Abstract
Canonical autophagy is positively regulated by the Beclin 1/phosphatidylinositol 3-kinase class III (PtdIns3KC3) complex that generates an essential phospholipid, phosphatidylinositol 3-phosphate (PtdIns(3)P), for the formation of autophagosomes. Previously, we identified the human WIPI protein family and found that WIPI-1 specifically binds PtdIns(3)P, accumulates at the phagophore and becomes a membrane protein of generated autophagosomes. Combining siRNA-mediated protein downregulation with automated high through-put analysis of PtdIns(3)P-dependent autophagosomal membrane localization of WIPI-1, we found that WIPI-1 functions upstream of both Atg7 and Atg5, and stimulates an increase of LC3-II upon nutrient starvation. Resveratrol-mediated autophagy was shown to enter autophagic degradation in a noncanonical manner, independent of Beclin 1 but dependent on Atg7 and Atg5. By using electron microscopy, LC3 lipidation and GFP-LC3 puncta-formation assays we confirmed these results and found that this effect is partially wortmannin-insensitive. In line with this, resveratrol did not promote phagophore localization of WIPI-1, WIPI-2 or the Atg16L complex above basal level. In fact, the presence of resveratrol in nutrient-free conditions inhibited phagophore localization of WIPI-1. Nevertheless, we found that resveratrol-mediated autophagy functionally depends on canonical-driven LC3-II production, as shown by siRNA-mediated downregulation of WIPI-1 or WIPI-2. From this it is tempting to speculate that resveratrol promotes noncanonical autophagic degradation downstream of the PtdIns(3)P-WIPI-Atg7-Atg5 pathway, by engaging a distinct subset of LC3-II that might be generated at membrane origins apart from canonical phagophore structures.
Introduction
Macroautophagy (hereafter referred to as autophagy) is an evolutionary, highly conserved bulk degradation mechanism responsible for a constitutive turnover of cytoplasmic material including long-lived proteins and cellular organelles.Citation1,Citation2 Central to the process of autophagy is the activity of at least 18 core autophagy proteinsCitation3 (Atg proteins) functionally involved in the generation of autophagosomes, unique multimembrane vesicles that sequester the cytoplasmic cargo.Citation4 Autophagosome formation is initiated from preformed structures (phagophore) and requires the activity of two ubiquitin-like conjugation systems specific for autophagy, the Atg12–5 (Atg16L complex) and the LC3 (Atg8) conjugation system, both of which require Atg7.Citation5 Completed autophagosomes acquire acidic hydrolases by fusing with lysosomes, and subsequently, the cytoplasmic cargo is degraded in vesicles termed autolysosomes.Citation6
Tight control of basal autophagy is crucial to maintain cellular homeostasis as both reduced and elevated autophagy levels have recently been correlated with age-related human pathologies, such as cancer and neurodegeneration.Citation1,Citation7 However, the complexity of signaling events that control autophagy levels is insufficiently understood. Even so, the activity of phosphatidylinositol 3-kinase class III (PtdIns3KC3) in complex with Beclin 1 (Atg6), Atg14 and p150,Citation8 that generates the phosphoinositide PtdIns(3)P, represents an evolutionarily conserved positive regulatory circuit which controls autophagosome formation (canonical autophagy). Likewise, in a wide range of eukaryotic cellular systems, TOR complex activity has been demonstrated to inhibit the onset of autophagy.Citation9 Crosstalk between the positive regulation of autophagy by PtdIns3KC3 and the negative regulation by TOR should involve RAG GTPases that connect amino acid signals and TOR activity.Citation10,Citation11
In this context, resveratrol, an antioxidant agent mainly concentrated in red grapes, was shown to induce autophagy independent of Beclin 1Citation12 and most likely through AMPK-mediated mTOR inhibition.Citation13 Further Beclin 1-independent entries to autophagic degradation have been reported,Citation14-Citation22 however molecular details on how such noncanonical autophagic pathways stimulate autophagosome formation have not been analyzed in further detail. In order to contribute to this discussion we here investigated the involvement of the PtdIns(3)P effector WIPI-1 (and WIPI-2) that we identified earlierCitation23 to function downstream of PtdIns(3)P generating events, such as the activity of the Beclin 1/PtdIns3KC3 complex, during the initiation of autophagosome formation.Citation23-Citation27 By freeze-fracture immuno-electron microscopy we recently found that WIPI-1 localizes at membranes of the endoplasmic reticulum (ER) and the plasma membranes (PM) upon the induction of autophagy, suggesting that these membranes provide sources for autophagosomal membranes.Citation28 Further autophagic factors, such as Atg14, DFCP1 and WIPI-2, have been identified to localize close to the ER,Citation24,Citation26,Citation29,Citation30 suggesting that ER membrane sites represent mammalian phagophore structures. Moreover, WIPI-1 (and WIPI-2) becomes a membrane protein of generated autophagosomal vesicles, most likely through its specific binding to PtdIns(3)P.Citation28
By siRNA-based assays, we showed here that LC3 lipidation depends on the function of the PtdIns(3)P effector WIPI-1, and that WIPI-1 acts upstream of both autophagosomal ubiquitin-like conjugation systems, scored by modulating Atg7 and Atg5 protein levels. Basal autophagy was reflected by the number of cells (depending on the cell line approximately between 5–30% of the cells) that display WIPI-1 and WIPI-2 puncta, marking phagophores and autophagosomes, and that display Atg12 puncta, marking phagophore structures only. Upon nutrient starvation the number of WIPI-1, -2B, -2D and Atg12 puncta-positive cells significantly increased, but resveratrol treatment did not promote puncta formation above the basal level. However, the amount of lipidated LC3 (LC3-II) and membrane localized GFP-LC3 significantly increased upon resveratrol treatment, as well as the number of autophagosomal vesicles (AVi) scored by quantitative electron microscopy. Based on lysosomal inhibition assays, this situation should not reflect the inhibition of autophagy, but the induction of autophagy, as suggested by previous reports.Citation12,Citation31 During the cellular response to resveratrol exposure canonical PtdIns(3)P-WIPI-Atg7-Atg5 pathway components might become engaged to produce LC3-II for noncanonical autophagosome generation.
Results
Resveratrol promotes an increase of LC3-II, GFP-LC3 puncta and multilayered autophagosomal vesicles
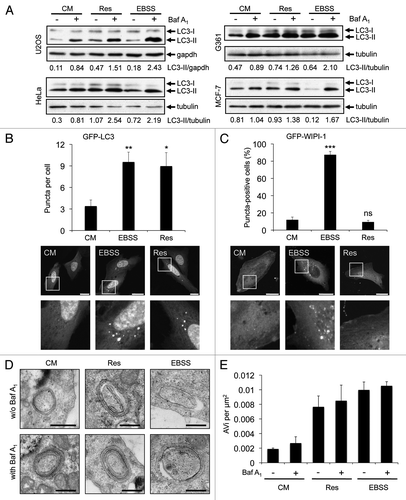
The intracellular localization of the lipidated form of LC3Citation32 that is conjugated to phosphatidylethanolamine, LC3-II, has been widely used to visualize autophagosomal membranes by fluorescence microscopy (LC3 puncta formation) or to monitor the autophagosomal flux by LC3-II protein abundance in correlation to a loading control as tubulin or gapdh from treatments in the presence or absence of lysosomal inhibitors.Citation32-Citation34 We investigated the effect of resveratrol on LC3-II protein abundance in four different human tumor cell lines (U2OS, HeLa, G361, MCF-7). To control for canonical autophagy induction we starved the cells by using EBSS, an amino acid- and serum-free culture medium with low glucose levels. To control for the autophagic flux we blocked lysosomal degradation of LC3-II with bafilomycin A1 (Baf A1). We found that resveratrol treatment increased the abundance of LC3-II in all four tumor cell lines tested (). Further, by transient expression of GFP-LC3 in U2OS cells, we confirmed that resveratrol treatment resulted in an increase in GFP-LC3 puncta per cell as demonstrated earlier in MCF-7 cellsCitation12 (). Next, we used quantitative WIPI-1 puncta-formation analysis, a new opportunity to assess autophagy by fluorescence microscopy in higher eukaryotes.Citation27 Since WIPI-1 specifically binds PtdIns(3)P, mainly generated by the Beclin 1/PtdIns3KC3 complex, and subsequently localizes at autophagosomal membranes, canonical PtdIns(3)P requirement for autophagosome formation can be investigated by quantifying the subcellular localization of WIPI-1. We observed that in contrast to the results shown in and , resveratrol did not increase the number of cells displaying GFP-WIPI-1 puncta in U2OS cells (), indicating that autophagic stimulation by resveratrol does not involve the PtdIns(3)P effector WIPI-1. In support, resveratrol treatment also did not stimulate endogenous WIPI-1 to form more punctate structures when compared with the basal amount of WIPI-1 puncta-positive cells in control settings in human G361 cells (data not shown). To further address the nature of these results, we asked whether or not resveratrol mediates an enrichment of double-membrane autophagosomes by conducting quantitative electron microscopy (EM) from stable GFP-WIPI-1 cells treated with control medium, resveratrol or EBSS in the presence (+) or absence (-) lysosomal inhibition (Baf A1). To control LC3 lipidation we monitored LC3-II protein abundance before the cells were subjected to EM (data not shown). We identified multilayered autophagosomal vesicular structures (AVi) (), and quantified the numbers of AVi by point-counting microscopyCitation35 (). When comparing conditions in which lysosomal degradation is blocked (by Baf A1) in contrast to non-Baf A1 conditions, only a minor increase in the number of autophagosomes was observed. Nevertheless, the increase of AVs upon nutrient starvation (EBSS) was comparable with previous reports using an equivalent technique.Citation36,Citation37 Clearly, resveratrol treatment also resulted in an increase of AVi ().
