanisms of how the protein stability and the transcriptional activity of MEF2A are regulated in cells, especially under oxidative stress, remain unknown. It is likely that the elucidation of the exact mechanisms by which MEF2A and other components in its physiological or pathological pathway are regulated could facilitate our understanding of neurodegeneration. In the present study, we show that MEF2A is a novel substrate for CMA, and although mild oxidative stress has been shown to induce CMA,Citation24 intense oxidative stress conditions that induce lysosomal destabilization lead to the CMA disruption and accumulation of nonfunctional MEF2A.
Results
MEF2A degradation is mediated through the autophagy-lysosome but not the ubiquitin-proteasome pathway
The ubiquitin-proteasome and autophagy-lysosome pathways are the 2 main routes of protein degradation in eukaryotes. To investigate whether MEF2A is degraded by the ubiquitin-proteasome pathway, HEK 293T cells were exposed to a specific proteasome inhibitor MG132 at different dosages. As shown in , inhibition of proteasome by 0.5 or 1 μM of MG132 for 24 h had no apparent effect on MEF2A protein levels. Unexpectedly, 2 μM of MG132 not only failed to induce accumulation of the MEF2A protein, but rather resulted in a slight reduction of its protein level. This reduction was even more pronounced with higher dosages of MG132 at 5 to 15 μM (Fig. S1A). To further confirm it, we tested the effects of MG132 in 2 human neuroblastoma cell lines. MG132 at 50 μM also decreased MEF2A level in SH-SY5Y cells (Fig. S1B), although it had no effect in SK-N-SH cells (). HIF1A/HIF-1α, a well-known substrate for the ubiquitin-proteasome degradation pathway, was used as a positive control. HIF1A protein levels were significantly increased in a dose-dependent manner with 0.5 to 2 μM of MG132 treatment, indicating that MG132 was functional in these cells at the determined dosages (). To further verify the effect of proteasome pathway inhibition on MEF2A protein level, HEK 293T cells were treated with another proteasome inhibitor, ALLN (N-acetyl-Leu-Leu-Nle-CHO). The addition of ALLN at 10 to 50 μM also showed no apparent effect on MEF2A protein levels. Addition of another proteasome inhibitor, lactacystin at 2.5 or 5 μM, showed no significant effect on MEF2A protein levels as well, and even at 10 μM it resulted in only a slight reduction (). To further assess whether lactacystin also affected MEF2A transcription, semiquantitative or quantitative RT-PCR analysis was performed. Lactacystin treatment resulted in no significant change in MEF2A mRNA levels (Fig. S1C). Meanwhile, the cytotoxicity induced by lactacystin treatment was assessed by the MTT assay. As shown in Figure S1D, lactacystin at 2.5 μM did not cause any apparent cytotoxicity, while when the dosage of lactacystin was higher than 5 μM, it decreased cell viability to around 80% in 24 h. Although a slight decrease in cell viability was observed upon 5 μM of lactacystin treatment, it had no significant effect on the protein level of MEF2A. These findings suggest that the ubiquitin-proteasome pathway seems not to be responsible for promoting MEF2A degradation in the cell types we examined.
Figure 1. MEF2A is degraded via the autophagy-lysosome but not the ubiquitin-proteasome pathway. (A–C) MEF2A is not degraded by the proteasome pathway. HEK 293T (A), SH-N-SH (B) cells were treated with proteasome inhibitor MG132 at different dosages for 24 h and the MEF2A protein levels were analyzed by western blotting. HIF1A, a well-known proteasome substrate, was included as a control in (A). (C) HEK 293T cells were treated with the proteasome inhibitor ALLN or lactacystin at different dosages for 24 h and the MEF2A protein levels were analyzed by western blotting. (D) Baf A1 induces accumulation of MEF2A protein. SK-N-SH cells were treated with or without Baf A1 for 24 h. The levels of MEF2A were determined by western blot. MEF2A levels were normalized against ACTB. (E and F) Lysosome inhibitors cause MEF2A accumulation. SK-N-SH cells were treated with different dosages of NH4Cl (E) or chloroquine (F) for 24 h. The levels of MEF2A were determined by immunoblotting. Bottom graph shows relative MEF2A levels that were normalized to ACTB (n = 3). *P < 0.05, **P < 0.01. (G) SK-N-SH cells were treated with Baf A1 (25 nM), NH4Cl (50 mM) and chloroquine (50 μM) for different times (upper). The relative protein levels of MEF2A were quantified by the band density scanning of western blot images and plotted against time (h) after treatment with different lysosome inhibitors (lower). *P < 0.05, +P < 0.05, #P < 0.05, ##P < 0.01 vs. 0 h. Ctrl, control; SC, solvent control.
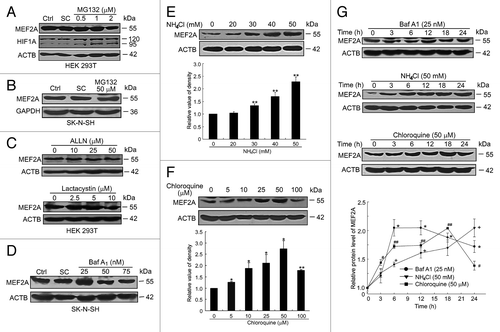
We next examined the role of the lysosome pathway in regulating MEF2A levels using different lysosome inhibitors. Twenty-five nanometers of bafilomycin A1 (Baf A1), a specific inhibitor of vacuolar-type proton ATPases that inhibits acidification and protein degradation in lysosomes, resulted in the accumulation of MEF2A, in neuronal cells, although higher doses of Baf A1 (up to 50 or 75 nM) had no evident effect on the levels of MEF2A (). Ammonium chloride (NH4Cl), another blocker of lysosomal proteolysis, dose-dependently increased MEF2A levels. (). Another lysosome inhibitor, chloroquine, at 5 to 50 μM also caused a dose-dependent increase in the levels of MEF2A in SK-N-SH cells (). MEF2A accumulation was also time dependent as shown in , in which cells were treated with the above lysosome inhibitors at the indicated concentrations (Baf A1 25 nM, NH4Cl 50 mM, chloroquine 50 μM for up to 24 h). MFE2A level in Baf A1 treated cells was steadily increased by 12 h but was reduced afterwards, while in chloroquine-treated cells it was increased to the maximum level at 18 h and then declined by 24 h. Importantly, at the 24 h time point, the protein levels in these 2 groups were still higher than the basal levels. In the NH4Cl-treated group, MEF2A levels kept on increasing within 24 h.
Taken together, these results suggest that the degradation of MEF2A is dependent on lysosomal, but not on proteasomal mechanisms.
Macroautophagy is not involved in MEF2A degradation
Three types of autophagy have been identified in mammals, macroautophagy, microautophagy, and CMA. Since macroautophagy was considered the major type of autophagy, we first examined whether MEF2A degradation in lysosomes was mediated by the macroautophagy pathway. The class III phosphatidylinositol 3-kinase (PtdIns3K) plays an important role in the regulation of macroautophagy, and 3-methyladenine (3-MA), a well-characterized class III PtdIns3K inhibitor, is an inhibitor of macroautophagy through blocking autophagosome formation. We treated SK-N-SH cells with 10 mM of 3-MA for 12 or 24 h and observed no effect of 3-MA on the steady-state levels of MEF2A (). In addition, wortmannin (at 100 nM) and LY294002 (at 10 μM), 2 inhibitors of both class I phosphoinositide 3-kinase and class III PtdIns3K that inhibit macroautophagic sequestration and protein degradation, also had no appreciable effects on the protein levels of MEF2A at various time points (). These results confirm that inhibition of macroautophagy does not alter MEF2A protein levels.
Figure 2. Macroautophagy is not involved in MEF2A degradation. (A–D) SK-N-SH cells were treated with different macroautophagy inhibitors, 10 mM 3-MA (A), 100 nM wortmannin (B) and 10 μM LY294002 (C) or a stimulator, 100 nM rapamycin (D) for 12 or 24 h. The levels of MEF2A were determined by immunoblotting. Bottom graph shows relative MEF2A levels (n = 3). (E) Induction of autophagy in rapamycin-treated SK-N-SH cells. Cells were treated with 100 nM of rapamycin and different autophagy inhibitors (3-MA at 10 mM, wortmannin at 100 nM and LY294002 at 10 μM) for 24 h. Total cell lysates were subjected to western blot using antibody against LC3B. ACTB was used as a loading control. (F) Effect of macroautophagy inhibitor (100 nM of wortmannin) or stimulator (100 nM of rapamycin) on MEF2A level of HEK 293T cells. Results shown are representative of 3 independent experiments. Bottom graph shows relative MEF2A levels (n = 3). WM, wortmannin; LY, LY294002; Rapa, rapamycin.
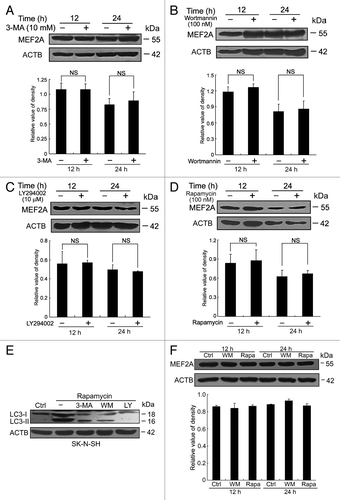
Moreover, MTOR is a kinase downstream of class I phosphoinositide 3-kinase whose activation suppresses autophagy. Rapamycin, which inhibits MTOR, activates macroautophagy in both yeast and mammalian cells. Treatment of SK-N-SH cells with rapamycin (100 nM) did not change MEF2A protein levels (), suggesting that macroautophagy activation has no significant effect on the protein levels of MEF2A.
In order to ensure the above-used macroautophagy activator or inhibitors were active in the cell, we examined the conversion of soluble LC3-I to lipid conjugated LC3-II, which is a well-established marker for macroautophagy, by western blot analysis under different conditions.Citation25 We found that the LC3-II level was negligible in the control cells in the absence of drugs, but its production was augmented by the treatment with rapamycin, the macroautophagy activator. Moreover, autophagy inhibitors (3-MA, wortmannin or LY294002) greatly inhibited the rapamycin-induced production of LC3-II (). Thus, rapamycin, 3-MA, wortmannin, and LY294002 were all functional in these cells at the determined concentrations. These findings were further validated using nonneuronal HEK 293T cells (), supporting the conclusion that the protein levels of MEF2A are barely affected by macroautophagy.
Serum deprivation accelerates degradation of MEF2A
We then sought to determine whether CMA, another form of autophagy, was involved in regulating MEF2A protein levels. Given that prolonged serum deprivation can activate CMA,Citation26 we assessed the effect of serum deprivation on the steady-state levels of MEF2A. As shown in , although one d of nutrient deprivation had no apparent effect on MEF2A levels in SK-N-SH cells, 3 or more d of serum starvation markedly decreased its protein levels. Similar results were obtained in the nonneuronal HEK 293T cells. Notably, the levels of MEF2A transcript were significantly induced but not reduced upon such treatments examined by semiquantitative or quantitative RT-PCR analysis (). These observations suggest that serum starvation-induced downregulation of MEF2A protein is most likely mediated through posttranscriptional mechanisms, which override its transcriptional upregulation.
