Abstract
The role of intracellular Ca2+ signaling in starvation-induced autophagy remains unclear. Here, we examined Ca2+ dynamics during starvation-induced autophagy and the underlying molecular mechanisms. Tightly correlating with autophagy stimulation, we observed a remodeling of the Ca2+ signalosome. First, short periods of starvation (1 to 3 h) caused a prominent increase of the ER Ca2+-store content and enhanced agonist-induced Ca2+ release. The mechanism involved the upregulation of intralumenal ER Ca2+-binding proteins, calreticulin and Grp78/BiP, which increased the ER Ca2+-buffering capacity and reduced the ER Ca2+ leak. Second, starvation led to Ins(1,4,5)P3R sensitization. Immunoprecipitation experiments showed that during starvation Beclin 1, released from Bcl-2, first bound with increasing efficiency to Ins(1,4,5)P3Rs; after reaching a maximal binding after 3 h, binding, however, decreased again. The interaction site of Beclin 1 was determined to be present in the N-terminal Ins(1,4,5)P3-binding domain of the Ins(1,4,5)P3R. The starvation-induced Ins(1,4,5)P3R sensitization was abolished in cells treated with BECN1 siRNA, but not with ATG5 siRNA, pointing toward an essential role of Beclin 1 in this process. Moreover, recombinant Beclin 1 sensitized Ins(1,4,5)P3Rs in 45Ca2+-flux assays, indicating a direct regulation of Ins(1,4,5)P3R activity by Beclin 1. Finally, we found that Ins(1,4,5)P3R-mediated Ca2+ signaling was critical for starvation-induced autophagy stimulation, since the Ca2+ chelator BAPTA-AM as well as the Ins(1,4,5)P3R inhibitor xestospongin B abolished the increase in LC3 lipidation and GFP-LC3-puncta formation. Hence, our results indicate a tight and essential interrelation between intracellular Ca2+ signaling and autophagy stimulation as a proximal event in response to starvation.
Introduction
Autophagy is an evolutionarily conserved process for delivery of cellular material, including long-lived proteins, lipids and organelles, to lysosomes for degradation. In this manner, cells can recycle building blocks essential for survival during stress responses like nutrient starvation.Citation1 Consequently, nutrient starvation leads to a stimulation of autophagy. In addition, basal autophagy prevents the accumulation of protein aggregates and damaged organelles, thereby maintaining cellular homeostasis (termed “quality control” autophagy).Citation2 Because of its importance in cellular life and death responses, an autophagic deficiency leads to various diseases.Citation3
Different ways of material delivery determine the various mechanisms of autophagy, e.g., microautophagy, chaperone-mediated autophagy and macroautophagy.Citation4 The latter process (hereafter referred to as autophagy) consists of the formation and elongation of a double-membranous structure (phagophore) into an entire vesicle (autophagosome), thereby enclosing cellular material. These vesicles eventually fuse with lysosomes to form autolysosomes. The degradation of the enclosed material is executed by the lysosomal enzymes. At the molecular level, many evolutionarily conserved genes, the ATG genes, regulate the different steps of this complex process, from the signaling to the final fusion.Citation1 One very important member is yeast ATG6, whose mammalian ortholog is the haploinsufficient tumor suppressor gene BECN1.Citation5 The protein Beclin 1 is known to dimerize and to interact with many proteins including Vps34, Vps15, UVRAG, Ambra1 and Bif1 to form the phosphatidylinositol (PtdIns) 3-kinase complex III.Citation6,Citation7 This complex phosphorylates PtdIns to PtdIns 3-phosphate [PtdIns(3)P] at the initial phagophore, which serves as a recruitment signal for other Atg proteins.Citation8 The presence of Beclin 1 is essential for the activity of this complex.Citation7 Hence, Beclin 1 depletion leads to autophagic deficiency.Citation9 Interestingly, Beclin 1 is a member of the pro-apoptotic BH3-only protein family, although it is not implicated in