Figure 1. Resveratrol promotes an increase of autophagosomes. U2OS, HeLa, G361 and MCF-7 cells were treated with 64 µM resveratrol (Res), nutrient-free medium (EBSS) or control medium (CM) for 3 h. Bafilomycin A1 (Baf A1) was added after 2 h as indicated (+). LC3-II protein abundance was determined and normalized over tubulin or gapdh. The quantification of one representative experiment (n = 3 for each cell line) is shown (A). U2OS cells were transiently transfected with GFP-LC3 (B) or GFP-WIPI-1 (C) and treated with 64 µM resveratrol (Res), nutrient-free medium (EBSS) or control medium (CM) for 3 h and images were acquired by confocal microscopy. The number of GFP-LC3 puncta per cell was determined from 33–36 individual cells (n = 3) by using Z-stack projections and ImagePro Plus 4.1 analysis software (B, upper panel). The number of GFP-WIPI-1 puncta-positive cells per treatment was determined from 400 individual cells (n = 4) by fluorescent microscopy (C, upper panel). The results were expressed as mean ± SD p-values: *p < 0.05; **p < 0.01; ***p < 0.001; ns: not significant. Scale bars: 20 µm. Stable GFP-WIPI-1 U2OS cells were treated with control medium (CM), 64 µM resveratrol (Res) or nutrient-free medium (EBSS) in the presence (+) or absence (-) of bafilomycin A1 (Baf A1). For EM analysis, ultrathin resin sections of different samples were used and representative images are shown (D). Scale bars: 400 nm. The number of multilayered autophagosomal vesicular structures (AVi) per square micrometer was determined for each treatment (E).
Resveratrol does not promote an increase of WIPI-1 puncta, prominent for canonical autophagy
In order to analyze the effect of resveratrol treatment on the localization of WIPI-1 in more detail, we used an automated high through-put procedure specific for automated acquisition (epifluoresence) and image analysis of GFP-WIPI-1 positive autophagosomal structures (WIPI-1 puncta-formation assay).Citation38 Using a stable GFP-WIPI-1 U2OS cells we either applied nutrient-free medium (EBSS) or resveratrol (32 μM, 64 μM, 128 μM) for treatments of 3, 24 or 48 h (). The analysis of up to 3,899 individual cells for each treatment from three independent experiments revealed that administration of resveratrol did not induce GFP-WIPI-1 puncta formation after 3, 24 or 48 h (). In higher concentrations or upon longer resveratrol treatments, the basal number of GFP-WIPI-1 puncta-positive cells was significantly reduced when compared with the control (). As expected, nutrient starvation profoundly increased the number of cells displaying prominent GFP-WIPI-1 puncta (), and lysosomal inhibition by Baf A1 treatment further significantly increased the number of GFP-WIPI-1 puncta-positive cells () and the number of GFP-WIPI-1 puncta per cell (data not shown). This effect was not observed in resveratrol-treated cells (). However, we confirmed that both nutrient starvation and resveratrol promote an increase of LC3-II in the GFP-WIPI-1 U2OS cell line (Fig. S1A and S1B). To further confirm proper canonical autophagic activity in the GFP-WIPI-1 U2OS cell line used here, we monitored both LC3-II protein abundance and WIPI-1 puncta formation upon autophagic stimulation by EBSS or rapamycin (Fig. S1B and S1C). As expected, LC3-II protein abundance increased when control treatment and EBSS or rapamycin treatments were compared (Fig. S1B). In line, GFP-WIPI-1 puncta-positive cells increased upon both rapamycin treatment (RM: 64.1% puncta-positive cells) and nutrient starvation (EBSS: 75.6% puncta-positive cells) and decreased upon wortmannin administration (WM: 9.1% puncta-positive cells) when compared with basal autophagic activity in control cells (CM: 26.2% puncta-positive cells) (Fig. S1C). Further compounds, etoposide and staurosporine, that have been shown to stimulate autophagy also increased the number of cells displaying GFP-WIPI-1 upon exposures of 3 h (Fig. S1D). Importantly, etoposide, a chemotherapeutic compound that induces DNA strand breaks by inhibiting topoisomerase II, was shown to induce a noncanonical Atg5-independent pathway for autophagic degradation that depends on the activity of the PtdIns3KC3 complex.Citation17 Both the administration of etoposide increased the level of cells displaying GFP-WIPI-1 puncta (to 48.7%) and staurosporine, a multiprotein kinase inhibitor and a strong inducer of apoptosis, also led to an increase of cells displaying GFP-WIPI-1 puncta (to 73.9%) (Fig. S1D). Hence these pro-apoptotic compounds found to modulate autophagy also stimulate WIPI-1 puncta formation.
Figure 2. WIPI-1 puncta formation is stimulated upon nutrient starvation but not upon resveratrol treatment. Stable GFP-WIPI-1 U2OS cells were treated with control medium (CM), 32 μM, 64 μM or 128 μM resveratrol (Res) or nutrient-free medium (EBSS) for 3, 24 or 48 h. GFP-WIPI-1 fluorescence images were automatically acquired and analyzed (A). The number of GFP-WIPI-1 puncta-positive cells was determined and each bar represents mean values of 1732–3899 individually analyzed cells (n = 3) ± SD (B). Stable GFP-WIPI-1 U2OS cells were treated with control medium (CM) or 64 μM resveratrol (Res) in the presence or absence of bafilomycin A1 (Baf A1) or nutrient-free medium (EBSS) for 3 h. Automated GFP-WIPI-1 puncta-formation analysis was conducted (n = 5, 16044–22479 individually analyzed cells) and expressed as the number of GFP-WIPI-1 puncta-positive cells ± SD (C). A time-course experiment was conducted by using the GFP-WIPI-1 U2OS cell line and the following treatments: control medium (CM), 64 μM resveratrol (Res), nutrient-free medium (EBSS), 100 μM LY294002 in control medium (LY294002), 64 μM resveratrol in nutrient-free medium (EBSS/Res) or 100 μM LY294002 in nutrient-free medium (EBSS/LY294002). Automated GFP-WIPI-1 puncta-formation analysis from up to 5000 individual cells per treatment (n = 3) was expressed as the number of GFP-WIPI-1 puncta-positive cells ± SD (D) or the number of GFP-WIPI-1 puncta per cell (E). p-values: *p < 0.05; **p < 0.01; ***p < 0.001; ns = not significant. Scale bars: 20 µm.