Figure 3. The effects of serum deprivation on MEF2A transcript and protein. (A) Serum withdrawal decreases MEF2A protein level. SK-N-SH and HEK 293T cells were treated with serum deprivation for the indicated times. The levels of MEF2A were determined by immunoblotting. (B) Semiquantitative and quantitative RT-PCR analyses of MEF2A mRNA. Total RNA isolated from SK-N-SH cells following serum deprivation was analyzed by semiquantitative (top: MEF2A and ACTB transcripts) and quantitative (bottom: levels of MEF2A transcript relative to ACTB transcript) RT-PCR. *P < 0.05. (C) Degradation rates of MEF2A were analyzed in SK-N-SH cells treated with 40 μg/ml of CHX in the presence or absence of serum for the indicated periods of time (top). The levels of MEF2A were normalized to that of ACTB expression, and the results showed the relative quantification as determined by optical densitometry (bottom). *P < 0.05, **P < 0.01.
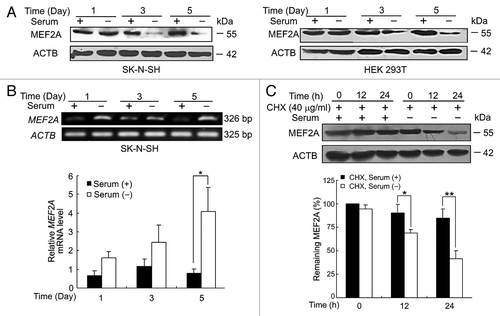
To determine whether this downregulation is due to protein turnover, SK-N-SH cells were treated with cycloheximide (CHX), an inhibitor of protein synthesis. Although the levels of MEF2A were not changed appreciably over 24 h in the presence of CHX at 40 μg/ml alone (Fig. S2), a slight reduction of MEF2A was detected at 12 h under the serum-deprivation condition in the presence of CHX. By 24 h, only < 42% of MEF2A protein remained. In contrast, MEF2A protein was degraded more slowly in the presence of serum, with over 85% of MEF2A protein remaining by 24 h (). These findings suggest that nutrition deprivation, which presumably activates the CMA pathway, accelerates MEF2A degradation.
Contribution of CMA to the degradation of exogenous overexpressed MEF2A in Neuro-2A cells
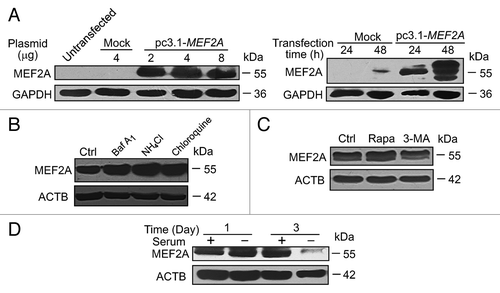
Our results so far indicate that CMA may participate in the degradation of endogenous MEF2A in the neuronal as well as nonneuronal cells. To investigate whether this finding could be extended to an exogenous overexpression system, we performed transient transfection of mouse neuroblastoma Neuro-2A cells with the full-length MEF2A plasmid. As shown in , neuro-2A cells transfected with 2 to 8 μg of pc3.1-MEF2A plasmid for 24 or 48 h showed a significantly higher expression of MEF2A protein as detected by western blot analysis. On the contrary, there was only a very low or undetectable level of MEF2A protein expression in the parental or empty vector-transfected control cells. Consistent with results obtained with endogenous MEF2A protein, different lysosome inhibitors, including Baf A1, NH4Cl, and chloroquine, caused an increase in the levels of exogenous overexpressed MEF2A (). However, using rapamycin and 3-MA, to respectively induce or inhibit macroautophagy in MEF2A-overexpressed cells, we did not observe any appreciable changes in MEF2A levels (). Furthermore, activation of CMA by serum deprivation over the course of 3 d led to a marked reduction in overexpressed-MEF2A levels in Neuro-2A cells (). These results suggest that CMA, at least in part, mediates MEF2A degradation.
Figure 4. Exogenous overexpressed MEF2A is degraded by CMA. (A) Overexpression of MEF2A in Neuro-2A cells. Neuro-2A cells were transfected with mock (pcDNA3.1) or pc3.1-MEF2A plasmid at different dosages or for different times. MEF2A protein levels were analyzed by western blotting and GAPDH was used as an internal control. (B) Lysosomal inhibitors cause exogenous overexpressed MEF2A accumulation. Neuro-2A cells transfected with the MEF2A-expressing vector were respectively treated with Baf A1 (25 nM), NH4Cl (50 mM) or chloroquine (50 μM) for 24 h. The levels of MEF2A were determined by immunoblotting. (C) Macroautophagy is not involved in degradation of exogenous overexpressed MEF2A. Neuro-2A cells transfected with the MEF2A-expressing vector were treated with a macroautophagy stimulator (100 nM of rapamycin) or an inhibitor (10 mM of 3-MA) for 24 h and MEF2A levels were determined by western blot. (D) Serum withdrawal decreases exogenous MEF2A protein. Neuro-2A cells transfected with the MEF2A-expressing vector cells were treated with serum deprivation for the indicated times. The levels of MEF2A were determined by immunoblotting.
Interactions of MEF2A with the key CMA regulators
The CMA pathway involves 2 key regulators, HSPA8 and LAMP2A. The HSPA8 chaperone and its cochaperones selectively recognize soluble cytosolic proteins containing a targeting motif, and target the protein cargo for binding to the lysosomal receptor LAMP2A for subsequent degradation in lysosomes.Citation27 To examine whether MEF2A interacts with HSPA8, we extracted total proteins from HEK 293T cells, and performed coimmunoprecipitation experiments with antibodies detecting the endogenous proteins. Immunoprecipitation (IP) with antibodies against MEF2A followed by immunoblotting with antibodies against HSPA8 demonstrated that MEF2A was coimmunoprecipitated with HSPA8 (). Reciprocally, to confirm that HSPA8 could bind with MEF2A, we expressed recombinant His-HSPA8 from E. coli. As shown in Figure S3A, following purification, we detected a polypeptide of ~70 kDa, the expected size of His-HSPA8. The His pull-down assay using this peptide showed that His-HSPA8 was able to pull down MEF2A ().
Figure 5. MEF2A interacts with the key CMA regulators. (A) MEF2A interacts with endogenous HSPA8. HEK 293T cell extracts were immunoprecipitated with IgG and anti-MEF2A antibody, respectively, and 10% of input extracts were analyzed by western blot with an anti-HSPA8 or anti-MEF2A antibody. (B) HSPA8 interacts with endogenous MEF2A. His affinity isolation assay was performed by incubating SK-N-SH cell lysates with His-HSPA8 or control protein. Bound MEF2A and 10% of input extracts were detected by immunoblotting with an anti-MEF2A or anti-HSPA8 antibody. Expressed His-HSPA8 protein was detected by anti-HSPA8 antibody. (C) Interaction of N-terminal MEF2A with HSPA8. GST affinity isolation assay was performed by incubating GST or GST-MEF2A-N fragments with SK-N-SH cell lysates. Bound HSPA8 and 10% input extracts were detected by immunoblotting with an anti-HSPA8 or anti-ACTB antibody. GST or GST-MEF2A-N fragments were detected by anti-GST antibody. (D) Degradation of MEF2A-∆N by serum deprivation. Neuro-2A cells were individually transfected with the full-length (MEF2A-WT, namely pc3.1-MEF2A) and the N-terminal deletion (MEF2A-∆N) constructs and cultured under serum deprivation for additional 2 d, and MEF2A levels in these cells were determined by western blot. (E) LAMP2A knockdown accumulates MEF2A. SK-N-SH cells were transfected with siRNAs targeting LAMP2A protein, or with negative control Luc-siRNA, and the lysates were collected 24 or 48 h later. LAMP2A knockdown efficiency and MEF2A levels were determined by western blot. (F) Effect of LAMP2A siRNA on endogenous LAMP2A, LAMP2B, LAMP2C, and MEF2A mRNA expression in SK-N-SH cells. SK-N-SH cells were individually transfected with Luc-siRNA, LAMP2A-siRNA-1 and -2. Twenty-four hours after treatment, the mRNAs were measured by RT-PCR; levels of ACTB expression are shown as an internal control.
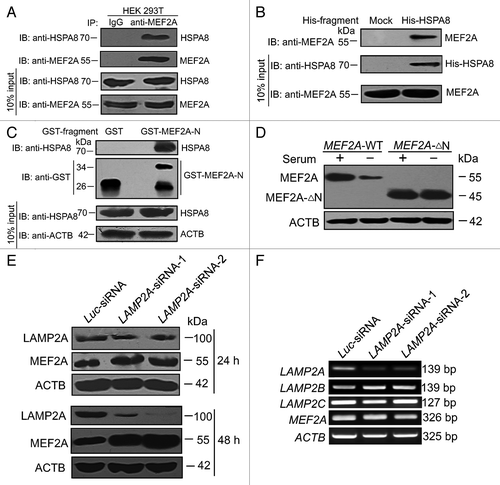
We then sought to map the HSPA8-interacting region in MEF2A. Cytosolic HSPA8 recognizes a conserved KFERQ-like motif in substrate proteins.Citation20 Analysis of the N terminus of MEF2A revealed the presence of several such motifs. We then performed GST affinity isolation assays with a GST-fused MEF2A N-terminal fragment (amino acid residues 1 to 86), GST-MEF2A-N. We incubated SK-N-SH cell lysates with purified GST-MEF2A-N proteins and performed both Coomassie brilliant blue staining and immunoblotting to detect bound HSPA8. As shown in Figure S3B and , HSPA8 interacted with GST-MEF2A-N, but not with GST alone. It should be pointed out that the purified GST-MEF2A-N proteins contained a considerable amount of polypeptides of incomplete length. One appealing possibility was that several rare codons appearing in the N-terminal part of this protein may terminate translation prematurely. Another possible explanation for this result was that GST-MEF2A-N was cleaved by endogenous proteases of bacteria after its expression. Moreover, we individually transfected the full-length (MEF2A-WT) and the N-terminal deletion (MEF2A-∆N) constructs into Neuro-2A cells, in which the endogenous MEF2A expression level was barely detectable. In contrast to wild-type MEF2A, the level of N-terminal truncated MEF2A (MEF2A-∆N) protein that lacked CMA recognition motifs was not significantly reduced by serum deprivation for 2 d (). These results obtained from either in vivo or in vitro suggest that MEF2A is recognized by the HSPA8 through its N terminus, and this interaction is required for subsequent lysosomal degradation through the CMA pathway.
LAMP2A is another key regulator in the CMA pathway. LAMP2A serves as the receptor on the lysosomal surface and its recognition is a rate-limiting step in CMA.Citation28 Interruption of LAMP2A results in a selective blockade of CMA.Citation29 We therefore examined the role of LAMP2A in MEF2A protein degradation using short hairpin RNAs (shRNAs) directed against the LAMP2A in SK-N-SH cells. Two LAMP2A siRNA expression plasmids (LAMP2A-siRNA-1 and -2) targeting different regions of human LAMP2A mRNA significantly downregulated the levels of LAMP2A protein as determined by western blot, as compared with the control firefly luciferase siRNA (Luc-siRNA). Knockdown of LAMP2A for 24 or 48 h, which presumably blocks the CMA pathway, concomitantly and time-dependently increased MEF2A levels in SK-N-SH cells (), in support of the role of CMA pathway in promoting MEF2A degradation. Because LAMP2B and LAMP2C are highly homologous to LAMP2A, the specificity of LAMP2A siRNAs was tested by RT-PCR. With treatment of LAMP2A-siRNA-1 and -2, the mRNA levels of endogenous LAMP2A in SK-N-SH cells were decreased significantly, whereas the mRNA levels of endogenous LAMP2B, LAMP2C and MEF2A were not influenced (). Therefore, the fact that endogenous MEF2A accumulates upon LAMP2A silencing provides further evidence that MEF2A is degraded in the lysosomes of neuronal cells via CMA.