apoptosis.Citation10 Its BH3 domain mediates an interaction with Bcl-2, Bcl-XL and Bcl-w,Citation11 hereby preventing the activation of the autophagic machinery. In this manner, besides their anti-apoptotic activity, Bcl-2-family members also inhibit autophagy. The Bcl-2-Beclin 1-protein complex is dynamically regulated by phosphorylation of Bcl-2 by c-Jun N-terminal protein kinase 1 (JNK1), or phosphorylation of Beclin 1 by death-associated protein kinase (DAPK).Citation12,Citation13 Also, other proteins like high motility group box 1 (HMGB1) or nutrient-deprivation autophagy factor 1 (NAF-1) modulate the binding of Beclin 1 to Bcl-2 and the onset of autophagy.Citation14,Citation15
Recently, several studies have implicated a role for intracellular Ca2+ signaling and inositol 1,4,5-trisphosphate (Ins(1,4,5)P3) receptors (Ins(1,4,5)P3Rs) in autophagy.Citation16 Ins(1,4,5)P3Rs are tetrameric ER-resident Ca2+ channels, which release Ca2+ from the ER to the cytosol in response to Ins(1,4,5)P3.Citation17 These ubiquitously expressed channels control a wide range of cellular processes, including cell development, proliferation and death.Citation18-Citation21 Downstream effects of intracellular Ca2+-release events depend on the spatiotemporal characteristics of the Ca2+ signal.Citation22 Recently, it has been shown that knockdown or chemical inhibition of Ins(1,4,5)P3Rs, or depletion of Ins(1,4,5)P3 induce autophagy.Citation23-Citation25 In this respect, Ins(1,4,5)P3Rs seem to be essential for a constitutive Ca2+ release from the ER to mitochondria to support mitochondrial bioenergetics.Citation26 As a result, depletion or inhibition of Ins(1,4,5)P3Rs will result in a decline in the Ca2+-dependent production of ATP, an increase in the AMP/ATP ratio and subsequent activation of the AMP-activated protein kinase (AMPK), leading to stimulation of autophagy.Citation26 Although this latter study reveals how intracellular, and particularly mitochondrial, Ca2+ signals suppress basal autophagy, other reports indicate that elevating cytosolic Ca2+ concentrations ([Ca2+]cyt) cause activation of autophagy.Citation27-Citation30 Finally, besides these functional links between Ca2+ and autophagy, Beclin 1 was recently shown to interact with the Ins(1,4,5)P3R.Citation31
However, the role of Ca2+ signaling during autophagy stimulation remains unclear. Therefore, we investigated (i) whether Ca2+ signaling is affected during the initial phase of starvation-induced autophagy, (ii) the molecular determinants underpinning the changes in intracellular Ca2+ signaling, and (iii) whether the changes in intracellular Ca2+ dynamics are essential for starvation-induced autophagy. We report here a remodeling of the Ca2+-signalosome during stimulation of autophagy at two levels: (i) an increased ER Ca2+-store content by increased levels of intralumenal Ca2+-binding proteins (CaBPs), and (ii) a sensitization of the Ins(1,4,5)P3R toward low and medium [Ins(1,4,5)P3] by enhanced binding of Beclin 1 to the Ins(1,4,5)P3R. This latter result suggests a novel role for Ins(1,4,5)P3R-Beclin 1 complexes in starvation-induced autophagy, in which Beclin 1 dynamically regulates Ins(1,4,5)P3R Ca2+-release activity.
Results
Starvation induces transient changes in Ca2+ dynamics, correlating with a transient stimulation of autophagy
We examined intracellular Ca2+ homeostasis and dynamics in HeLa cells that were exposed to starvation through incubation in HBSS for different time periods (2, 3 or 5 h). The ER Ca2+-store content was determined by measuring the Ca2+ released in response to 1 µM thapsigargin (TG), an irreversible inhibitor of the sarco- and endoplasmic reticulum Ca2+ ATPase (SERCA) pump, thereby resulting in a net Ca2+ leak from the ER.Citation32 Alternatively, we have used 10 µM ionomycin, a Ca2+ ionophore, to release all the stored Ca2+. EGTA (3 mM) was added to chelate all extracellular Ca2+. Importantly, the amplitude of the Fura-2 signal in response to TG or ionomycin was augmented after 2 