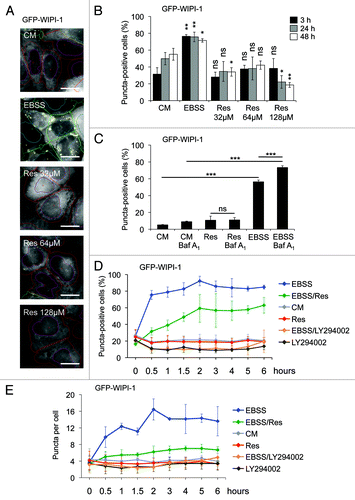
Based on this, we revisited the inhibitory effect of resveratrol on the formation of WIPI-1 puncta () and conducted a time-course experiment over 6 h (). We used EBSS to induce autophagy and LY294002 to inhibit the generation of PtdIns(3)P, hence WIPI-1 puncta formation. In addition, we also included co-treatments of resveratrol or LY294002 in nutrient-free medium (EBSS). Indeed, we demonstrated that resveratrol counteracts nutrient starvation-induced GFP-WIPI-1 puncta formation, measured either as the number of GFP-WIPI-1 puncta-positive cells () or the number of GFP-WIPI-1 puncta per individual cell (). As the treatment with EBSS is used to starve the cells by both amino acid and serum deprivation, we also conducted this experiment with control medium that contains amino acids but lacks serum (CM w/o FCS). The tendency that resveratrol counteracts GFP-WIPI-1 puncta formation was also observed in CM w/o serum (Fig. S2), but this effect was not as prominent compared with EBSS (), indicating that resveratrol counteracts amino acid starvation-induced canonical autophagy.
Evidence that WIPI-1 and WIPI-2B/2D display a redundant function in autophagy
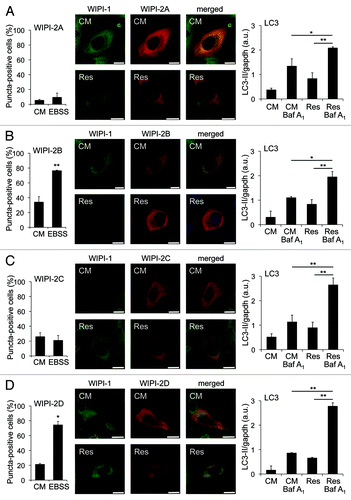
Our results warranted an investigation of whether human WIPI-2, the closest human homolog of WIPI-1Citation23 would respond to resveratrol treatments with puncta formation. Therefore, we transiently expressed either of the four GFP-WIPI-2 isoforms (WIPI-2A, -2B, -2C, -2D) in U2OS cells and quantified cells (minimum of 600 cells from seven independent experiments) displaying WIPI puncta upon resveratrol treatment in the presence or absence of lysosomal inhibition by confocal microscopy. Clearly, the presence of WIPI-2 puncta-positive cells did not increase upon resveratrol treatment (Fig. S3). Importantly, we identified that two of the WIPI-2 variants tested, WIPI-2B and WIPI-2D, responded to nutrient starvation with cytoplasmic puncta formation equivalent to WIPI-1 (, left panels). Further, we monitored the intracellular localization of coexpressed WIPI-1 (GFP-WIPI-1) with either of the WIPI-2 isoforms (myc-tagged WIPI-2A, -2B, -2C, -2D) upon resveratrol administration by confocal microscopy. Again, neither WIPI-1 nor WIPI-2 displayed puncta upon resveratrol treatment when coexpressed in the same cell (, middle panels). By quantitative LC3 lipidation analysis in the presence or absence of lysosomal inhibition we demonstrate that autophagy was properly induced by resveratrol treatment in WIPI-1/WIPI-2 coexpressing cells (, right panels). It was reported that endogenous WIPI-2 puncta colocalize with GFP-LC3 when cells were treated with resveratrol.Citation26 Our quantification of WIPI-2 puncta-positive cells suggests that this might reflect WIPI-2 puncta formation in basal conditions as approximately 20% of the cells displayed WIPI-2 puncta in control medium (, left panels, CM = control medium).
Figure 3. WIPI-2 puncta formation is not induced upon resveratrol treatment, but WIPI-2B and WIPI-2D form puncta upon nutrient starvation. U2OS cells were transiently tranfected with either of four GFP-WIPI-2 variants (GFP-WIPI-2A, -2B, -2C, -2D) and treated with control medium (CM) or nutrient-free medium (EBSS). The number of GFP-WIPI-2 puncta-positive cells was determined by fluorescence microscopy and mean values from 400–600 cells (n = 2–3) are presented ± SD (A–D, left panels). Stable GFP-WIPI-1 U2OS cells were transiently transfected with myc-tagged WIPI-2 (myc-WIPI-2A, -2B, -2C, -2D), treated with or without 64 µM resveratrol (Res) in control medium (CM) for 3 h followed by anti-myc/Alexa546 immunostaining and confocal microscopy (representative images, n = 2). Nuclei were stained with TO-PRO-3 (A–D, middle panels). In parallel, U2OS cells coexpressing myc-tagged WIPI-2 variants (myc-WIPI-2A, -2B, -2C, -2D) and GFP-WIPI-1, treated with or without 64 µM resveratrol (Res) in control medium (CM) for 3 h in the presence or absence of bafilomycin A1 (Baf A1) were subjected to LC3-II protein monitoring (A–D, right panels). Each bar represents mean values of three independent experiments ± SD p-values: *p < 0.05; **p < 0.01. Scale bars: 20 µm.
Resveratrol does not promote phagophore localization of endogenous Atg12
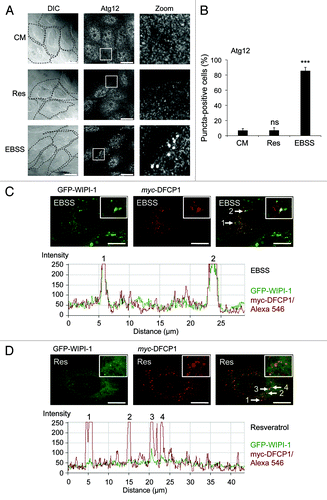
The Atg16L complex, composed of Atg12 conjugated to Atg5 and associated with Atg16,Citation5 functions downstream of WIPI-1.Citation24 Hence we asked whether or not the Atg16L complex is activated to localize at the phagophore upon resveratrol treatment. We treated U2OS cells with resveratrol and used EBSS as a positive control. We visualized the endogenous Atg16L complex by anti-Atg12 immunofluorescence (). Quantification of 600 cells from three independent experiments clearly shows, that upon canonical induction of autophagy by nutrient starvation (EBSS) the number of Atg12 puncta-positive cells strongly increase (to 85.3%) when compared with control cells (CM). However, cells positive for Atg12 puncta do not increase upon treatment with resveratrol for 3 h (; equivalent results were obtained in GFP-WIPI-1 U2OS cells, data not shown).
Figure 4. Increase of Atg12 puncta and WIPI-1/DFCP1 colocalization upon nutrient-starvation but not upon resveratrol treatment. U2OS cells were treated with 64 µM resveratrol (Res), nutrient-free medium (EBSS) or control medium (CM) for 3 h, followed by anti-Atg12/Alexa488 immunostaining in order to visualize the endogenous Atg16L complex by confocal microscopy (A). The number of Atg12 puncta-positive cells was determined (600 cells, n = 3) and each bar represents mean values ± SD (B). p-values: ***p < 0.001; ns = not significant. Stable GFP-WIPI-1 U2OS cells were transiently transfected with myc-DFCP1, treated with nutrient-free medium (EBSS) or with 64 µM resveratrol (Res) for 3 h followed by anti-myc/Alexa546 immunostaining. Representative confocal microscopy images (n = 3) from individual optical sections (upper panels) along with the corresponding intensity profiles (lower panels) are presented (C and D). Scale bars: 20 µm.
WIPI-1 colocalizes with DFCP1 under nutrient-free conditions but not upon resveratrol treatment
Because neither WIPI-1 nor WIPI-2 were stimulated to prominently localize at the phagophore upon resveratrol treatment, we asked whether or not a further PtdIns(3)P-binding protein involved in early steps of phagophore formation, DFCP1,Citation29 might respond to this treatment. We found that overexpression of myc-tagged DFPC1 in U2OS cells or the GFP-WIPI-1 U2OS cell line resulted in the localization of DFCP1 puncta (). Upon nutrient starvation-induced WIPI-1 puncta formation, DFCP1 and WIPI-1 partially colocalized in the majority of cells (). Upon resveratrol treatment, this partial colocalization was not observed, as the number of WIPI-1 puncta-positive cells was not elevated due to the treatment (). However, in the few GFP-WIPI-1 puncta-positive cells in control medium (CM) or upon resveratrol treatment, GFP-WIPI-1 and DFCP1 puncta also partially colocalized (data not shown).