Regulation of MEF2A degradation by oxidative stress
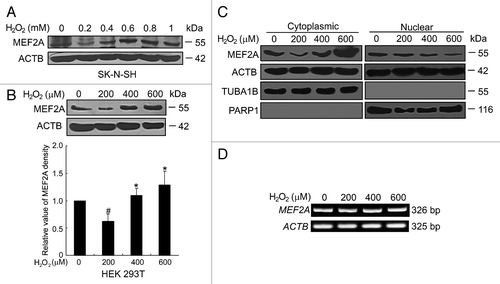
Oxidative stress is a major cause of neurodegenerative disorders. In order to verify whether oxidative stress could modulate the degradation of MEF2A, SK-N-SH cells were exposed to various concentrations of H2O2 (200 μM to 1 mM) for 24 h and MEF2A protein levels were examined by western blot. As shown in , 200 μM of H2O2 induced a significant reduction of MEF2A level, whereas 400 μM to 1 mM of H2O2 led to a dose-dependent accumulation of MEF2A proteins in SK-N-SH cells. Similar results were obtained in nonneuronal HEK 293T cells () or MEF2A-overexpressed Neuro-2A cells treated with 200 to 600 μM of H2O2 (Fig. S4A). We further examined the nuclear and cytoplasmic localization of MEF2A under oxidative stress in SK-N-SH cells. The mild oxidative stress with stimulation of 200 μM of H2O2 induced a significant reduction in the cytoplasmic MEF2A, but no apparent change in nuclear MEF2A level, whereas excessive oxidative stress with stimulation of 400 or 600 μM of H2O2 consistently reduced nuclear MEF2A but caused its concurrent accumulation in the cytoplasm (). To exclude the effects of oxidative stress on MEF2A gene expression, MEF2A mRNA level was assessed by RT-PCR. No significant change was detected in the levels of MEF2A transcript with 200 to 600 μM of H2O2 treatment for 24 h (). In addition, the effects of oxidative stress to the protein levels of other members of the MEF2 superfamily were detected. It seemed that the protein levels of both MEF2A and MEF2D were downregulated by mild oxidative stress, while excessive oxidative stress led to their protein accumulation. On the contrary, the protein levels of MEF2B and MEF2C were nearly not affected by oxidative stress (Fig. S4B). The reason for this discrepancy is not clear. Lysosome-targeting motifs, normally buried in the protein core, will be exposed after oxidative modification.Citation30 It is reasonable to expect a certain degree of difference in the protein structure and conformation between these 4 isoforms, which probably accounts for the different extent of exposure of CMA recognition motifs. Thus, the degradation of MEF2 proteins is regulated by oxidative stress in an isotype-specific manner.
Figure 6. Regulation of MEF2A degradation by oxidative stress. (A and B) Effects of H2O2 on the protein level of MEF2A. SK-N-SH (A) and HEK 293T (B) cells were treated without or with H2O2 (0 to 1.0 mM, 24 h) and protein levels were determined by western blot. Bottom graph shows the relative MEF2A levels normalized to ACTB (n = 3). #P < 0.05 vs. cells without H2O2 treatment; *P < 0.05 vs. 200 μM of H2O2 treatment. (C) Excessive oxidative induces increased accumulation of cytoplasmic MEF2A, which is accompanied by a decline of nuclear MEF2A. SK-N-SH cells were treated with 0 to 600 μM of H2O2 for 24 h followed by extraction of cytoplasmic and nuclear fractions. Equal quantities of samples from both fractions (50 μg per sample of cytoplasmic fraction and 25 μg per sample of nuclear fraction) were subjected to SDS-PAGE and western analysis with an anti-MEF2A, anti-ACTB (detecting both nuclear and cytoplasmic fractions), anti-TUBA1B (detecting cytoplasmic fraction), or anti-PARP1 antibody (detecting nuclear fraction). (D) MEF2A mRNA level is not affected by H2O2 treatment. SK-N-SH was exposed to different dosages of H2O2 (0 to 600 μM) for 24 h. Total RNA was prepared for RT-PCR analysis of MEF2A. The ACTB band is shown as a loading control.
CMA degradation of MEF2A under oxidative stress
Because we demonstrated above that MEF2A was degraded through the CMA pathway in basal conditions, we first examined whether CMA was also involved in its degradation under oxidative stress. LAMP2A has been suggested as the rate-limiting factor in CMA,Citation31 then a western blot assay was performed to detect the levels of LAMP2A at the cell lysosomal membrane. As shown in , mild oxidative stress (stimulation by 200 μM of H2O2) significantly increased LAMP2A levels compared with untreated controls, while under excessive oxidative stress (stimulation by 400 or 600 μM of H2O2), the levels of LAMP2A were significantly decreased to or even under the basal levels.
Figure 7. CMA degradation of MEF2A under oxidative stress. (A) Effects of H2O2 on the protein level of LAMP2A. SK-N-SH was exposed to different dosages of H2O2 (0 to 600 μM) for 24 h and protein levels were determined by western blot. *P < 0.05, **P < 0.01 vs. culture without H2O2. (B) Lysosomal binding of MEF2A. Lysosomes were isolated from the overnight-starved rat liver (left), or from H2O2-treated culture cells (middle). Lysosomes were coincubated with purified GST or GST-MEF2A fusion proteins. The presence of binding protein was determined by anti-GST immunoblotting. The LAMP2A protein levels were analyzed by western blotting and LAMP1 was shown as a loading control. Right panel shows the positions of GST or GST-MEF2A-N by Coommassie staining. (C) Degradation of MEF2A-∆N by oxidative stress. Neuro-2A cells were individually transfected with the full-length (MEF2A-WT, namely pc3.1-MEF2A) and the N-terminal deletion (MEF2A-∆N) constructs and cultured under 200 μM of H2O2 for 24 h. MEF2A levels in these cells were determined by western blot. (D) Proteasome and macroautophagy pathways are not implicated in MEF2A degradation under oxidative stress. SK-N-SH cells were treated with proteasome inhibitor MG132 (left) or macroautophagy inhibitor 3-MA (right) in the present of 200 μM of H2O2 for 24 h and the MEF2A protein levels were analyzed by western blotting.
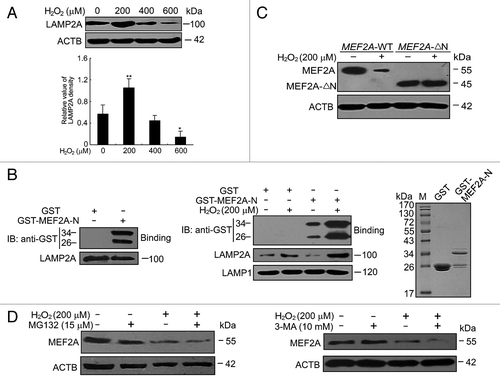
In order to further support the idea that CMA is involved in MEF2A degradation under oxidative stress, CMA activity in vitro using isolated lysosomes was measured. Lysosomes were purified from the overnight-starved rat liver, or from culture cells in response to mild oxidative stress. In line with our finding in intact cells, compared with untreated controls, LAMP2A level was increased in the purified lysosomes, which were isolated from cells in response to oxidative stress (, middle). This increase was selective for LAMP2A, since levels of LAMP1 (one of the other lysosomal membrane proteins) remained unchanged in both conditions. To show that lysosomes directly regulate MEF2A, lysosomal-binding assay was also performed using GST or GST-MEF2A-N as a substrate and detected by anti-GST immunoblotting. Compared with the control peptide, GST, only GST-MEF2A-N, which contained a KFERQ-like motif, could be bound by isolated lysosomes (, left). More importantly, the amount of GST-MEF2A-N bound to lysosomal membrane was significantly higher (about a 4-fold increase) in lysosomes from H2O2-treated cells (, middle). The right panel shows the positions of GST or GST-MEF2A-N by Coommassie Brilliant Blue staining (, right). On the contrary, the mild oxidative stress failed to accelerate the degradation of MEF2A-∆N (lacked CMA recognition motifs) as compared with wild-type MEF2A ().
To exclude the possibility of proteasome and macroautophagy pathways implication in MEF2A degradation under oxidative stress, these 2 pathways were individually inhibited by MG132 or 3-MA under oxidative stress. It should be noted that proteasome inhibition by MG132 (15 μM) did not accumulate but somehow decreased the protein level of MEF2A (, left), similarly to its pharmacological effects on MEF2A protein level under basal conditions (Fig. S1A). Additionally, inhibition of macroautophagy by 3-MA (10 mM) treatment accelerated the MEF2A reduction induced by mild oxidative stress (, right). These results confirmed that both proteasome and macroautophagy pathways were not implicated in MEF2A degradation under oxidative stress conditions. Furthermore, to investigate whether macroautophagy was also affected under oxidative stress, the changes in LC3 amounts were monitored by western blotting. It should be noticed that, no LC3-II bands were detected in the control or low-dose of H2O2 treated cells, whereas the amount of LC3-II was significantly increased upon high dosage of H2O2 treatment (Fig. S5). These findings indicate that despite those changes in macroautophagy in our cell model, the fact that MEF2A levels did not increase but rather decrease upon treatment with 3-MA suggest that macroautophagy was not responsible for the degradation of the protein under mild oxidative stress.
Because substrates selectively degraded by CMA are directly translocated across lysosomal membrane,Citation19 we next investigated whether excessive oxidative stress could induce lysosomal rupture/permeabilization, thus leading to the blockade of CMA degradation of MEF2A. Two lysosomotropic fluorescence probes, LysoTracker Red and acridine orange (AO), were used to determine lysosomal membrane stabilization on the basis of either low pH or proton trapping. As shown in , mild oxidative stress (stimulation by 200 μM of H2O2) showed no significant influence on lysosome pH, whereas excessive oxidative stress (stimulation by 400 or 600 μM of H2O2) dramatically decreased the numbers of LysoTracker-stained intact lysosomes in SK-N-SH cells as examined under the fluorescence microscope. Consistent results were also obtained using flow cytometry. Lysosomal destabilization was detected in cells exposed to extensive oxidative stress, as evidenced by an increased number of “pale cells”(cells with reduced numbers of LysoTracker and AO-accumulating lysosomes), whereas this change was very weak in mild oxidative stress treated groups (). Fluorescence microscopy also indicated that cells treated with H2O2 upon 400 μM had marked decrease of red fluorescence of lysosomes and exhibited predominantly green fluorescence over the cytosol regions, with only few red vesicles as compared with the control cells (Fig. S6).
Figure 8. Excessive oxidative stress induces lysosome rupture. SK-N-SH cells were treated in the absence or presence of H2O2 (0 to 600 μM) for 24 h. Cells were then visualized by bright-field microscopy or stained with LysoTracker Red (50 nM) or Hoechst 33342 (10 μg/ml) and subjected to immunofluorescence imaging. Scale bar: 25 μm.