or 3 h of starvation, but declined again after 5 h (). Similar transient changes in Ca2+ release were observed when the cells were exposed to low (0.3 µM) and high (100 µM) doses of ATP (). ATP binding to its metabotropic purinergic receptor leads to Ins(1,4,5)P3 production and subsequent Ins(1,4,5)P3R-mediated Ca2+ release from the ER into the cytosol. A similar increase in Ca2+ signaling was observed upon starvation of two noncancer cell lines, mouse embryonic fibroblasts (MEF cells; Fig. S1A) or a mouse L fibroblast cell line overexpressing Ins(1,4,5)P3R1 (L15 cells; Fig. S1C), two cell lines frequently used to study Ins(1,4,5)P3R-dependent signaling.Citation33-Citation35 Interestingly, in all cell types, these early transient changes in Ca2+ signaling correlated well with the stimulation of autophagy, monitored by following the lipidation of LC3, the mammalian ortholog of yeast Atg8, via western blotting (, Fig. S1B and S1D). The lipidated form (LC3-II) migrates faster on a gel (around 16 kDa) than the native protein (LC3-I, 18 kDa).Citation36 The detection of LC3-I, reported to be less immunoreactive and less stable than LC3-II,Citation37 was only visible after longer exposures (data not shown). In all experiments, autophagy was quantified as the ratio of LC3-II over GAPDH, the loading control, as recommended.Citation36,Citation37 The presence of LC3-II was maximal between the first and third hour after starvation in HeLa cells (), as previously described,Citation38,Citation39 while this occurred after 5 h in MEF cells (Fig. S1B) and after around 1.5 h of starvation in L15 cells (Fig. S1D). The transient effect of LC3 lipidation was confirmed by quantification of GFP-LC3 puncta in HeLa cells transfected with a GFP-LC3 construct (). Similar to the results obtained by LC3-II-western blotting, 3 h of starvation provoked a maximal amount of autophagic cells (). The number of autophagic cells in control conditions was relatively high (~35%) (), a feature not observed with LC3-II-western blotting (). This difference is probably due to the transfection procedure, which is known to provoke autophagy in basal conditions.Citation37,Citation40
Figure 1. Transient changes in Ca2+ dynamics after starvation correlate with changes of the LC3-II levels. (A) Measurements of cytosolic Ca2+ signals, displayed as normalized Fura-2 ratio (R/R0), showing the effect of 1 µM thapsigargin (TG) or 10 µM ionomycin in intact HeLa cells with (2, 3, 5 h) or without (0 h) starvation. Representative recordings of the Fura-2 ratio after addition of TG (left panel) or ionomycin (right panel) in cells pretreated with HBSS for 0, 2, 3 or 5 h are shown. Forty-five seconds prior to addition of TG or ionomycin, EGTA (3 mM) was given as indicated. (B) Measurements of cytosolic Ca2+ signals, displayed as normalized Fura-2 ratio (R/R0), after addition of 0.3 µM (left panel) or 100 µM (right panel) ATP in intact HeLa cells with (2, 3, 5 h) or without (0 h) starvation. (C) Representative LC3-II western blots of HeLa cells starved for the indicated time period (0–7 h). (D) GFP-LC3-puncta formation in HeLa cells transfected with GFP-LC3 constructs. Cells were starved (3 h or 5 h) or not (0 h), as indicated. Left: Representative pictures. The scale bar represents 20 µm. Right: Quantification of autophagic cells. Only cells displaying more than 10 puncta were considered autophagic (n = 3). (E) Analysis of XBP-1-mRNA splicing in cells pretreated with HBSS (0–5 h) or 2 µg/ml tunicamycin (TM; 24 h, 48 h); uXBP-1 = unspliced XBP-1; sXBP-1 = spliced XBP-1. (F) Representative caspase 3 western blots of HeLa cells starved for the indicated time (0–5 h) or treated with 1 µM staurosporine (STS) for 5 h. (G) XTT-assay for measurement of cell viability, after incubation in HBSS for the indicated time period or treatment with 1 µM staurosporine for 5 h (n = 4). The cell viability is expressed as the absorbance at 490 nm minus the absorbance at 630 nm (A490-A630). *p < 0.05; **p < 0.01 (paired t-test).