Cellular PtdIns(3)P availability is not inhibited upon resveratrol treatment
Next we asked whether resveratrol influences the overall availability of PtdIns(3)P within the cell. We marked the endosomal PtdIns(3)P pool by using a stable GFP-2xFYVE Citation39,Citation40U2OS cell line upon resveratrol, wortmannin or LY294002 treatments for 3 h (Fig. S4). Using automated fluorescent image acquisition and analysis we did not find a prominent difference in the amount of GFP-2xFYVE positive signals per cell upon resveratrol treatment when compared with control cells, but upon LY294002 treatment as expected (Fig. S4A). This strongly indicates that PtdIns(3)P generation is not blocked by resveratrol. In line, the colocalization of endogenous EEA1, a marker for early endosomes,Citation39 and GFP-2xFYVE did not change upon resveratrol treatment (Fig. S4B). GFP-2xFYVE signals occupy 84% of early endosomes in control and resveratrol-treated cells (Fig. S4B). Thus, our results show that resveratrol does not block PtdIns(3)P generation and availability. However, upon both nutrient-starvation and resveratrol treatment a decrease in the amount of GFP-2xFYVE puncta per cell was observed (Fig. S4C).
Partial wortmannin-insensitivity of resveratrol-mediated LC3 response
Next, we investigated whether resveratrol-mediated LC3-II and GFP-LC3 puncta increase occurs when PtdIns(3)P generation is blocked. We co-treated stable GFP-WIPI-1 U2OS cells with wortmannin and either resveratrol or EBSS (). The quantification of LC3 protein abundance from six independent experiments showed that the level of LC3-II significantly increased in cells that have been co-treated with resveratrol and wortmannin in the presence of bafilomycin A1. In contrast, wortmannin treatment in the presence of bafilomycin A1 did not lead to an increase in LC3-II protein as expected, and co-treatment of EBSS and wortmannin also completely abolished an LC3-II increase (). However, compared with resveratrol treatment alone, cells that have been co-treated with resveratrol and wortmannin showed also a significant reduction in the level of LC3-II (). To confirm the results obtained from LC3-II protein monitoring, we generated a stable GFP-LC3 U2OS cell line and used this cell line for automated high through-put GFP-LC3 puncta analysis. Again, we co-treated GFP-LC3 U2OS cells with wortmannin and resveratrol or EBSS in the presence or absence of lysosomal inhibition (with Baf A1) (). Co-treatment of resveratrol and wortmannin significantly increases GFP-LC3 puncta per cell compared with wortmannin treatment alone or co-treatment of EBSS and wortmannin (). However, upon resveratrol/wortmannin co-treatment the number of GFP-LC3 puncta per cell was significantly reduced compared with resveratrol treatment alone (). In addition, the increase of LC3 puncta per cell in cells co-treated with resveratrol and wortmannin compared with the same conditions when lysosomal degradation is blocked (by Baf A1) is not as strong as in the cells stimulated with resveratrol alone (). Thus, our results provide evidence that resveratrol-mediated autophagy is partially wortmannin sensitive. However, resveratrol still stimulated an increase of LC3-II and GFP-LC3 in the presence of wortmannin.
Figure 5. Resveratrol-promoted increase of LC3-II protein abundance and GFP-LC3 puncta is partially insensitive to wortmannin treatment. Stable GFP-WIPI-1 U2OS cells were treated with control medium (CM), resveratrol (Res), wortmannin (WM), resveratrol plus wortmannin (Res/WM), nutrient-free medium (EBSS) or nutrient-free medium plus wortmannin (EBSS/WM) for 3 h. Bafilomycin A1 (Baf A1) was added after 2 h as indicated (+). LC3-II protein abundance was determined and normalized over tubulin. Each bar represents mean values ± SD (n = 6) (A). Stable GFP-LC3 U2OS cells were treated as above and subjected to automated fluorescence image acquisition (B) and analysis (C). Each bar represents mean values ± SD from the analysis of 1553–3207 individual cells (n = 3). p-values: *p < 0.05; **p < 0.01; ***p < 0.001; ns: not significant. Scale bars: 20 µm.
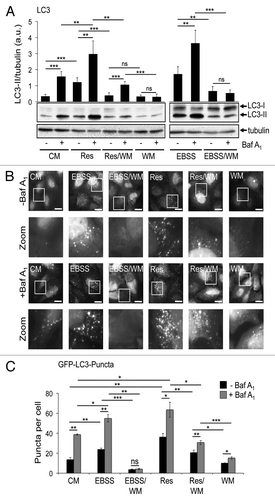
Atg7 and Atg5 dependent LC3 response upon nutrient starvation and resveratrol treatment
Next, we analyzed whether or not canonical LC3 lipidation is necessary for the increase of both LC3-II and GFP-LC3 puncta. We employed m5-7 cells, mouse embryonic fibroblasts from Atg5 knockout mice harboring a doxycycline-repressable integrated Atg5 cDNA.Citation41 Clearly, using this cell line in the absence of doxycycline the Atg12–5 conjugate was formed and subsequently LC3-II (). As expected, upon nutrient starvation (EBSS) the amount of LC3-II increased (). In line with our previous findings in this study, resveratrol treatment also resulted in an increase of LC3-II (). This effect was almost nullified upon doxycycline-mediated shutdown of Atg5 expression in both, the presence and absence of lysosomal inhibition (). To further investigate the dependency on Atg5 in the resveratrol-mediated LC3 response, we downregulated Atg5 by transient siRNA transfection and quantified LC3-II protein abundance (). Nutrient starvation-induced LC3-II production was decreased when Atg5 was downregulated, as expected. In line with results shown in , LC3-II levels also decreased in resveratrol-treated cells in the presence of siAtg5 (). Upon downregulation of Atg7, the level of the Atg12–5 conjugate (please note that Atg5 is mainly detected in the Atg12–5 conjugated form) also decreased () as expected.Citation5 As a result, LC3-II protein abundance decreased in the presence of siAtg7 either using control treatment (CM), nutrient-free medium (EBSS) or resveratrol (Res) (). From this it became clear that canonical-driven Atg7/Atg5 dependent LC3-II production is a prerequisite for the effect of resveratrol on LC3. However, resveratrol treatment did not promote a prominent phagophore localization of Atg12, reflecting the localization of the Atg12–5/Atg16L complex, but Atg5 is functionally required for the resveratrol-mediated LC3 response.
Figure 6. Resveratrol-mediated autophagy depends on Atg7 and Atg5. Mouse embryonic fibroblasts (m5-7 cells) were treated with 64 µM resveratrol (Res), nutrient-free medium (EBSS) or control medium (CM) for 3 h and subjected to quantitative anti-LC3 western blotting (n = 7) (A). Doxycycline (1 µg/ml) was added to the culture medium in order to shut down Atg5 expression in m5-7 cells. Representative western blots (n = 3) detecting Atg5 (Atg12–5 conjugate), LC3 or tubulin upon treatments with control medium (CM), 64 µM resveratrol (Res) or nutrient-free medium (EBSS) for 3 h are shown; bafilomycin A1 (Baf A1) was added after 2 h as indicated (B). Stable GFP-WIPI-1 U2OS cells were transfected with control, Atg5 or Atg7 siRNAs for 72 h and treated with control medium (CM), 64 µM resveratrol (Res) or nutrient-free medium (EBSS) for 3 h. Representative downregulation of Atg5 (Atg12–5 conjugate) or Atg7 protein is shown and LC3-II protein abundance was normalized over tubulin and quantified (n = 3) (C). p-values: *p < 0.05; ***p < 0.001.