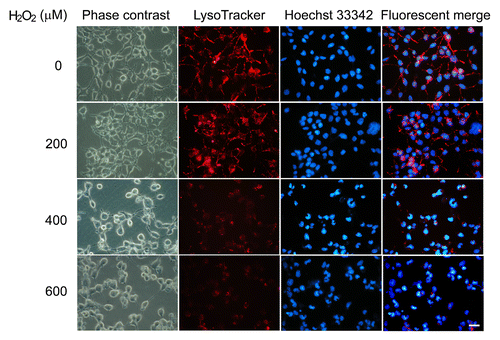
Figure 9. Lysosomal rupture assayed with the AO-uptake and LysoTracker Red-uptake methods. SK-N-SH cells were treated in the absence or presence of H2O2 (0 to 600 μM) for 24 h. Lysosomes stained by LysoTracker Red (50 nM) and acridine orange (5 μg/ml) were quantified by flow cytometry.
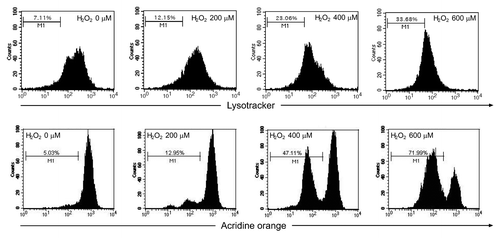
Collectively, these observations support the idea that the upregulated LAMP2A levels are responsible for the accelerated CMA degradation of MEF2A under mild oxidative stress, while extensive oxidative stress causes a decline in lysosomal stability, which might explain the inhibition of MEF2A degradation.
Inhibition of MEF2A degradation by extensive oxidative stress decreases DNA-binding and transcriptional activity of MEF2A
The MEF2 family members play critical roles in diverse cellular processes including neuronal survival, mainly depending on their transcriptional activity.Citation32 We therefore investigated how different degrees of oxidative stress regulate MEF2A transcriptional activity. We transiently transfected SK-N-SH cells with a MEF2A-dependent luciferase reporter construct and exposed them to various dosages of H2O2. As demonstrated in and , 200 μM of H2O2 treatment increased MEF2A activity from 3.8 to 4.6 relative luciferase activity despite a reduction in MEF2A protein level. 400 and 600 μM of H2O2 decreased MEF2A activity to 3.1 and 1.5 relative luciferase activity, respectively, despite an accumulation of total MEF2A. These results indicate that although excessive oxidative stress promotes MEF2A accumulation, it induces the loss of its transcriptional activity.
Figure 10. Impairment of MEF2A functions after blockade of CMA with excessive oxidative stress. (A) Effect of H2O2 on MEF2A transactivation activity. HEK 293T cells were transfected with a MEF2A-responsive luciferase reporter and pRL-TK. Twelve hours after transfection, the cells were cultured in the absence or presence of H2O2 (0 to 600 μM) for an additional 12 h. The firefly and renilla luciferase were measured, respectively, and the firefly luciferase activity was normalized to respective renilla luciferase. #P < 0.05 vs. mt-MEF2A reporter transfection alone; **P < 0.01 vs. wt-MEF2A reporter transfection alone. (B) Inhibition of MEF2A transactivation activity by lysosome inhibitors. HEK 293T cells were transfected with a MEF2A-responsive luciferase reporter and pRL-TK. Twelve h after transfection, the cells were respectively treated with chloroquine, Baf A1 or NH4Cl for an additional 12 h, and MEF2A transactivation activity was measured by MEF2A-dependent reporter assay as described in (A). #P < 0.05 vs. mt-MEF2A reporter transfection alone; *P < 0.05 vs. wt-MEF2A reporter transfection alone. (C) Effect of LAMP2A knockdown on MEF2A transactivation activity. HEK 293T cells were cotransfected with Luc-siRNA or Lamp2A-siRNA-1 and a MEF2A-responsive luciferase reporter and pRL-TK. Twenty-four hours after transfection, the cells were treated with or without 200 μM of H2O2 for an additional 12 h, and MEF2A transactivation activity was measured by MEF2A-dependent reporter assay. ##P < 0.01 vs. mt-MEF2A reporter transfection alone; **P < 0.01 vs. wt-MEF2A reporter transfection alone. (D) Effect of H2O2 on MEF2A DNA binding activity. DNA-binding activity of MEF2A in the cytoplasmic or nuclear fractions of SK-N-SH cells was assessed by EMSA 24 h after treatment with different dosages of H2O2 (0 to 600 μM). Upper arrow indicates the specific MEF2A-probe complex, lower arrow indicates the free probes. (E) Effects of MEF2A on the survival of neuron cells treated with H2O2. Neuro-2A cells transfected with mock vector (pcDNA3.1) or pc3.1-MEF2A plasmid were exposed to different concentrations of H2O2 for 24 h. Viable cells were identified by the MTT assay. * P < 0.05, **P < 0.01. (F) Neuro-2A cells transfected with mock vector (pcDNA3.1), pc3.1-MEF2A or MEF2A-∆N plasmid were exposed to 200 μM of H2O2 for 24 h. Viable cells were identified by the MTT assay. *P < 0.05, **P < 0.01. Mock-vector transfected cells were considered as 100% in (E and F).
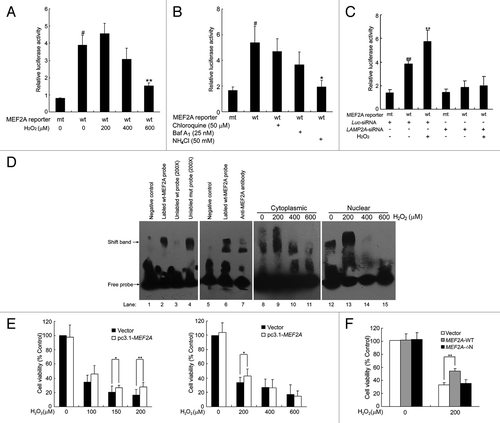
To investigate whether inhibition of MEF2A degradation affects its transcriptional activity, SK-N-SH cells transfected with MEF2A-mut-luc or MEF2A-wt-luc were treated with lysosome inhibitors. The luciferase assay showed that chloroquine, Baf A1, and ammonium chloride, which neutralized the lysosomal pH and thus inhibited MEF2A degradation (), markedly reduced MEF2A-dependent reporter activity (). Moreover, the changes of MEF2A transactivation in LAMP2A knockdown cells were also determined. As shown in , when CMA was impaired as a consequence of knocking down LAMP2A under oxidative stress, MEF2A transcriptional activity significantly decreased, indicating the impairment of MEF2A function after blockade of CMA. These results clearly indicate that whenever MEF2A degradation is disrupted at the LAMP2A recognition step or in the lysosome-mediated proteolysis step, it will reduce the transcriptional activity of the accumulated MEF2A protein.
To further assess whether the accumulated MEF2A retains its DNA binding activity, we performed an electrophoretic mobility shift assay (EMSA). As shown in , DNA-protein complexes were detected when wild-type labeled MEF2A probes were used (lane 2). These complexes were competitively blocked by a 200-fold excess of unlabeled specific probes, but not by 200-fold of mutant MEF2A probe (lane 3 and 4). Supershift assays showed that the MEF2A antibody induced a supershift band that was accompanied by the disappearance of the original protein-DNA complex (lane 7), indicating that MEF2A in cell lysates bound specifically to the DNA probe. Upon mild oxidative stress with 200 μM of H2O2, both cytoplasmic and nuclear MEF2A bound to the DNA probe at a far higher rate than the controls did (lane 8 and 9, 12 and 13). In contrast, there were fewer or even no detectable DNA-protein complexes using cytoplasmic or nuclear proteins extracted from cells treated with 400 or 600 μM of H2O2 (lane 10 and 11, 14 and 15). These results suggest that the loss of MEF2A transcriptional activity upon excessive oxidative stress, at least partially, is due to the loss of its DNA binding activity.
Furthermore, we investigated the role of MEF2A in H2O2-induced cell damage. Compared with the respective mock vector-transfected cells, overexpression of MEF2A attenuated cell viability loss induced by low dosage of H2O2 (100 to 200 μM), whereas under excessive oxidative stress situation (> 400 μM H2O2), MEF2A overexpression not only failed to protect neuron cells but rather led to a slight reduction of its cell viability (). Moreover, the viability of the neurons expressing the N-terminal deletion MEF2A mutant under oxidative stress was determined. As shown in , MEF2A-∆N, which lacked CMA recognition and DNA binding motifs, failed to alleviate H2O2-induced cell damage as wild-type MEF2A did. These data showed that the N terminus of MEF2A is necessary for its neuronal protection function.
Taken together, our data suggests that mild oxidative stress upregulates the DNA binding and transcription activity of MEF2A and it plays a protective role against oxidative stress-induced cell damage, while extensive oxidative stress inhibits MEF2A degradation and concomitantly decreases its transcriptional activity. More importantly, the nonfunctional accumulated MEF2A itself maybe toxic to neurons.
Oxidative stress-induced N-terminal HDAC4 cleavage product (HDAC4-NT) represses MEF2A activity
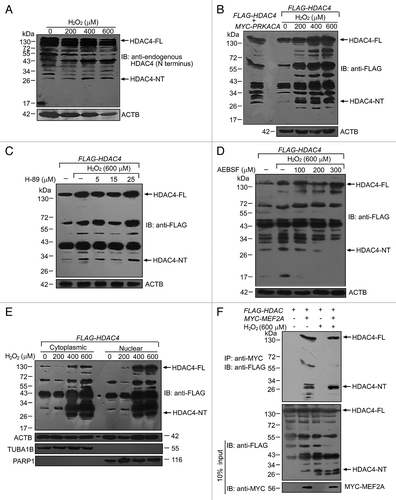
Our above results suggest that extensive oxidative stress not only induces the accumulation of total MEF2A but also preferentially promotes its cytoplasmic localization (). However, the reduction of nuclear localization of MEF2A under excessive oxidative conditions does not seem to be sufficient to account for the striking loss of DNA-binding activity of nuclear MEF2A (, lane 14 and 15), and fails to explain the reduced DNA-binding activity of cytoplasmic MEF2A (, lane 10 and 11). Recently, an N-terminal HDAC4 cleavage product (HDAC4-NT) was identified to selectively repress MEF2A activity in a PRKACA/PKA (protein kinase, cAMP-dependent, catalytic, α, PKA)-dependent manner.Citation33 To explore the mechanistic basis of the inhibitory effect of excessive oxidative stress on MEF2A transcriptional activity, we investigated whether HDAC4 could also be proteolytically processed to HDAC4-NT that represses MEF2A in response to oxidative stress. We exposed SK-N-SH cells to different dosages of H2O2 (0 to 600 μM), and examined the presence of HDAC4-NT using an antibody to the N terminus of HDAC4, in western blot. As shown in , an endogenous basal level of HDAC4-NT (~28 kDa) was detected in untreated control cells and a pronounced increase was observed after H2O2 treatment, especially in the excessive oxidative stress-treated groups. To further test whether oxidative stress can induce proteolysis of exogenous expressed HDAC4, HEK 293T cells were transfected with FLAG-HDAC4 plasmid followed by H2O2 treatment. Different dosages of H2O2 treatment from 200 to 600 μM also caused an obvious production of a protein band with the same size as the HDAC4-NT occurred in our positive control cells, which were the cells cotransfected with MYC-PRKACA and FLAG-HDAC4, and therefore possessing an active PRKACA signaling (). We then tested the role of the PRKACA pathway in HDAC-NT production in response to extensive oxidative stress by treating FLAG-HDAC4 transfected cells with H-89, a potent and selective inhibitor of PRKACA, in the presence of 600 μM of H2O2. As shown in , the PRKACA inhibitor failed to block HDAC4 cleavage stimulated by excessive oxidative. However, AEBSF, a serine protease inhibitor, effectively prevented H2O2-induced HDAC4-NT production at the concentrations > 200 μM (), suggesting that lysosomal serine proteases released under oxidative stress could effectively trigger proteolytic cleavage of HDAC4 in a PRKACA-independent manner. We further examined the subcellular localization of HDAC-NT, and observed the presence of HDAC-NT in both the cytoplasm and nucleus in the cells transfected with FLAG-HDAC4 and treated with H2O2 (). To further verify the interaction between HDAC4 and MEF2A under basal or excessive oxidative stress conditions, we performed immunoprecipitation experiments in which cell lysates were obtained from FLAG-HDAC4 and/or MYC-MEF2A plasmids transfected 293T cells treated with 600 μM of H2O2. As shown in , MEF2A interacted with both the full-length and N-terminal cleavage product of HDAC4 whenever under basal or stress conditions. To be noticed, compared with basal conditions, more HDAC-NT was sequestered by MEF2A under stressed conditions, verifying the increased cleavage of HDAC-4 by H2O2 treatment. This result raises a possibility that under excessive oxidative stress conditions, the production of HDAC-NT in the nucleus represses the transcriptional activity of nuclear MEF2A.