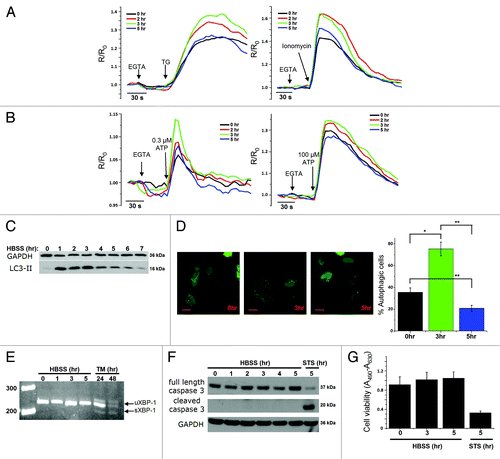
To examine whether other processes, like the adaptive ER-stress response or apoptosis, were involved in this early cellular response toward starvation, we monitored XBP-1-mRNA splicing, an early event in the IRE1-dependent ER-stress pathway as an adaptive event in the unfolded protein response,Citation41 and caspase-3 cleavage, a critical event in the execution of apoptosis. While tunicamycin-treated (2 μg ml−1, 24–48 h) cells displayed a clear increase in XBP-1-mRNA splicing, 5 h of starvation did not lead to an adaptive ER-stress response (). Similarly, while staurosporine (1 μM, 5 h) led to a clear increase in caspase-3 cleavage, 5 h of starvation did not (). In agreement with these observations, we found that these starvation conditions did not influence the total cell viability, as indicated by the XTT assay (). Therefore, our experimental conditions suggest a correlation between changes in intracellular Ca2+ dynamics and the stimulation of autophagy.
Increased ER Ca2+-store content in response to starvation is mediated by elevated levels of ER Ca2+-binding proteins and a reduction in the ER Ca2+-leak rate
Next, we focused on HeLa cells, either not starved or starved for 3 h, to evaluate the effects on the Ca2+-store content. Therefore, we calibrated the F340/F380 ratio obtained for 0 h and 3 h starvation in to [Ca2+]cyt after using the fluorescence ratios in the presence of high [EGTA], a Ca2+ buffer (Rmin), and in the presence of saturating [Ca2+] (Rmax) determined in each experiment. This procedure completely excludes that the observed changes in fluorescent signal in starved HeLa cells are artifacts caused by bleaching or changes in cell shape, caused by HBSS administration. This revealed a significantly increased Ca2+ release (~1.4 fold) into the cytosol in cells starved for 3 h and treated with either TG or ionomycin (). Similar observations were made with an independent experimental approach, using unidirectional 45Ca2+-flux assays in saponin-permeabilized HeLa cells starved for 3 h as compared with non-starved cells. Here, we determined the Ca2+-store content by measuring the Ca2+-ionophore-releasable Ca2+. The addition of 10 µM A23187 caused a significantly larger release of 45Ca2+ in cells pretreated for 3 h with HBSS ().
Figure 2. Increased Ca2+-store content caused by upregulation of Ca2+-binding proteins (CaBP) and decreased ER Ca2+-leak rate during starvation. (A) Fura-2 measurements, calibrated for [Ca2+]cyt in HeLa cells with (3 h) or without (0 h) starvation, after addition of 1 µM TG (left) or 10 µM ionomycin (right). (B) Quantitative analysis of the increase of [Ca2+]cyt (Δ[Ca2+]cyt; assessed by subtracting resting value from peak value) after addition of TG (left) or ionomycin (right) in cells with (3 h) or without (0 h) starvation (n = 7). (C) Unidirectional 45Ca2+-flux experiments in permeabilized cells pretreated with (3 h) or without (0 h) HBSS. Left: Ca2+ content is shown as a function of time of efflux. The single arrow indicates the addition of 10 µM A23187 (triangles); the double arrow represents the amount of Ca2+ released by A23187. Right: Quantification of the Ca2+ released by A23187 (n = 8). (D) Western blot analysis of SERCA2b from lysates of cells pretreated with HBSS for 0, 1, 3 or 5 h. Upper: Representative blots. Lower: Quantitative analysis of protein/GAPDH ratio normalized to control (0 h) conditions (n = 4). (E) Western blot analysis for CaBP proteins: Grp78/BiP (left) and calreticulin. (F) Decrease in ER Ca2+ content as a function of time in permeabilized cells pretreated with (3 h) or without (0 h) HBSS. Traces were normalized and expressed as percentage of the initial Ca2+ content. The ER Ca2+-leak rate is presented as the decline of the ER 45Ca2+-store content as a function of time plotted on a logarithmic scale (n = 4). *p < 0.05; **p < 0.01; ***p < 0.001 (paired t-test).