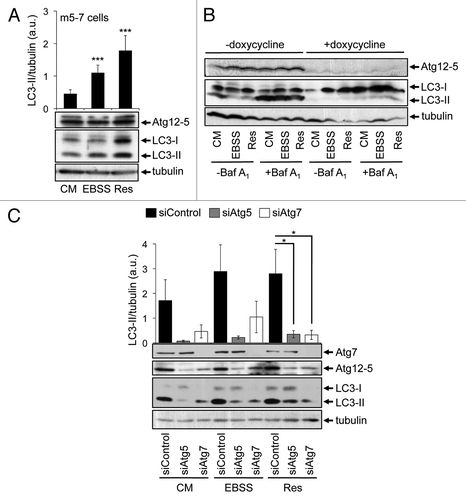
WIPI-1 functions upstream of Atg7 and Atg5 and is required for the nutrient starvation and resveratrol-mediated LC3-II increase
We addressed the question of whether or not WIPI-1 or WIPI-2 might also be functionally required for the LC3 response, despite the lack of induced phagophore localization. Using human G361 cells that express a prominent level of endogenous WIPI-1 and WIPI-2, we transiently downregulated either WIPI-1 or WIPI-2 by siRNA-mediated transfections and provide a representative result from three independent experiments (). Prominently, upon WIPI-1 downregulation basal LC3-II protein levels decreased, found by control medium (CM) treatments in the presence and absence of lysosomal inhibition (). In line, the increase of LC3-II upon nutrient starvation (EBSS) was also lowered when WIPI-1 was downregulated, further demonstrating that WIPI-1 is functionally required for canonical LC3 lipidation (, right panel). Clearly, LC3-II protein levels also decreased when siWIPI-1 transfected cells were treated with resveratrol, showing that WIPI-1 is indeed functionally required for the resveratrol-mediated LC3 response (, left panel). This result was also achieved by using siWIPI-2; in addition, downregulation of WIPI-1 might have a more prominent effect on LC3 lipidation as downregulation of WIPI-2 (, right panel). This result demonstrates, that WIPI-1 is functionally required for both canonical and resveratrol-mediated autophagy.
Figure 7. Functional requirement of WIPI-1 and WIPI-2 for LC3 lipidation upon both nutrient starvation and resveratrol treatment. Human G361 cells were transfected with control, WIPI-1 or WIPI-2 siRNA for 72 h and treated with control medium (CM), 64 µM resveratrol (Res) or nutrient-free medium (EBSS) for 3 h. Bafilomycin A1 (Baf A1) was added after 2 h as indicated (+). Representative western blot results (A) along with the quantification of EBSS and resveratrol treatments (B) was shown. Stable GFP-WIPI-1 U2OS cells were transfected with control, Atg5 or Atg7 siRNA for 72 h, treated as above and subjected to automated GFP-WIPI-1 image acquisition and analysis (C). Each bar represents mean values of GFP-WIPI-1 puncta-positive cells (up to 2500 individual cells, n = 3) ± SD p-values: *p < 0.05; **p < 0.01; ***p < 0.001; ns = not significant.
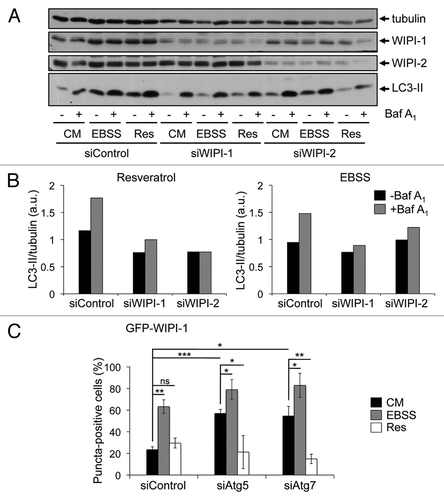
Because it was shown that WIPI-1 should function upstream of Atg5,Citation24 we downregulated Atg5 or Atg7 in the GFP-WIPI-1 cell line and measured the number of GFP-WIPI-1 puncta-positive cells in an automated fashion. Strikingly, the number of GFP-WIPI-1 puncta-positive cells significantly increased upon either Atg5 or Atg7 downregulation in both control medium (CM) and nutrient-free medium (EBSS) (), suggesting that autophagosome formation from WIPI-1 positive initial phagophore-like structures is blocked. Resveratrol treatment did not promote GFP-WIPI-1 puncta formation in cells transfected with either siAtg5 or siAtg7 (), in fact, the numbers of GFP-WIPI-1 puncta-positive cells decreased as shown above (). These results provide evidence that WIPI-1 is functionally required for LC3 lipidation by acting upstream of Atg7 and Atg5, but WIPI-1 (and WIPI-2) does not respond to the resveratrol treatment with induced phagophore localization as it occurs upon canonical autophagy-triggering treatments ( and Fig. S1).
Discussion
Generation of the phosphoinositide PtdIns(3)P is essential to the process of autophagy. The human PtdIns(3)P effector protein WIPI-1, specifically binds PtdIns(3)P upon the initiation of the autophagic pathway and subsequently accumulates at the phagophore, initial autophagosomal membranes positive for the Atg16L complex and LC3.Citation23,Citation24,Citation27 Consequently, WIPI-1 becomes a membrane protein of multilayered autophagosomes, localizing at inner and outer membranes.Citation28 We began to characterize the PtdIns(3)P-dependent localization of human WIPI-1 at autophagosomal membranes (WIPI-1 puncta-formation assay) stimulated by canonical induction of autophagy, such as nutrient starvation and mTOR inhibition.Citation15,Citation23,Citation27,Citation38,Citation42 Here, we further characterized WIPI-1 by using our recently established automated high through-put WIPI-1 puncta image acquisition and analysis procedureCitation38 and found that WIPI-1 regulates LC3 lipidation upstream of both autophagosomal ubiquitin-like conjugation systems. From this, PtdIns(3)P-dependent membrane localization of WIPI-1 should be functionally required for canonical phagophore and autophagosome formation, maybe by defining the autophagosomal membrane source and by permitting the recruitment of the ubiquitin-like conjugation systems for LC3 lipidation (LC3-II).
Mainly by monitoring LC3 lipidation, early evidence was provided that resveratrol induces autophagy in tumor cells.Citation12,Citation13 Here, we investigated the autophagy-inducing properties of resveratrol in further human tumor cell lines (U2OS, G361, HeLa, MCF-7) and also found that resveratrol promoted an increase of LC3-II. Further, by quantitative electron microscopy we found that the number of autophagosomes increased upon resveratrol treatment for 3 h. Because it was reported that resveratrol treatment induces Beclin 1/PtdIns3KC3-independent autophagyCitation12 we asked whether or not the PtdIns(3)P-effector WIPI-1 and the downstream Atg16L complex would participate in resveratrol-mediated autophagy.
Clearly, our results show that resveratrol does not trigger a PtdIns(3)P-dependent accumulation of WIPI-1 at autophagosomal membranes above basal level. In line with this, we also found that the Atg16L complex was not promoted to localize at autophagosomal membranes upon resveratrol treatment. We confirmed that this notion is not due to a decrease of cellular PtdIns(3)P upon resveratrol treatment, and that WIPI-2, the closest human WIPI-1 homolog does not compensate for the lack of increased WIPI-1 localization at autophagosomal membranes in this setting. However, PtdIns(3)P-dependency during resveratrol-mediated autophagy was implied by analyzing the effect of wortmannin administration in the presence of resveratrol which reduced both the amount of LC3-II protein and the number of GFP-LC3 puncta per cell. However, under circumstances of PtdIns(3)P inhibition (resveratrol/wortmannin co-treatment) resveratrol still promoted an increase of the autophagic flux, measured by both LC3-II protein increase and GFP-LC3 puncta formation. Hence resveratrol-mediated autophagy is in part sensitive to PtdIns(3)P inhibition, but PtdIns(3)P inhibition does not abolish resveratrol-mediated autophagy.
During the course of this study, these results initially indicated that resveratrol-mediated autophagy might partly induce autophagosome formation by targeting yet unidentified factors under circumstances of blocked PtdIns(3)P signaling, thereby bypassing WIPI-1/WIPI-2 and the Atg16L complex. In addition, by analyzing the localization of coexpressed WIPI-1 and DFCP1 we found that both proteins colocalize in part whenever WIPI-1 forms punctate structures; in the majority of starved cells this partial colocalization is prominent because WIPI-1 puncta formation is induced, in control settings and upon resveratrol treatment the majority of cells lack WIPI-1 puncta and here, only the few cells that display WIPI-1 puncta (basal level) partially colocalize with DFCP1. In addition, we did not find an increased number of DFCP1 puncta alone upon resveratrol treatment. From this it does not appear that WIPI-2 or DFCP1 compensate for the WIPI-1 PtdIns(3)P-effector activity at phagophores during resveratrol treatment.