Figure 11. Oxidative stress induces the N-terminal HDAC4 cleavage product (HDAC4-NT). (A) Oxidative stress induced endogenous proteolysis of HDAC4. SK-N-SH cells were exposed to different dosages of H2O2 for 24 h and cell lysates were immunoblotted with an antibody directed against endogenous HDAC4. HDAC4-NT was indicated with a molecular mass of ~28 kDa. HDAC4-FL, full-length of HDAC4. (B) Oxidative stress induced proteolysis of exogenous expressed HDAC4. HEK 293T cells were transfected with FLAG-HDAC4 alone or cotransfected with MYC-PRKACA. Twenty-four hours after transfection, cells were exposed to different dosages of H2O2 for an additional 24 h. N-terminal FLAG-tagged HDAC4 protein was blotted with anti-Flag antibody. (C and D) Oxidative stress induced HDAC4-NT production was not affected by PRKACA inhibitor-H89, but by serine protease inhibitor-AEBSF. HEK 293T cells were transfected with FLAG-HDAC4 followed by 600 μM of H2O2 treatment for 24 h in the presence of the inhibitor. HDAC4-FL and -NT were detected by western blot analysis. The effects of increasing concentrations of H-89 (5 to 25 μM in C) or AEBSF (100 to 300 μM in D) are shown. (E) HDAC4-NT was detected both in the cytosol and nucleus. FLAG-HDAC4 transfected 293T cells were treated with 200 to 600 μM of H2O2 for 24 h followed by extraction of cytoplasmic and nuclear fractions. HDAC4-FL and -NT were detected by western blot analysis. (F) The interaction between MEF2A and HDAC4. HEK 293T cells were transfected with FLAG-HDAC4 alone or cotransfected with MYC-MEF2A. Twenty-four hours after transfection, cells were treated with or without 600 μM of H2O2 for an additional 24 h. MYC-tagged MEF2A protein was immunoprecipitated with anti-MYC beads, and blotted with anti-FLAG. 10% of input extracts were analyzed by western blot with anti-FLAG and anti-MYC antibodies.
Discussion
Studies of primary neuronal cultures have indicated that MEF2A regulates the expression of key factors involved in neuronal survival and differentiation.Citation9 In neurons, MEF2D, which is one of the 4 isoforms of MEF2, can be turned over by caspase-dependent cleavageCitation34 and by CMA under basal conditions.Citation23 However, whether and how different neuronal stress signals, especially oxidative stress, regulate the protein levels of other MEF2 isoforms, in particular MEF2A, is still unclear.
First, we provide solid experimental evidence supporting that MEF2A is selectively degraded by CMA, which includes: 1) MEF2A accumulation when lysosomal proteolytic activity is blocked (); 2) No changes in MEF2A protein levels when macroautophagy is activated or inhibited (); 3) increased degradation rates during serum deprivation when CMA is activated (; ); 4) interaction with cytosolic HSPA8 (); 5) presence of a N-terminal KFERQ-like motif (); 6) increased intracellular MEF2A levels upon LAMP2A silencing (). Our novel findings presented in this study demonstrate that MEF2A is a substrate for CMA
CMA constitutes part of the mechanisms of the cellular response to stresses, such as prolonged starvation, neurotoxins, and oxidative stress.Citation19 In our study, we observed that mild oxidative stress induced a rapid degradation of MEF2A, while excessive oxidative stress led to an accumulation of MEF2A (). As reported by a previous study, during mild oxidative stress, a higher rate of CMA is mediated by the enhanced ability of mutual recognition between substrates and lysosomes.Citation24 Indeed, we have observed an upregulation of LAMP2A in both intact cells and isolated lysosomes under mild oxidative stress (). To be mentioned, the possible implication of the proteasome and macroautophagy pathways in MEF2A degradation under this condition, was excluded (). These findings and the additional lysosomal binding assay () provide further confirmation that MEF2A is a CMA substrate whether in basal or oxidative stress conditions.
CMA activity is severely impaired during aging, which involves extensive oxidative stress, and part of the accumulation of oxidized proteins observed in old organisms may result from the malfunctioning of CMA.Citation35 In our study, an obvious lysosomal rupture/permeabilization was observed under excessive oxidative stress, which seems to be one of the possibilities for the inhibition of MEF2A degradation. There are alternative possibilities. It is known that excessive oxidative stress can induce amino acid modification, such as deamidation and oxidation.Citation36 It is possible that the N-terminal KFERQ-like motif of MEF2A is modified under this condition, and cannot be targeted for lysosomal degradation. To be mentioned, up to now very little is known about how nonfunctional lysosomes are eliminated. It is reasonable for us to hypothesize that excessive oxidative stress may activate macroautophagy to sequester the “leaking” lysosomes. Our observation that a significant change of LC3 amount was induced by high dosages of H2O2 treatment supported this idea (Fig. S5).
However, there was another puzzling and interesting observation that the accumulated MEF2A protein seemed to be nonfunctional as assessed by luciferase reporter assay or EMSA assay (). The result of reduced transcriptional activity upon treatment with chloroquine, Baf A1, or NH4Cl () was expected as these protease inhibitors will prevent degradation of the protein in lysosomes but not its translocation to lysosomes, so it is expected that if the protein is no longer in the cytosol it will not have transcriptional activity, but what is really interesting is that silencing of LAMP2A () has a similar effect, because in this case the protein remains in the cytosol. These results indicate that once MEF2A degradation is disrupted, the accumulated MEF2A proteins may lose its transcriptional activity. Moreover, in the EMSA assays, both cytoplasmic and nuclear MEF2A lost its DNA-binding capacity when CMA was blocked by excessive oxidative stress. Since in EMSA assays, MEF2A proteins presented in the prepared cytoplasmic and nuclear fractions could access DNA freely independent of its subcellular localization, it is possible that disruption of CMA degradation induces MEF2A modification or its interaction with other proteins that inhibits its DNA-binding activity. Interestingly, a similar result was reported that accumulated cytoplasmic MEF2D was also nonfunctional when high levels of SNCA disrupted its CMA degradation.Citation23 The precise mechanism which renders MEF2 isoforms incapable of binding to DNA upon disruption of CMA remains to be elucidated.
The regulation of MEF2 function is quite complicated, and is coordinated at multiple levels. For example, MEF2 could be phosphorylated by positive regulators such as MAPK14/p38α13 and MAPK7/ERK5,Citation37 as well as the negative regulator cyclin-dependent kinase 5.Citation34 MEF2 could also be regulated by acetylationCitation14 and sumoylation.Citation15 However, all these modifications mentioned above modulate MEF2 transactivation activity through its C’-transactivation domain. To our knowledge, HDAC4 is the only protein so far that has been verified to negatively regulate MEF2 activity by interacting directly with its DNA binding domain.Citation16,Citation38 Besides the fact that the full-length HDAC4 acts as a negative regulator of MEF2, the N-terminal HDAC4 cleavage product (HDAC4-NT) has been recently reported to selectively repress MEF2 activity in the nucleus in a PRKACA-dependent manner.Citation33 However, there is other evidence that PRKACA directly phosphorylates MEF2 at Thr20 to increase MEF2 DNA binding activity.Citation39 Therefore, the effect of the cAMP-PRKACA pathway on MEF2 activity is somewhat conflicting. In fact, PRKACA signaling could be activated by H2O2 treatment.Citation40 We speculated here that PRKACA activity may be differentially regulated in response to different degrees of oxidative stress signals. Under mild oxidative stress, PRKACA is activated and mainly executes its positive regulatory role on MEF2, whereas excessive stress enhances the binding of PRKACA to HDAC4 and the consequent proteolytic cleavage. The former hypothesis is supported by our results that 200 μM of H2O2 treatment indeed stimulated the transcriptional activity of MEF2A as determined both by luciferase reporter or EMSA assays (). However, our results do not support the latter one. In our study, HDAC4-NT production was not significantly decreased by the PRKACA inhibitor H-89, even though high dosage of H-89 (25 μM) was used (its usual working concentration is from 10 to 30 μM). Therefore, other mechanisms independent of PRKACA activation are responsible for HDAC4-NT production in our study. Moreover, in our study, the elevation of full-length HDAC4 was observed both in the cytoplasmic and nuclear fractions of the samples treated with high H2O2 concentrations (HDAC4-FL bands in ), which is in accordance with other studies showing that direct oxidation also results in accumulation of HDACs in the cytosol.Citation41 The full-length HDAC4 protein in combination with its cleavage products HDAC4-NT may have a more potent repressive effect on MEF2A under excessive oxidative stress. Another possibility that could not be ruled out is that the accumulated MEF2A protein itself undergoes structural and conformational changes that result in the loss of its DNA binding ability. Furthermore, in our study it seems like the accumulated nonfunctional MEF2A itself is “toxic” to neurons (). The specific mechanism remains to be further elucidated.
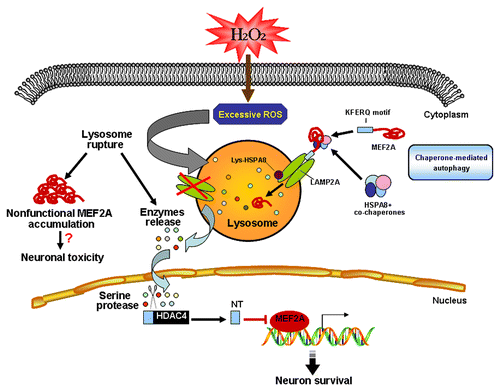
Collectively, our study provides evidence that MEF2A is degraded through CMA in basal conditions. Upon excessive oxidative stress, defective CMA that is likely due to lysosomal rupture, leads to the accumulation of nonfunctional MEF2A, which is transcriptionally inactive. Under this condition, the MEF2 repressor, HDAC4-NT, is produced through proteolysis mediated by lysosomal serine-proteases in a PRKACA-independent manner. The presence of nuclear HDAC4-NT could at least partially account for the transcriptional inactivity of accumulated MEF2A (). Of note, lysosome dysfunction is the link between the blockade of CMA degradation of MEF2A and its activity loss. Therapies targeting preservation of lysosome function may ameliorate age-related neurodegenerative diseases or other lysosome-related disorders. Several endogenous inhibitors of lysosomal membrane permeabilization, such as HSPA8,Citation42 have been identified, however, thus far no specific chemical inhibitors have been developed. Future investigations focusing on lysosome protection may open a new window for treating or preventing neurodegenerative diseases.