![Figure 2. Increased Ca2+-store content caused by upregulation of Ca2+-binding proteins (CaBP) and decreased ER Ca2+-leak rate during starvation. (A) Fura-2 measurements, calibrated for [Ca2+]cyt in HeLa cells with (3 h) or without (0 h) starvation, after addition of 1 µM TG (left) or 10 µM ionomycin (right). (B) Quantitative analysis of the increase of [Ca2+]cyt (Δ[Ca2+]cyt; assessed by subtracting resting value from peak value) after addition of TG (left) or ionomycin (right) in cells with (3 h) or without (0 h) starvation (n = 7). (C) Unidirectional 45Ca2+-flux experiments in permeabilized cells pretreated with (3 h) or without (0 h) HBSS. Left: Ca2+ content is shown as a function of time of efflux. The single arrow indicates the addition of 10 µM A23187 (triangles); the double arrow represents the amount of Ca2+ released by A23187. Right: Quantification of the Ca2+ released by A23187 (n = 8). (D) Western blot analysis of SERCA2b from lysates of cells pretreated with HBSS for 0, 1, 3 or 5 h. Upper: Representative blots. Lower: Quantitative analysis of protein/GAPDH ratio normalized to control (0 h) conditions (n = 4). (E) Western blot analysis for CaBP proteins: Grp78/BiP (left) and calreticulin. (F) Decrease in ER Ca2+ content as a function of time in permeabilized cells pretreated with (3 h) or without (0 h) HBSS. Traces were normalized and expressed as percentage of the initial Ca2+ content. The ER Ca2+-leak rate is presented as the decline of the ER 45Ca2+-store content as a function of time plotted on a logarithmic scale (n = 4). *p < 0.05; **p < 0.01; ***p < 0.001 (paired t-test).](/cms/asset/5ec2d4ab-5051-4692-aa21-ce1415158a16/kaup_a_10917909_f0002.gif)
To elucidate the underlying molecular mechanisms responsible for the increased Ca2+-store content, we evaluated different parameters related to ER function. ER stress and subsequent ER remodeling were already excluded (). Hence, we analyzed the levels of several proteins important in cellular Ca2+ handling: the ER Ca2+ pump SERCA and the ER CaBPs, Grp78/BiP and calreticulin (CRT). The level of SERCA2b, the major isoform in these cells, was detected using an antibody specific for SERCA2b and was found to rapidly decline (-47 ± 5%) after 1 h of starvation (). A decrease in SERCA2b can obviously not account for the increased Ca2+-store content. In contrast, Grp78/BiP and CRT levels were significantly elevated after 3 h of starvation (Grp78/BiP: +38 ± 10%; CRT: +46 ± 8%; ). A similar increase was observed in mouse L15 cells (Fig. S1E). Hence, an increased Ca2+-buffering capacity can contribute to the increased ER Ca2+-store content after 3 h of starvation. Finally, we assessed the unidirectional ER Ca2+-leak rate by measuring the rate of decline in the normalized Ca2+ content as a function of time, plotted on a logarithmic scale, as previously described.Citation42 In cells starved for 3 h, the unidirectional ER Ca2+-leak rate was clearly reduced in comparison to the leak rate in control conditions ().
Taken together, the increased Ca2+-store content is likely due to an upregulation of intraluminal CaBPs concomitant with a decline in the ER Ca2+-leak rate.
Starvation induces Ins(1,4,5)P3R sensitization
Since changes in the steady-state ER Ca2+ content likely affect agonist-induced Ca2+ signals, we examined the response of non-starved and starved HeLa cells toward ATP. indicates that Fura-2 fluorescence (excitation ratio 340 nm/380 nm) in response to ATP was enhanced in starved cells. We verified whether these effects were confirmed after calibration of the Fura-2 fluorescence toward [Ca2+]cyt, which indicated a significant increase in the Ca2+ response to low (~1.4 fold) and high (~1.6 fold) [ATP] in cells starved for 3 h ().