Based on the above results we wished to address the question of whether or not WIPI-1 is functionally required for resveratrol-mediated autophagy, despite the lack of induced phagophore accumulation. Clearly, WIPI-1 downregulation resulted in decreased LC3 lipidation and we further showed that WIPI-1 acts upstream of both Atg7 and Atg5 to permit LC3 lipidation. Importantly, both starvation-induced and resveratrol-mediated increase in LC3-II was decreased when WIPI-1 was downregulated by transient siRNA transfections, strongly suggesting that WIPI-1 is functionally required to generate LC3-II for the resveratrol response. It was found that resveratrol-mediated autophagy depends on Atg7 and Atg5,Citation12,Citation31 and in line with this we also provide evidence that LC3-II lipidation upon resveratrol treatment depends on both Atg7 and Atg5. From this it became clear that although resveratrol administration did not promote an induced formation of phagophore structures, the WIPI-1-Atg7/5 pathway is functionally required to produce LC3-II. This result explains the effect that resveratrol treatment is in part sensitive to wortmannin, since it blocks the generation of PtdIns(3)P which is required for the localization of WIPI-1 at autophagosomal membranes.
From this we suggest that resveratrol might use LC3-II, generated by the PtdIns(3)P-WIPI-Atg7-Atg5 pathway for the formation of noncanonical autophagosomes. Considering the different membrane localization of WIPI-1 during autophagy, ER, plasma membrane and on autophagosomes,Citation28 an attractive explanation might be that resveratrol-mediated, noncanonical autophagy employs LC3-II produced via the WIPI-1 route at membranes different from canonical phagophore structures, e.g., plasma membrane. We also found that resveratrol significantly reduced the induction of newly formed WIPI-1 positive phagophores/autophagosomes, indicating that the resveratrol-mediated response counteracts canonical autophagosome formation. This could explain why the cellular response to resveratrol treatment triggers both autophagosome formation and cell death under some circumstances.Citation31 In fact, the proposed effect of resveratrol on longevityCitation43 might be due to an early elimination of transformed cells through redirecting canonical autophagy to noncanonical proteolysis that results in cell death (Fig. S5). In this regard it will be important to understand whether or not compounds such as resveratrol could be employed to subduct the cytoprotective function of autophagy in tumor cells. In addition, it will become an important issue to investigate the supply of PtdIns(3)P for autophagosome formation in more detail, as our results indicate that under some circumstances, canonical Beclin 1-mediated PtdIns3KC3 engagement might be overcome.
Materials and Methods
Compounds
Resveratrol, rapamycin, wortmannin, LY294002, staurosporine and etoposide were purchased from Sigma Aldrich (R5010, R0395, W1628, L9908, S4400, E1383, respectively), bafilomycin A1 from AppliChem (A7823).
Antibodies and dyes
The following primary antibodies were used in this study: anti-WIPI-1 (Abnova, H00055062-M02), anti-WIPI-2 (Abgent, AP9559b), anti-LC3 (NanoTools, 0231-100/LC3-5F10), anti-Atg5 (Cosmo Bio Co., CAC-TMD-PH-AT5), anti-Atg12 (Abgent, AP1816a), anti-Atg7 (Cell Signaling Technology, 2631), anti-myc raised in mouse or rabbit (Santa Cruz, sc-40 and sc-789), anti-EEA1 (BD Biosciences, 610456), anti-GAPDH (Hytest, 5G4), anti-tubulin (Sigma Aldrich, T5168). Anti-mouse IgG Alexa Fluor 546, anti-rabbit IgG Alexa Fluor 488, anti-rabbit IgG Alexa Fluor 546 were purchased from Molecular Probes (A11003, A11008, A11010, respectively). Anti-rabbit IgG-HRP from Cell Signaling Technology (7074) and anti-mouse IgG-HRP from GE Healthcare (NA931V). TO-PRO-3 was purchased from Invitrogen (T3605) and DAPI from Applichem (A4099).
cDNA constructs
GFP-LC3 was provided by Tamotsu Yoshimori (Osaka University). GFP-WIPI-1 was described earlier.Citation27 GFP-WIPI-2A and -2C were generated from initial GFP-WIPI-2 (GFP-WIPI-2B, GFP-WIPI-2D) isolatesCitation23 according to NCBI NP_056425, NP_057087, NP_001028690, NP_001028691. Myc-WIPI-2 (-2A, -2B, -2C, -2D) constructs were generated by subcloning the WIPI-2 cDNA variants into pCMV-Tag3 (HindIII/ EcoRI). Myc-DFCP1 was generated by cloning DFCP1 cDNA (imaGenes, IRATp970A0374D) into pCMV-Tag3b (BamHI/XhoI). Construct integrities were confirmed by PCR, automated DNA sequencing and by protein expression analysis.
siRNAs
Atg5 siRNA, Atg7 siRNA and the corresponding control siRNA were obtained from Cell Signaling Technology (6345, 6604, 6568, respectively). WIPI-1 siRNAs (SR310458A, B, C), WIPI-2 siRNAs (SR308719A, B, C) and the corresponding control siRNA (SR30004) were purchased from Origene.
Media and supplements
DMEM and OPTI.MEM were obtained from Invitrogen (31966, 51985, respectively), EBSS from Sigma Aldrich (E2888). The following supplements for DMEM were used: Fetal calf serum (PAA, A15-043), penicillin/streptomycin (Invitrogen, 15140), plasmocin (Invivogen, ant-mpp), doxycycline (Sigma Aldrich, D9891), G418 (Invitrogen, 11811).
Cell culture
Human U2OS, G361 and HeLa, cells (all from ATCC, HTB-96, CRL-1424, CCL-2, respectively), MCF-7 cells (Evelyne May/CEA, CNRS) and m5-7 cells (Noboru Mizushima, Tokyo Medical and Dental University) were cultured in DMEM, 10% FCS, 100 U/ml penicillin/100 μg/ml streptomycin, 5 μg/ml plasmocin. Downregulation of Atg5 protein expression in m5-7 cells was achieved by using 1 µg/ml doxycycline in the culture medium.Citation41 Stable GFP-WIPI-1 U2OSCitation38 or GFP-2xFYVE U2OS cell line (GE Healthcare) were cultured in the above culture medium in the presence of 0.6 mg/ml G418.
DNA transfection
The following transfection reagents were used in this study: Lipofectamine 2000 (Invitrogen, 11668), Promofectin (Promokine, PK-CT-2000-100) or DEAE-Dextran (Sigma Aldrich, D9885). Routinely, transient transfections were conducted in six-well plates by using 3 µg DNA and 5 µl Promofectin according to the manufacturer's protocol. Stable GFP-LC3 cell lines were generated by using 4 µg DNA and 10 µl Lipofectamine 2000 for the transfection of U2OS cells in six-well plates, followed by G418 selection (1 mg/ml). Transient transfection of the myc-DFCP1 plasmid into stable GFP-WIPI-1 U2OS cells was conducted by applying the standard DEAE-dextran procedure with 2 µg DNA and 15 µl DEAE-Dextran.Citation44
siRNA transfection
Stable GFP-WIPI-1 U2OS cells were transfected with 20 nM control siRNA, Atg5 siRNA or Atg7 siRNA and 1 µl Lipofectamine RNAiMAX (Invitrogen, 11668) in 24-well plates according to the manufacturer’s protocol. After 48 h a second transfection was conducted for a further incubation period of 24 h. G361 cells were transfected with 60 nM control siRNA, a mix of three unique WIPI-1 siRNAs (each 20 nM), or a mix of 3 unique WIPI-2 siRNAs (each 20 nM) combined with 5 µl Lipofectamine RNAiMAX in six-well plates. After 36 h a second transfection procedure was conducted and cells were incubated for a further period of 36 h.
Autophagy assays
Assays were performed in control medium (DMEM) supplemented with or without 10% FCS as described in the text. Treatments were conducted for the indicated time periods (generally for 3 h) by administration of EBSS, resveratrol (32 µM, 64 µM or 128 µM), wortmannin (233 nM), rapamycin (300 nM), staurosporine (100 nM), etoposide (5 µM), or LY294002 (100 µM or 50 µM). Co-treatments were also conducted as follows: resveratrol (64 µM) plus wortmannin (233 nM), EBSS plus wortmannin (233 nM), EBSS plus resveratrol (64 µM), EBSS plus LY294002 (100 µM). In order to inhibit the autophagic flux, 200 nM bafilomycin A1 was added either throughout the treatment period, or for the last hour of treatment as indicated.