Figure 12. Schematic model of the regulation of MEF2A degradation under physiological or pathological conditions. MEF2A is degraded through the CMA pathway in basal conditions. Excessive oxidative stress, which is induced by neurotoxins such as H2O2, leads to lysosomal rupture. Such lysosome destabilization disrupts the CMA degradation of MEF2A and accounts for the accumulation of nonfunctional MEF2A, which is transcriptionally inactive. Under this condition, the MEF2 repressor, HDAC4-NT, is produced through proteolysis mediated by lysosomal serine proteases in a PRKACA-independent way. The presence of nuclear HDAC4-NT could at least partially account for the transcriptional inactivity of accumulated MEF2A.
Materials and Methods
Antibodies and reagents
Anti-MEF2A (sc-313), anti-MEF2B (sc-101097), anti-MEF2C (sc-365862), anti-MEF2D (sc-271153), anti-ACTB (β-actin, sc-47778) and goat anti-mouse (sc-2005) or rabbit (sc-2004) IgG-HRP were purchased from Santa Cruz Biotechnology. Anti-HIF1A (HIF-1α, NB100-105) was purchased from Novus Biologicals. Anti-LC3B (L7543), anti-LAMP1 (L1418), N-acetyl-Leu-Leu-Nle-CHO (ALLN, A6185), lactacystin (L6785), ammonium chloride (A0171), chloroquine diphosphate salt (C6628), 3-methyladenine (M9281), 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride (AEBSF, A8456), EZview™ Red Anti-c-MYC Affinity Gel, lysosomal isolation kit (LYSISO1) and acridine orange (318337) were purchased from Sigma Aldrich. Anti-HSPA8 (HSC70, ab51052) and anti-LAMP2A (ab18528) was purchased from Abcam. Anti-PARP1 (AP102), anti-TUBA1B (α-tubulin, AT819), anti-FLAG (AF519), anti-MYC (AM926), anti-HDAC4 (AH388), Alexa Fluor 488 goat anti-rabbit IgG (A0423), MG132 (S1748), cycloheximide (S1560), rapamycin (S1842), wortmannin (S1952), LY294002 (S1737), LysoTracker Red (C1046) and H-89 (S1643) were purchased from Beyotime Biothechnology. Anti-GAPDH (KC-5G4) was purchased from Shanghai Kangchen. Bafilomycin A1 (ALX-380-030-C100) was purchased from Alexis Biochemicals. Protein A/G plus-Agarose beads (sc-2003) was purchased from Santa Cruz. Protease inhibitor mixture cocktail (PI003) was purchased from Shanghai Shenergy Biocolor Biothechnology. All primers and oligonucleotides for shRNA or luciferase reporter plasmid constructions were synthesized by Shangon Biotech. All other chemicals were purchased from Sigma Aldrich.
Cell culture, transfection, and treatments
SK-N-SH, SH-SY5Y, Neuro-2A, and HEK 293T cells were all obtained from the Institute of Biochemistry and Cell Biology (Shanghai, China). Two human neuroblastoma cell lines, SK-N-SH and SH-SY5Y, and mouse neuroblastoma Neuro-2A cells were maintained in MEM supplemented with 10% fetal calf serum, 100 U/ml penicillin and 100 μg/ml streptomycin in a humid atmosphere of 5% CO2 and 95% air at 37 °C. Human embryonic kidney 293T (HEK 293T) cells were cultured in high-glucose DMEM. Cells were transfected with plasmids by Lipofectamine 2000 transfection reagent (Invitrogen, 11668) according to the manufacturer’s instructions. To deprive cells from serum, cells were extensively washed 3 times with 1 × PBS before replacing with fresh medium without serum. Mild and excessive oxidative stresses were induced in culture cells by supplementation of the culture medium with different dosages of H2O2 (200 μM to 1 mM) for 24 h.
Plasmid construction
The MEF2A coding region was amplified from total RNA isolated from SH-SY5Y cells using the following primers, 5′-TAAGGGCCCA GCATGGGGCG GAAGAAAATA CAAATCAC-3′ and 5′-TAGGTACCTT AGGTCACCCA CGCGTCCA-3′ with LA Taq DNA polymerase (TaKaRa, RR02MQ). The cloned cDNA sequence had 4 different sites in the nucleotide sequence from those in GenBank, which led to 2 nonsense amino acid transitions and 2 sense transitions, Arg79 to Gly and Val379 to Ala. The human MEF2A cDNA was inserted into the pcDNA3.1(-) ApaI/KpnI cloning site to generate pc3.1-MEF2A. MEF2A-∆N was amplified from pc3.1-MEF2A with primers 5′-TAAGGGCCCA GCATGACTTT AAGAAAGAAA GGCCT-3′ and 5′-TAGGTACCTT AGGTCACCCA CGCGTCCA-3′ and cloned into pcDNA3.1(-) vector. GST-MEF2A-N was also amplified from pc3.1-MEF2A with primers 5′- CGGGATCCAT GGGGCGGAAG AAAATACAAA-3′ and 5′-ACGCGTCGAC TTACTCAACA ATATCCGAGT TGGTTCTGCT-3′ and inserted into the pGEX-4T-1 (GE Healthcare, 28-9545-49) BamHI/SalI cloning site. The entire open reading frame of MEF2A was amplified by PCR from pc3.1-MEF2A and was subcloned into pcDNA3-MYC to generate MYC-MEF2A. Plasmid encoding His-HSPA8 (pEASY-E2-HSPA8) was kindly provided by Dr Jianhua Ming (Nanjing Agricultural University). FLAG-HDAC4 (HDAC4 containing N-terminal FLAG tag) and MYC-PRKACA were kind gifts from Johannes Backs (University of Heidelberg, Heidelberg, Germany).
Construction of human LAMP2A shRNA plasmids
To construct shRNAs targeting human LAMP2A (NM_002294.2), the following oligonucleotides were designed. LAMP2A-siRNA-1 sense, 5′- GATCCGACTG CAGTGCAGAT GATTCAAGAG ATCATCTGCA CTGCAGTCTT TTTGGAAGAA TTCA-3′ and LAMP2A-siRNA-1 antisense, 5′-AGCTTGAATT CTTCCAAAAA GACTGCAGTG CAGATGATCT CTTGAATCAT CTGCACTGCA GTCG-3′ (targeting region: 1279 to 1295); LAMP2A-siRNA-2 sense, 5′-GATCCGCACC ATCATGCTGG ATATTTCAAG AGAATATCCA GCATGATGGG CTTTTTGGAA GAATTCA-3′ and LAMP2A-siRNA-2 antisense, 5′-AGCTTGAATT CTTCCAAAAA GCACCATCAT GCTGGATATT CTCTTGAAAT ATCCAGCATG ATGGTGCG-3′ (targeting region: 1383 to 1401). The control-scrambled shRNA was constructed by insertion of a similar structure but encoding a nonsense minigene with no homology to any known sequences in human and mouse genomes. Luciferase-siRNA (Luc-siRNA) was also used as a scrambled control. The sequences for Luc-siRNA were as follows: Luc-siRNA sense, 5′-GATCCCGCTT ACGCTGAGTA CTTCGATTCA AGAGATCGAA GTACTCAGCG TAAGTTTTTG GAAGAATTCA-3′ and Luc-siRNA antisense, AGCTTGAATT CTTCCAAAAA AGCTTCATGA GTCGCATTCT CTCTTGAATC GAAGTACTCA GCGTAAGCGG-3′. Each pair of sense and antisense strands described above was annealed and ligated into the BamHI/HindIII site of Pgenesil-1 (Yrbio). Recombinant chimeric plasmids were checked by restriction enzyme analysis (EcoRI) and sequencing (Invitrogen).
RT-PCR and real-time PCR
Total RNA was isolated with Tripure reagent (Roche, 11667165001). cDNA was synthesized from 2 μg of RNA with oligo(dT)18 primers using M-MLV reverse transcriptase (TaKaRa, 2641Q) according to the manufacturer’s instructions. Primer sequences of MEF2A, LAMP2A, LAMP2B, LAMP2C and ACTB for RT-PCR were as follows: MEF2A forward: 5′-GCAATGCAGG TGGGATGT-3′ and MEF2A reverse: 5′-AGCAAGAGGT TGAGTGGC-3′; LAMP2A forward: 5′-TCAAGACTGC AGTGCA-3′ and LAMP2A reverse: 5′-CTAAAATTGCTCATAT-3′; LAMP2B forward: 5′-CCAAGAGTGT TCGCTG-3′ and LAMP2B reverse: 5′-TTACAGAGTC TGATAT-3′; LAMP2C forward: 5′-TGACTCTGAC CTCAACT-3′ and LAMP2C reverse: 5′-TTACACAGAC TGATAA-3′; ACTB forward: 5′-GCCGGGACCT GACTGACTAC-3′ and ACTB reverse: 5′-CGGATGTCCA CGTCACACTT-3′.
Real-time PCR was performed on an ABI Prism 7500 system (Applied Biosystems, USA), using SYBR Green I dye (Applied Biosystems, 4309155) and then calculated by means of the comparative Ct method (2-∆∆Ct) with the expression of ACTB as an internal control. The primers used were: MEF2A forward: 5′-ACTATCGGAG GAAGAGGA-3′ and MEF2A reverse: 5′-TTGTAGGCAG TCGGCATT-3′; ACTB forward: 5′-CGACAGGATG CAGAAGGAGA-3′ and ACTB reverse: 5′-CGTCATACTC CTGCTTGCTG-3′.
Coimmunoprecipitation
Cells were harvested using the SP-Lytics IP extraction buffer (Shanghai Shenergy Biocolor, LY008) containing 50 mM TRIS-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40 (Shanghai Sangon Biotech, NDB0385), 0.25% sodium deoxycholate (Shanghai Sangon Biotech, D0613) and a complete protease inhibitor cocktail, and kept on ice for 10 min. One mg of cell lysates in 500 μl IP extraction buffer were precleared with protein A/G plus-agarose for 30 min at 4 °C, incubated with 4 μg of anti-MEF2A primary antibody or normal rabbit immunoglobin G (IgG; Santa Cruz, sc-2027) at 4 °C overnight with gentle agitation, followed by incubation and with protein A/G plus agarose for another 3 h. After 5 washes of agarose pellets with IP extraction buffer, bound proteins were extracted with 1 × SDS sample buffer by heating at 100 °C for 5 min and subjected to SDS-PAGE followed by immunoblotting with antibodies against HSPA8, MEF2A, HDAC4 or FLAG-tag. For cell lysate containing MYC-tagged protein, it was treated with 50 μl of EZview™ Red Anti-c-MYC Affinity Gel (Sigma) or protein A/G agarose, respectively, according to the manufacturer’s instructions.