Figure 3. Sensitization of the Ins(1,4,5)P3R toward Ins(1,4,5)P3 during starvation. (A) Mean traces of Fura-2 measurements, calibrated for [Ca2+]cyt in HeLa cells with (3 h) or without (0 h) starvation, after addition of 0.3 µM (full line) or 100 µM (dashed line) ATP. (B) Quantitative analysis of the increase of [Ca2+]cyt (Δ[Ca2+]cyt; assessed by subtracting resting value from peak value) after addition of 0.3 µM (left) or 100 µM (right) ATP (n = 7). (C) Unidirectional 45Ca2+-flux experiments in permeabilized cells pretreated with (3 h) or without (0 h) HBSS. Mean fractional 45Ca2+ release (%/2 min) is shown as a function of time. The effect of 0.7 µM Ins(1,4,5)P3 (circles), 10 µM A23187 (triangles) or no addition (squares) are shown. The arrow indicates the addition of Ins(1,4,5)P3 or A23187. (D) Quantitative analysis of the Ins(1,4,5)P3-induced 45Ca2+ release relative to the A23187-induced 45Ca2+ release for the indicated concentrations of Ins(1,4,5)P3 in cells pretreated with (3 h) or without (0 h) HBSS (n = 8). **p < 0.01; N.S. not significant (paired t-test).
![Figure 3. Sensitization of the Ins(1,4,5)P3R toward Ins(1,4,5)P3 during starvation. (A) Mean traces of Fura-2 measurements, calibrated for [Ca2+]cyt in HeLa cells with (3 h) or without (0 h) starvation, after addition of 0.3 µM (full line) or 100 µM (dashed line) ATP. (B) Quantitative analysis of the increase of [Ca2+]cyt (Δ[Ca2+]cyt; assessed by subtracting resting value from peak value) after addition of 0.3 µM (left) or 100 µM (right) ATP (n = 7). (C) Unidirectional 45Ca2+-flux experiments in permeabilized cells pretreated with (3 h) or without (0 h) HBSS. Mean fractional 45Ca2+ release (%/2 min) is shown as a function of time. The effect of 0.7 µM Ins(1,4,5)P3 (circles), 10 µM A23187 (triangles) or no addition (squares) are shown. The arrow indicates the addition of Ins(1,4,5)P3 or A23187. (D) Quantitative analysis of the Ins(1,4,5)P3-induced 45Ca2+ release relative to the A23187-induced 45Ca2+ release for the indicated concentrations of Ins(1,4,5)P3 in cells pretreated with (3 h) or without (0 h) HBSS (n = 8). **p < 0.01; N.S. not significant (paired t-test).](/cms/asset/b3d3608f-21ff-44df-a654-9f33221334d3/kaup_a_10917909_f0003.gif)
To assess the role of the Ins(1,4,5)P3R in this enhanced agonist-induced Ca2+ signaling, we examined the Ins(1,4,5)P3R Ca2+-flux properties in saponin-permeabilized HeLa cells, in which the nonmitochondrial Ca2+ stores have been loaded with 45Ca2+ to steady-state. This approach, performed in 12-well plates, allows a direct access to the Ins(1,4,5)P3R and a very accurate assessment of its Ca2+-release activity under unidirectional Ca2+-efflux conditions in the absence of SERCA Ca2+-uptake activity and of mitochondrial Ca2+ fluxes.Citation43,Citation44 Importantly, Ins(1,4,5)P3 is added when the ER Ca2+ stores still contain ~50% of their maximal level, precluding any significant Ins(1,4,5)P3R inhibition by store depletion.Citation45 Here, we directly compared in paired experiments untreated cells and cells starved for 3 h before experimental analysis. In agreement with the results obtained in intact cells, the Ins(1,4,5)P3-induced Ca2+ release was potentiated after 3 h starvation (). To reveal whether Ins(1,4,5)P3Rs have been sensitized toward Ins(1,4,5)P3 independently of the increased ER Ca2+ store, we normalized the values obtained for the Ins(1,4,5)P3R-dependent Ca2+ release relative to ER Ca2+-store content (as given by the Ca2+-ionophore-releasable Ca2+). The data presented in show that Ins(1,4,5)P3-induced Ca2+ release relative to the total releasable Ca2+ at that moment, was increased in HBSS-pretreated cells for submaximal doses of Ins(1,4,5)P3 (≤ 3 µM), but not for saturating doses (30 µM) (). This indicates that the Ins(1,4,5)P3R is sensitized during early autophagy stimulation triggered by 3 h of starvation.