Confocal microscopy
Fixed cells (3.7% paraformaldehyde) were prepared for direct GFP fluorescence or indirect immunofluorescence (overexpressed myc-tagged WIPI-2 isoforms or DFCP1, or endogenous Atg12) by using an LSM 510 microscope (Zeiss) and a 63 × 1.4 DIC Plan-Apochromat oil-immersion objective as previously described.Citation23 Routinely, 10–20 optical slices (0.5 µm) were acquired and final images prepared from projections of 1–5 optical slices. To obtain intensity profiles, individual optical slices were used. The number of GFP-LC3 puncta per cell was determined by using projections of three optical slices and the ImagePro-Plus 4.1 software. As previously described, GFP-WIPI-1 or Atg12 puncta-positive cells were counted by fluorescence microscopy (up to 400 cells for each treatment from three to four independent experiments).
Automated GFP image acquisition and analysis.Citation38
Stable GFP-WIPI-1, GFP-LC3 or GFP-2xFYVE U2OS cells were plated on 96-well (or 24-well) plates and assayed as indicated. Using the In Cell Analyzer 1000 (GE Healthcare) acquisition mode, automated images were acquired with a Nikon 40x Planfluor objective. To visualize DAPI stained nuclei the excitation filter D360/40 and the emission filter HQ460/40M, and to visualize GFP the excitation filter S475/20 and the emission filter HQ535/50M were used. Using the In Cell Analyzer 1000 Workstation 3.4 software specific analysis protocols were established for acquired fluorescence as follows. Applying the Dual Area Object Analysis GFP-WIPI-1 puncta were detected (sensitivity set to 5) by Inclusions and the Multiscale Top Hat algorithms and expressed as the number of GFP-WIPI-1 puncta per cell. A further stringent decision tree including Intensity (> 200) and Inclusion/Cell Intensity (> 1.15) was applied and defined the number of GFP-WIPI-1 puncta-positive cells. Using the Dual Area Object Analysis GFP-LC3 or GFP-2xFYVE puncta were detected by Inclusions and the Multiple Top Hat algorithms. Here, the sensitivity was set to 40 and the number of puncta per cell was identified. Images of GFP-2x-FYVE U2OS cells stained with EEA1/Alexa 546 were acquired using the In Cell Analyzer 1000 (GE Healthcare) and 30 cells (from 3–4 fields) were analyzed by using the ImagePro-Plus 4.1 software.
Immunoblotting
Cells were washed with PBS and lysed either in TBS/1% Triton-X100 or in 750 mM Aminocaproic acid, 50 mM Bis-Tris, 0.5 mM EDTA, 1% Triton-X100. Soluble fractions (centrifugation: 14000 rpm, 10 min, 4°C) were supplemented with Laemmli loading buffer, subjected to SDS-PAGE and blotted on to a PVDF membrane (Millipore, IPVH00010). Alternatively, total protein extracts were generated by adding boiling Laemmli buffer to the cells followed by chromatin shearing with a 23G needle. Signal intensities of standard ECL detections were quantified using the Personal Densitometer SI (Molecular Dynamics). For quatification of protein abundance, LC3-II signal intensities were normalized over tubulin or gapdh.
Quantitative electron microscopy
Upon treatments with control medium (CM), 64 µM resveratrol (RM) or nutrient-free medium (EBSS) for 3 h, GFP-WIPI-1 U2OS cells were fixed in 2.5% glutaraldehyde for 2 h, embedded in agarose and treated with 1% osmium tetroxide and 1% potassium ferricyanide for 90 min. on ice. After washing and staining with 1% aqueous uranyl acetate (60 min.), afar blocks were dehydrated in a graded series of ethanol and embedded in exposy resin. Ultrathin resin sections were analyzed using a LEO 906 transmission electron microscope.Citation27 Sections from two different epoxy resin blocks (per sample) were analyzed (total of 72–87 cells per treatment). Multilayered autophagosomal vesicles (AV) were identified and counted at a magnification of 16,700x, and representative images were taken. Overviews of the counted cells were generated (400× magnification) and a raster (25 µm2) was applied for point counting.Citation35,Citation37
Statistical analysis
Statistical significance was evaluated using two-tailed heteroscedastic t-testing and p-values were calculated.
| Abbreviations: | ||
| WIPI | = | WD-repeat protein interacting with phosphoinositides |
| PtdIns(3)P | = | phosphatidylinositol 3-phosphate |
| LC3 | = | microtubule-associated protein 1 light chain 3 |
| PtdIns3KC3 | = | phosphatidylinositol 3-kinase class III |
| mTOR | = | mammalian target of rapamycin |
| RAG | = | Ras-related GTP-binding protein |
| FYVE | = | Fab1 YOTB Vac1p EEA1 domain |
| EEA1 | = | early endosome antigen 1 |
| EBSS | = | Earl’s balanced salt solution |
Additional material
Download Zip (4.3 MB)Acknowledgments
We thank Noboru Mizushima for providing the m5-7 cells and Tamotsu Yoshimori for sharing the pEGFP-LC3 construct. We acknowledge technical assistance from Sabine Ruckerbauer and Boris Jakuschkin. The Codogno laboratory received grant support from institutional fundings from INSERM, University Paris-Sud 11 and grants from the ANR (Agence Nationale de la Recherche and INCa (Institut National du Cancer). The Proikas-Cezanne laboratory received support from the Ministry of Science, Research and Arts Baden-Wuerttemberg (Landesgraduiertenförderung, predoctoral fellowship to M.M.); and grants from the Federal Ministry for Education and Science (BMBF, BioProfile) and the German Research Society (DFG, SFB 773) to T.P.-C.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069 - 75; http://dx.doi.org/10.1038/nature06639; PMID: 18305538
- Weidberg H, Shvets E, Elazar Z. Biogenesis and Cargo Selectivity of Autophagosomes. Annu Rev Biochem 2010; http://dx.doi.org/10.1146/annurev-biochem-052709-094552PMID: 21548784
- Klionsky DJ, Cregg JM, Dunn WA Jr., Emr SD, Sakai Y, Sandoval IV, et al. A unified nomenclature for yeast autophagy-related genes. Dev Cell 2003; 5:539 - 45; http://dx.doi.org/10.1016/S1534-5807(03)00296-X; PMID: 14536056
- Kawamata T, Kamada Y, Kabeya Y, Sekito T, Ohsumi Y. Organization of the pre-autophagosomal structure responsible for autophagosome formation. Mol Biol Cell 2008; 19:2039 - 50; http://dx.doi.org/10.1091/mbc.E07-10-1048; PMID: 18287526
- Ohsumi Y, Mizushima N. Two ubiquitin-like conjugation systems essential for autophagy. Semin Cell Dev Biol 2004; 15:231 - 6; http://dx.doi.org/10.1016/j.semcdb.2003.12.004; PMID: 15209383
- Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science 2004; 306:990 - 5; http://dx.doi.org/10.1126/science.1099993; PMID: 15528435
- Meijer AJ, Codogno P. Signalling and autophagy regulation in health, aging and disease. Mol Aspects Med 2006; 27:411 - 25; http://dx.doi.org/10.1016/j.mam.2006.08.002; PMID: 16973212
- Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell 2008; 19:5360 - 72; http://dx.doi.org/10.1091/mbc.E08-01-0080; PMID: 18843052
- Meijer AJ, Codogno P. Regulation and role of autophagy in mammalian cells. Int J Biochem Cell Biol 2004; 36:2445 - 62; http://dx.doi.org/10.1016/j.biocel.2004.02.002; PMID: 15325584
- Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL. Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol 2008; 10:935 - 45; http://dx.doi.org/10.1038/ncb1753; PMID: 18604198
- Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008; 320:1496 - 501; http://dx.doi.org/10.1126/science.1157535; PMID: 18497260