Western blotting
Western blots were performed as previously reported.Citation43
GST and His affinity isolation assays
In GST pulldown assays, GST and GST fusion proteins were expressed in BL21 Escherichia coli using the pGEX vector system, and crude bacterial lysates were prepared by sonication in 1 × PBS containing protease inhibitor mixture. After sonication, 8 ml of bacterial lysis supernatant fraction was added to 50 μl of preequilibrated glutathione-sepharose beads (Novagen, 70541-3) and incubated for 30 min at room temperature (RT) on a rotating platform. Then the beads were washed 5 times with GST binding/wash buffer (4.2 mM Na2HPO4, 2 mM KH2PO4, 140 mM NaCl, 10 mM KCl, pH 7.2). To each 30 μl aliquot of beads carrying GST-fusion protein or GST control, ~3 mg of SK-N-SH cell lysates and wash buffer with 1% BSA were added to make the final volume of 500 μl. Incubation was continued for 1 h with gentle mixing at RT. The beads were then washed 5 times in binding/wash buffer, resuspended in 1 × SDS sample buffer, and heated at 100 °C for 5 min. Bound proteins were resolved by SDS-PAGE and subjected to immunoblotting.
For His affinity isolation assays, fusion proteins were affinity-purified on His-Bind Resin (Novagen, 69670-3). Briefly, after a cell lysate precleaning step to reduce nonspecific binding to the agarose beads, 100 μl of resin immobilized with fusion proteins (≈ 500 μg) or His-tag control were incubated with ~4 mg of SK-N-SH cell lysates in binding buffer (0.5 M NaCl, 20 mM TRIS-HCl, 5 mM imidazole, pH 7.9) for 1 h with gentle mixing at RT. After 3 washes with binding buffer, Ni2+-bound complexes were eluted from the resin and denatured with 1 × SDS sample buffer. Proteins complexes were resolved by SDS-PAGE and subjected to immunoblotting.
Luciferase reporter gene assay
A reporter plasmid containing the MEF2 DNA-binding sequence (pGL3-MEF2A-wt) was engineered by inserting the sequence 5′- CGCGTCGACG GGCTATTTTT AGGGCCGGAT CC-3′ into the MluI/BglII site of the pGL-promoter plasmid DNA (Promega, E176A), upstream of an SV40 minimal promoter and a luciferase gene. The mutant sequence 5′- CGCGTCGACG GGCGATTTTT CGGGCCGGAT CC-3′, which does not bind to MEF2 proteins,Citation44 was used to generate pGL3-MEF2A-mut. For the luciferase assay, HEK 293T (3 × 105) cells were plated in 6-well plates and transfected with 2 μg of plasmids expressing MEF2A-wt-luc or MEF2A-mut-luc (containing firefly luciferase gene) and 500 ng of TK-Renilla-luc (containing renilla luciferase gene), using Lipofectamine 2000. Cells were harvested at 24 h posttransfection, and luciferase activities were measured with Dual-luciferase assay kits (Promega, E1910) according to the manufacturer’s protocol. Reporter gene activity was determined by normalization of relevant firefly luciferase to renilla luciferase activity.
Electrophoretic mobility shift assay (EMSA)
Nuclear extracts were prepared from SK-N-SH cells transfected with the pc3.1-MEF2A plasmid using NE-PER nuclear and cytoplasmic extraction reagents (Pierce, 78835) according to the manufacturer’s instructions. Synthetic reverse complimentary oligonucleotides with 5′-biotin (Sangon Biotech) were annealed to generate double-stranded oligonucleotides as probes. The following double-stranded oligonucleotides were used (only top strands are shown): 5′-TCGGACTGTT ACTAAAAATA GCACACCTG-3′ (MEF2A wild-type probe); 5′-TCGGACTGTT ACGAGCGATC GCACACCTG-3′ (MEF2A mutant probe, the underlined nucleotides indicate sites of mutation). EMSA was performed according to the instructions provided with the LightShift Chemiluminescent EMSA kit (Pierce, 20148). Briefly, the lysates (10 μg of nuclear extract or 20 μg of cytoplasmic lysates) were incubated at room temperature for 20 min in the presence of 2 μl of binding buffer, 1 μl glycerol, 0.1 μmol MgCl2, 1 μl NP40, 1 μl Poly (dI.dC) and 0.5 pmol of biotinylated probe in a reaction volume of 20 μl. Then 5 μl of 5 × loading buffer was added and the samples were electrophoresed through a 6.5% nondenaturing polyacrylamide gel in 0.5 × TBE at 4 °C. For competition experiments, unlabeled wild-type oligonucleotide was added in 200 × molar excess prior to the addition of the biotinylated probe. To identify the transcription factor present in the DNA protein complex using a supershift assay, the nuclear extracts were incubated with MEF2A antibody (Santa Cruz, sc-313) at room temperature for 20 min before incubation with labeled probe.
Lysosomal stability assessment
Cells were assessed for lysosomal stability using acridine orange uptake assays and LysoTracker Red-uptake.Citation45 Lysosomal membrane permeabilization was determined by staining cells with AO, a lysosomotropic dye which normally concentrates in lysosomes to produce red fluorescence.Citation46 Permeabilization of lysosomal membranes would result in a leakage of AO into the cytosol, thus producing a decrease of red fluorescence and an increase of green fluorescence. Cells were grown on coverslips and exposed to H2O2. After treatment, cells were washed once with PBS and stained with AO (1 μM) at 37 °C for 15 min. Excess AO was washed away with PBS and cells were immediately examined under a fluorescence microscope (Olympus, X51, Japan). For flow cytometry assay, cells were probed with AO (5 μg/ml) for 15 min at 37 °C and then washed with PBS once for immediate detection. Red lysosomal fluorescence of 10,000 cells per sample was determined by flow cytometry using the FL3 channel.
The pH changes in the lysosome were measured with LysoTracker Red dye by fluorescence microscopy or flow cytometry. LysoTracker Red, which is colorless at physiological pH, has red fluorescence at the acidic pH present in late endosomes and lysosomes. Following drug treatment, the medium was aspirated. Cells were incubated in D-Hanks’ solution containing LysoTracker Red (50 nM) at 37 °C for 15 min protected from light, and then washed twice with serum-free medium. Cells stained with LysoTracker dyes were then examined under a fluorescence microscope (Olympus, X51). For flow cytometric assay, cells were probed with 50 nM of LysoTracker Red and then washed with PBS once for immediate lysosome detection. Red fluorescence (lysosome) was detected through a 563 to 607 nm band-pass filter (FL2 channel) using a FACSCalibur flow cytometer (Becton Dickinson, USA).
Isolation of lysosomes
Lysosomes were purified from an overnight starved rat liver, or from culture cells using a lysosome isolation kit (Sigma) according to the manufacturer’s instructions. Briefly, livers or cells were washed 3 times with cold PBS and homogenized in the extraction buffer. After centrifuge serially at 1,000 and 20,000 g, pellet was collected and separated by density gradient centrifugation (150,000 g for 4 h). Lysosomal fractions were collected and the integrity of the lysosomes was assessed using the Neutral Red dye.
Binding of substrate protein by isolated lysosomes
For the binding assay, isolated lysosomes were coincubated with purified GST or GST-MEF2A-N fusion proteins for 20 min at 37 °C and washed for 4 times with cold PBS. After that samples were centrifuged at 20,000 g for 5 min at 4 °C, pellets were subjected to western blotting with antibodies against GST.
Statistical analysis
All experiments were performed with at least 3 independent biological replicates. For western blots, one representative blot is shown in figures; quantifications were performed from 3 independent experiments and expressed as mean ± SD. Statistical evaluations were performed with the Student t test when 2 value sets were compared. P < 0.05 was considered to be statistically significant.
| Abbreviations: | ||
| 3-MA | = | 3-methyladenine |
| ACTB | = | actin, beta |
| ALLN | = | N-acetyl-Leu-Leu-Nle-CHO |
| AO | = | acridine orange |
| Baf A1 | = | bafilomycin A1 |
| CHX | = | cycloheximide |
| CMA | = | chaperone-mediated autophagy |
| GAPDH | = | glyceraldehyde-3-phosphate dehydrogenase |
| HDAC4 | = | histone deacetylase 4 |
| HDAC4-NT | = | N terminal HDAC4 cleavage product |
| HIF1A | = | hypoxia-inducible factor 1, alpha subunit (basic helix-loop-helix transcription factor) |
| HSPA8/HSC70 | = | heat shock cognate protein 70 kDa protein 8 |
| LAMP | = | lysosomal-associated membrane protein |
| LC3 | = | microtubule-associated protein 1 light chain 3 (MAP1LC3) |
| MADS family | = | MCM1, Agamous, Deficiens and SRF |
| MAPK7/ERK5 | = | mitogen-activated protein kinase 7 |
| MAPK14/p38 | = | mitogen-activated protein kinase 14 |
| MEF2 | = | myocyte enhancer factor 2 |
| MPTP | = | 1-methyl-4-phenyl-1, 2, 5, 6-tetrahydropyridine |
| MTOR | = | mechanistic target of rapamycin |
| MYOD1 | = | myogenic differentiation 1 |
| PARP1 | = | poly (ADP-ribose) polymerase 1 |
| PtdIns3K | = | phosphatidylinositol 3-kinase |
| PRKACA/PKA | = | protein kinase, cAMP-dependent, catalytic, alpha |
| SNCA | = | synuclein, alpha |
| TUBA1B | = | tubulin, alpha 1b |
Additional material
Download Zip (405.6 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr Johannes Backs (University of Heidelberg, Heidelberg, Germany) for providing us with FLAG-HDAC4 and MYC-PRKACA plasmids. We are also grateful to Mr Xincan Zhao, Ms Fei Song (Jiangnan University, Wuxi, China) and Mr Lingjie Zhang (University of Toronto) for their technological assistance. This study was supported by the grants from the National Natural Science Foundation of China (Nos. 30870727, 90913023, 81173070 and 91229109), the Ministry of Health Foundation of China (W201304), the Natural Science Foundation of Jiangsu Province (No. BK2010156), the Science and Research Foundation of Health Bureau of Jiangsu Province (No. H2010027), and the Public Service Platform for Science and Technology Infrastructure Construction Project of Jiangsu Province (BM2012066).
References
- Naya FJ, Olson E. MEF2: a transcriptional target for signaling pathways controlling skeletal muscle growth and differentiation. Curr Opin Cell Biol 1999; 11:683 - 8; http://dx.doi.org/10.1016/S0955-0674(99)00036-8; PMID: 10600704
- Olson EN, Perry M, Schulz RA. Regulation of muscle differentiation by the MEF2 family of MADS box transcription factors. Dev Biol 1995; 172:2 - 14; http://dx.doi.org/10.1006/dbio.1995.0002; PMID: 7589800
- Potthoff MJ, Olson EN. MEF2: a central regulator of diverse developmental programs. Development 2007; 134:4131 - 40; http://dx.doi.org/10.1242/dev.008367; PMID: 17959722
- Black BL, Olson EN. Transcriptional control of muscle development by myocyte enhancer factor-2 (MEF2) proteins. Annu Rev Cell Dev Biol 1998; 14:167 - 96; http://dx.doi.org/10.1146/annurev.cellbio.14.1.167; PMID: 9891782
- Molkentin JD, Olson EN. Combinatorial control of muscle development by basic helix-loop-helix and MADS-box transcription factors. Proc Natl Acad Sci U S A 1996; 93:9366 - 73; http://dx.doi.org/10.1073/pnas.93.18.9366; PMID: 8790335
- McDermott JC, Cardoso MC, Yu YT, Andres V, Leifer D, Krainc D, Lipton SA, Nadal-Ginard B. hMEF2C gene encodes skeletal muscle- and brain-specific transcription factors. Mol Cell Biol 1993; 13:2564 - 77; http://dx.doi.org/10.1128/MCB.13.4.2564; PMID: 8455629
- Heidenreich KA, Linseman DA. Myocyte enhancer factor-2 transcription factors in neuronal differentiation and survival. Mol Neurobiol 2004; 29:155 - 66; http://dx.doi.org/10.1385/MN:29:2:155; PMID: 15126683
- Shalizi AK, Bonni A. brawn for brains: the role of MEF2 proteins in the developing nervous system. Curr Top Dev Biol 2005; 69:239 - 66; http://dx.doi.org/10.1016/S0070-2153(05)69009-6; PMID: 16243602
- She H, Mao Z. Regulation of myocyte enhancer factor-2 transcription factors by neurotoxins. Neurotoxicology 2011; 32:563 - 6; http://dx.doi.org/10.1016/j.neuro.2011.05.019; PMID: 21741404
- Li M, Linseman DA, Allen MP, Meintzer MK, Wang X, Laessig T, Wierman ME, Heidenreich KA. Myocyte enhancer factor 2A and 2D undergo phosphorylation and caspase-mediated degradation during apoptosis of rat cerebellar granule neurons. J Neurosci 2001; 21:6544 - 52; 21/17/6544 PMID: 11517243
- Burton TR, Dibrov A, Kashour T, Amara FM. Anti-apoptotic wild-type Alzheimer amyloid precursor protein signaling involves the p38 mitogen-activated protein kinase/MEF2 pathway. Brain Res Mol Brain Res 2002; 108:102 - 20; http://dx.doi.org/10.1016/S0169-328X(02)00519-3; PMID: 12480183
- Smith PD, Mount MP, Shree R, Callaghan S, Slack RS, Anisman H, Vincent I, Wang X, Mao Z, Park DS. Calpain-regulated p35/cdk5 plays a central role in dopaminergic neuron death through modulation of the transcription factor myocyte enhancer factor 2. J Neurosci 2006; 26:440 - 7; http://dx.doi.org/10.1523/JNEUROSCI.2875-05.2006; PMID: 16407541
- Yang SH, Galanis A, Sharrocks AD. Targeting of p38 mitogen-activated protein kinases to MEF2 transcription factors. Mol Cell Biol 1999; 19:4028 - 38; PMID: 10330143
- Ma K, Chan JK, Zhu G, Wu Z. Myocyte enhancer factor 2 acetylation by p300 enhances its DNA binding activity, transcriptional activity, and myogenic differentiation. Mol Cell Biol 2005; 25:3575 - 82; http://dx.doi.org/10.1128/MCB.25.9.3575-3582.2005; PMID: 15831463
- Riquelme C, Barthel KK, Liu X. SUMO-1 modification of MEF2A regulates its transcriptional activity. J Cell Mol Med 2006; 10:132 - 44; http://dx.doi.org/10.1111/j.1582-4934.2006.tb00295.x; PMID: 16563226
- Miska EA, Karlsson C, Langley E, Nielsen SJ, Pines J, Kouzarides T. HDAC4 deacetylase associates with and represses the MEF2 transcription factor. EMBO J 1999; 18:5099 - 107; http://dx.doi.org/10.1093/emboj/18.18.5099; PMID: 10487761
- Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006; 443:780 - 6; http://dx.doi.org/10.1038/nature05291; PMID: 17051204
- Hale AN, Ledbetter DJ, Gawriluk TR, Rucker EB 3rd. Autophagy: regulation and role in development. Autophagy 2013; 9:951 - 72; http://dx.doi.org/10.4161/auto.24273; PMID: 24121596
- Cuervo AM. Chaperone-mediated autophagy: selectivity pays off. Trends Endocrinol Metab 2010; 21:142 - 50; http://dx.doi.org/10.1016/j.tem.2009.10.003; PMID: 19857975
- Dice JF. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem Sci 1990; 15:305 - 9; http://dx.doi.org/10.1016/0968-0004(90)90019-8; PMID: 2204156
- Wing SS, Chiang HL, Goldberg AL, Dice JF. Proteins containing peptide sequences related to Lys-Phe-Glu-Arg-Gln are selectively depleted in liver and heart, but not skeletal muscle, of fasted rats. Biochem J 1991; 275:165 - 9; PMID: 2018472
- Kaushik S, Bandyopadhyay U, Sridhar S, Kiffin R, Martinez-Vicente M, Kon M, Orenstein SJ, Wong E, Cuervo AM. Chaperone-mediated autophagy at a glance. J Cell Sci 2011; 124:495 - 9; http://dx.doi.org/10.1242/jcs.073874; PMID: 21282471
- Yang Q, She H, Gearing M, Colla E, Lee M, Shacka JJ, Mao Z. Regulation of neuronal survival factor MEF2D by chaperone-mediated autophagy. Science 2009; 323:124 - 7; http://dx.doi.org/10.1126/science.1166088; PMID: 19119233
- Kiffin R, Christian C, Knecht E, Cuervo AM. Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell 2004; 15:4829 - 40; http://dx.doi.org/10.1091/mbc.E04-06-0477; PMID: 15331765
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19:5720 - 8; http://dx.doi.org/10.1093/emboj/19.21.5720; PMID: 11060023
- Cuervo AM, Knecht E, Terlecky SR, Dice JF. Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. Am J Physiol 1995; 269:C1200 - 8; PMID: 7491910
- Cuervo AM, Wong E. Chaperone-mediated autophagy: roles in disease and aging. Cell Res 2014; 24:92 - 104; http://dx.doi.org/10.1038/cr.2013.153; PMID: 24281265
- Bandyopadhyay U, Sridhar S, Kaushik S, Kiffin R, Cuervo AM. Identification of regulators of chaperone-mediated autophagy. Mol Cell 2010; 39:535 - 47; http://dx.doi.org/10.1016/j.molcel.2010.08.004; PMID: 20797626
- Massey AC, Kaushik S, Sovak G, Kiffin R, Cuervo AM. Consequences of the selective blockage of chaperone-mediated autophagy. Proc Natl Acad Sci U S A 2006; 103:5805 - 10; http://dx.doi.org/10.1073/pnas.0507436103; PMID: 16585521
- Kiffin R, Bandyopadhyay U, Cuervo AM. Oxidative stress and autophagy. Antioxid Redox Signal 2006; 8:152 - 62; http://dx.doi.org/10.1089/ars.2006.8.152; PMID: 16487049
- Cuervo AM, Dice JF. Regulation of lamp2a levels in the lysosomal membrane. Traffic 2000; 1:570 - 83; http://dx.doi.org/10.1034/j.1600-0854.2000.010707.x; PMID: 11208145
- Mao Z, Bonni A, Xia F, Nadal-Vicens M, Greenberg ME. Neuronal activity-dependent cell survival mediated by transcription factor MEF2. Science 1999; 286:785 - 90; http://dx.doi.org/10.1126/science.286.5440.785; PMID: 10531066
- Backs J, Worst BC, Lehmann LH, Patrick DM, Jebessa Z, Kreusser MM, Sun Q, Chen L, Heft C, Katus HA, et al. Selective repression of MEF2 activity by PKA-dependent proteolysis of HDAC4. J Cell Biol 2011; 195:403 - 15; http://dx.doi.org/10.1083/jcb.201105063; PMID: 22042619
- Tang X, Wang X, Gong X, Tong M, Park D, Xia Z, Mao Z. Cyclin-dependent kinase 5 mediates neurotoxin-induced degradation of the transcription factor myocyte enhancer factor 2. J Neurosci 2005; 25:4823 - 34; http://dx.doi.org/10.1523/JNEUROSCI.1331-05.2005; PMID: 15888658
- Koga H, Kaushik S, Cuervo AM. Protein homeostasis and aging: The importance of exquisite quality control. Ageing Res Rev 2011; 10:205 - 15; http://dx.doi.org/10.1016/j.arr.2010.02.001; PMID: 20152936
- Gracy RW, Talent JM, Zvaigzne AI. Molecular wear and tear leads to terminal marking and the unstable isoforms of aging. J Exp Zool 1998; 282:18 - 27; http://dx.doi.org/10.1002/(SICI)1097-010X(199809/10)282:1/2<18::AID-JEZ5>3.0.CO;2-Q; PMID: 9723162
- Barsyte-Lovejoy D, Galanis A, Clancy A, Sharrocks AD. ERK5 is targeted to myocyte enhancer factor 2A (MEF2A) through a MAPK docking motif. Biochem J 2004; 381:693 - 9; http://dx.doi.org/10.1042/BJ20031940; PMID: 15132737
- Wang AH, Bertos NR, Vezmar M, Pelletier N, Crosato M, Heng HH, Th’ng J, Han J, Yang XJ. HDAC4, a human histone deacetylase related to yeast HDA1, is a transcriptional corepressor. Mol Cell Biol 1999; 19:7816 - 27; PMID: 10523670
- Wang X, Tang X, Li M, Marshall J, Mao Z. Regulation of neuroprotective activity of myocyte-enhancer factor 2 by cAMP-protein kinase A signaling pathway in neuronal survival. J Biol Chem 2005; 280:16705 - 13; http://dx.doi.org/10.1074/jbc.M501819200; PMID: 15735306
- Brennan JP, Bardswell SC, Burgoyne JR, Fuller W, Schröder E, Wait R, Begum S, Kentish JC, Eaton P. Oxidant-induced activation of type I protein kinase A is mediated by RI subunit interprotein disulfide bond formation. J Biol Chem 2006; 281:21827 - 36; http://dx.doi.org/10.1074/jbc.M603952200; PMID: 16754666
- Ago T, Liu T, Zhai P, Chen W, Li H, Molkentin JD, Vatner SF, Sadoshima J. A redox-dependent pathway for regulating class II HDACs and cardiac hypertrophy. Cell 2008; 133:978 - 93; http://dx.doi.org/10.1016/j.cell.2008.04.041; PMID: 18555775
- Nylandsted J, Gyrd-Hansen M, Danielewicz A, Fehrenbacher N, Lademann U, Høyer-Hansen M, Weber E, Multhoff G, Rohde M, Jäättelä M. Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J Exp Med 2004; 200:425 - 35; http://dx.doi.org/10.1084/jem.20040531; PMID: 15314073
- Guo WJ, Zhang YM, Zhang L, Huang B, Tao FF, Chen W, Guo ZJ, Xu Q, Sun Y. Novel monofunctional platinum (II) complex Mono-Pt induces apoptosis-independent autophagic cell death in human ovarian carcinoma cells, distinct from cisplatin. Autophagy 2013; 9:996 - 1008; http://dx.doi.org/10.4161/auto.24407; PMID: 23580233
- Coso OA, Montaner S, Fromm C, Lacal JC, Prywes R, Teramoto H, Gutkind JS. Signaling from G protein-coupled receptors to the c-jun promoter involves the MEF2 transcription factor. Evidence for a novel c-jun amino-terminal kinase-independent pathway. J Biol Chem 1997; 272:20691 - 7; http://dx.doi.org/10.1074/jbc.272.33.20691; PMID: 9252389
- Yuan XM, Li W, Dalen H, Lotem J, Kama R, Sachs L, Brunk UT. Lysosomal destabilization in p53-induced apoptosis. Proc Natl Acad Sci U S A 2002; 99:6286 - 91; http://dx.doi.org/10.1073/pnas.092135599; PMID: 11959917
- Zelenin AV. Fluorescence microscopy of lysosomes and related structures in living cells. Nature 1966; 212:425 - 6; http://dx.doi.org/10.1038/212425a0; PMID: 5339137