- Scarlatti F, Maffei R, Beau I, Codogno P, Ghidoni R. Role of non-canonical Beclin 1-independent autophagy in cell death induced by resveratrol in human breast cancer cells. Cell Death Differ 2008; 15:1318 - 29; http://dx.doi.org/10.1038/cdd.2008.51; PMID: 18421301
- Puissant A, Robert G, Fenouille N, Luciano F, Cassuto JP, Raynaud S, et al. Resveratrol promotes autophagic cell death in chronic myelogenous leukemia cells via JNK-mediated p62/SQSTM1 expression and AMPK activation. Cancer Res 2010; 70:1042 - 52; http://dx.doi.org/10.1158/0008-5472.CAN-09-3537; PMID: 20103647
- Bhutia SK, Kegelman TP, Das SK, Azab B, Su ZZ, Lee SG, et al. Astrocyte elevated gene-1 induces protective autophagy. Proc Natl Acad Sci USA 2010; 107:22243 - 8; http://dx.doi.org/10.1073/pnas.1009479107; PMID: 21127263
- Gao P, Bauvy C, Souquere S, Tonelli G, Liu L, Zhu Y, et al. The Bcl-2 homology domain 3 mimetic gossypol induces both Beclin 1-dependent and Beclin 1-independent cytoprotective autophagy in cancer cells. J Biol Chem 2010; 285:25570 - 81; http://dx.doi.org/10.1074/jbc.M110.118125; PMID: 20529838
- Lépine S, Allegood JC, Park M, Dent P, Milstien S, Spiegel S. Sphingosine-1-phosphate phosphohydrolase-1 regulates ER stress-induced autophagy. Cell Death Differ 2011; 18:350 - 61; http://dx.doi.org/10.1038/cdd.2010.104; PMID: 20798685
- Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, et al. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009; 461:654 - 8; http://dx.doi.org/10.1038/nature08455; PMID: 19794493
- Scarlatti F, Maffei R, Beau I, Ghidoni R, Codogno P. Non-canonical autophagy: an exception or an underestimated form of autophagy?. Autophagy 2008; 4:1083 - 5; PMID: 18849663
- Smith DM, Patel S, Raffoul F, Haller E, Mills GB, Nanjundan M. Arsenic trioxide induces a beclin-1-independent autophagic pathway via modulation of SnoN/SkiL expression in ovarian carcinoma cells. Cell Death Differ 2010; 17:1867 - 81; http://dx.doi.org/10.1038/cdd.2010.53; PMID: 20508647
- Tian S, Lin J, Jun Zhou J, Wang X, Li Y, Ren X, et al. Beclin 1-independent autophagy induced by a Bcl-XL/Bcl-2 targeting compound, Z18. Autophagy 2010; 6:1032 - 41; http://dx.doi.org/10.4161/auto.6.8.13336; PMID: 20818185
- Wong CH, Iskandar KB, Yadav SK, Hirpara JL, Loh T, Pervaiz S. Simultaneous induction of non-canonical autophagy and apoptosis in cancer cells by ROS-dependent ERK and JNK activation. PLoS ONE 2010; 5:e9996; http://dx.doi.org/10.1371/journal.pone.0009996; PMID: 20368806
- Zhu JH, Horbinski C, Guo F, Watkins S, Uchiyama Y, Chu CT. Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyl-4-phenylpyridinium-induced cell death. Am J Pathol 2007; 170:75 - 86; http://dx.doi.org/10.2353/ajpath.2007.060524; PMID: 17200184
- Proikas-Cezanne T, Waddell S, Gaugel A, Frickey T, Lupas A, Nordheim A. WIPI-1alpha (WIPI49), a member of the novel 7-bladed WIPI protein family, is aberrantly expressed in human cancer and is linked to starvation-induced autophagy. Oncogene 2004; 23:9314 - 25; http://dx.doi.org/10.1038/sj.onc.1208331; PMID: 15602573
- Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 2010; 6:764 - 76; http://dx.doi.org/10.4161/auto.6.6.12709; PMID: 20639694
- Jeffries TR, Dove SK, Michell RH, Parker PJ. PtdIns-specific MPR pathway association of a novel WD40 repeat protein, WIPI49. Mol Biol Cell 2004; 15:2652 - 63; http://dx.doi.org/10.1091/mbc.E03-10-0732; PMID: 15020712
- Polson HE, de Lartigue J, Rigden DJ, Reedijk M, Urbe S, Clague MJ, et al. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 2010; 6:506 - 22; http://dx.doi.org/10.4161/auto.6.4.11863; PMID: 20505359
- Proikas-Cezanne T, Ruckerbauer S, Stierhof YD, Berg C, Nordheim A. Human WIPI-1 puncta-formation: a novel assay to assess mammalian autophagy. FEBS Lett 2007; 581:3396 - 404; http://dx.doi.org/10.1016/j.febslet.2007.06.040; PMID: 17618624
- Proikas-Cezanne T, Robenek H. Freeze-fracture replica immunolabelling reveals human WIPI-1 and WIPI-2 as membrane proteins of autophagosomes. J Cell Mol Med 2011; 15:2007-10.
- Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol 2008; 182:685 - 701; http://dx.doi.org/10.1083/jcb.200803137; PMID: 18725538
- Matsunaga K, Morita E, Saitoh T, Akira S, Ktistakis NT, Izumi T, et al. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J Cell Biol 2010; 190:511 - 21; http://dx.doi.org/10.1083/jcb.200911141; PMID: 20713597
- Morselli E, Marino G, Bennetzen MV, Eisenberg T, Megalou E, Schroeder S, et al. Spermidine and resveratrol induce autophagy by distinct pathways converging on the acetylproteome. J Cell Biol 2011; 192:615 - 29; http://dx.doi.org/10.1083/jcb.201008167; PMID: 21339330
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19:5720 - 8; http://dx.doi.org/10.1093/emboj/19.21.5720; PMID: 11060023
- Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy 2007; 3:542 - 5; PMID: 17611390
- Rubinsztein DC, Cuervo AM, Ravikumar B, Sarkar S, Korolchuk V, Kaushik S, et al. In search of an “autophagomometer”. Autophagy 2009; 5:585 - 9; http://dx.doi.org/10.4161/auto.5.5.8823; PMID: 19411822
- Bahmer FA, Schwichtenberg U. Efficient estimation of ulcer and wound area by point counting. Phlebologie 2000; 29:33 - 6
- Demarchi F, Bertoli C, Copetti T, Tanida I, Brancolini C, Eskelinen EL, et al. Calpain is required for macroautophagy in mammalian cells. J Cell Biol 2006; 175:595 - 605; http://dx.doi.org/10.1083/jcb.200601024; PMID: 17101693
- Eskelinen EL, Schmidt CK, Neu S, Willenborg M, Fuertes G, Salvador N, et al. Disturbed cholesterol traffic but normal proteolytic function in LAMP-1/LAMP-2 double-deficient fibroblasts. Mol Biol Cell 2004; 15:3132 - 45; http://dx.doi.org/10.1091/mbc.E04-02-0103; PMID: 15121881
- Grotemeier A, Alers S, Pfisterer SG, Paasch F, Daubrawa M, Dieterle A, et al. AMPK-independent induction of autophagy by cytosolic Ca2+ increase. Cell Signal 2010; 22:914 - 25; http://dx.doi.org/10.1016/j.cellsig.2010.01.015; PMID: 20114074
- Gaullier JM, Ronning E, Gillooly DJ, Stenmark H. Interaction of the EEA1 FYVE finger with phosphatidylinositol 3-phosphate and early endosomes. Role of conserved residues. J Biol Chem 2000; 275:24595 - 600; http://dx.doi.org/10.1074/jbc.M906554199; PMID: 10807926
- Gaullier JM, Simonsen A, D'Arrigo A, Bremnes B, Stenmark H, Aasland R. FYVE fingers bind PtdIns(3)P. Nature 1998; 394:432 - 3; http://dx.doi.org/10.1038/28767; PMID: 9697764
- Hosokawa N, Hara Y, Mizushima N. Generation of cell lines with tetracycline-regulated autophagy and a role for autophagy in controlling cell size. FEBS Lett 2006; 580:2623 - 9; http://dx.doi.org/10.1016/j.febslet.2006.04.008; PMID: 16647067
- Vergne I, Roberts E, Elmaoued RA, Tosch V, Delgado MA, Proikas-Cezanne T, et al. Control of autophagy initiation by phosphoinositide 3-phosphatase Jumpy. EMBO J 2009; 28:2244 - 58; http://dx.doi.org/10.1038/emboj.2009.159; PMID: 19590496
- Morselli E, Maiuri MC, Markaki M, Megalou E, Pasparaki A, Palikaras K, et al. Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis 2010; 1:e10; http://dx.doi.org/10.1038/cddis.2009.8; PMID: 21364612
- Cullen BR. Use of eukaryotic expression technology in the functional analysis of cloned genes. Methods Enzymol 1987; 152:684 - 704; http://dx.doi.org/10.1016/0076-6879(87)52074-2; PMID: 3657593