Beclin 1 is essential for Ins(1,4,5)P3R sensitization during starvation, independently of its Bcl-2-binding site
To investigate whether the interaction of Beclin 1 with the Ins(1,4,5)P3R is responsible for Ins(1,4,5)P3R sensitization, we examined whether Ins(1,4,5)P3R-Beclin 1 complexes were formed in HeLa cells during the initial response toward starvation. HeLa cells predominantly express Ins(1,4,5)P3R3 (57%) and Ins(1,4,5)P3R1 (28%).Citation46 Using western blot analysis, we analyzed the presence of Beclin 1 in immunopurified Ins(1,4,5)P3R1 and Ins(1,4,5)P3R3 complexes. This analysis revealed an interaction between Beclin 1 and the Ins(1,4,5)P3R, which became much more pronounced in cells starved for 3 h (), indicating that Ins(1,4,5)P3R-Beclin 1-complex formation is dynamic and is enhanced during stimulation of autophagy. It should be noted that in some experiments this interaction could even not be observed in control conditions (). In experiments in which the Ins(1,4,5)P3R-Beclin 1 complex could be measured in control conditions, the interaction was ~4 times higher after 3 h of starvation (). Moreover, we found a transient profile for Beclin 1 binding to Ins(1,4,5)P3R1 during the first 5 h of starvation, which was similar to the transient change in LC3 lipidation ().
Figure 4. Beclin 1 binding to Ins(1,4,5)P3Rs is enhanced during starvation and directly targets the suppressor domain. (A) Co-immunoprecipitation experiments of Beclin 1 with Ins(1,4,5)P3R1 and Ins(1,4,5)P3R3 from lysates of HeLa cells pretreated with (3 h) or without (0 h) HBSS. Representative blots are shown of co-immunoprecipitation experiments with Ins(1,4,5)P3R1 and Ins(1,4,5)P3R3 and immunoblot for Beclin 1 (upper four panels). IgG = negative control (samples of both 0 h and 3 h were analyzed with the same result); IN = input. The double lines indicate that the lanes were taken from another part of the same gel. Lower three panels represent the levels of the indicated proteins analyzed by western blotting. (B) Quantitative analysis of the Beclin 1/Ins(1,4,5)P3R1 (upper) or Beclin 1/Ins(1,4,5)P3R3 (lower) ratio in the immunoprecipitate in samples where binding in basal conditions was detectable (n = 3). (C) Representative co-immunoprecipitation experiment of Beclin 1 with Ins(1,4,5)P3R1 from lysates of HeLa cells starved (3 h or 5 h) or not (0 h). IgG = negative control; IN = input. Lower three panels represent the levels of the indicated proteins analyzed by western blotting analysis. (D) Pull-down experiments with GST-fused domains of Ins(1,4,5)P3R1 and Ins(1,4,5)P3R3 and purified Beclin 1. Ligand-binding domain (1–604); suppressor domain (1–225); Ins(1,4,5)P3-binding core (226–604). Representative example is shown of a pull-down experiment. The GST-fusion proteins were visualized with SYPRO Orange, while Beclin 1 was detected using anti-Beclin 1. The asterisks indicate the intact GST-Ins(1,4,5)P3R fragments. The double lines indicate that the lanes were taken from another part of the same gel. A Beclin 1 degradation product can be observed in some lanes, as is indicated by the arrow. IN = input. (E) Comparative analysis of the amount of Beclin 1 present in the pull-down experiments, normalized to control conditions (GST) (n = 4). *p < 0.05 (paired t-test).
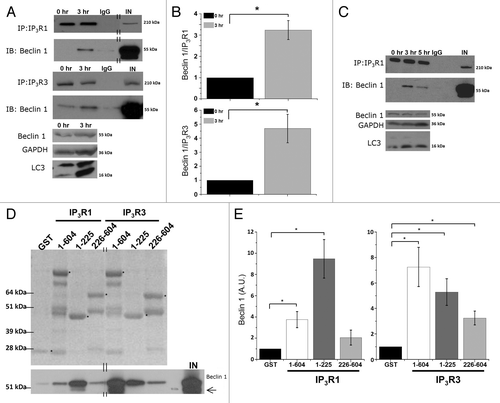
To map the Beclin 1-binding site on the Ins(1,4,5)P3R, we performed pull-down experiments using purified Beclin 1 and equal amounts of Ins(1,4,5)P3R domains fused to GST. Recombinant Beclin 1 was purified as a GST-fusion protein in BL21 (DE3) E. coli, and afterwards the N-terminal GST tag was removed using PreScission protease (Fig. S2C). We examined the Beclin 1-binding properties of the functional domains of the N terminus of the Ins(1,4,5)P3R (the complete ligand-binding domain (LBD; a.a. 1-604), which consists of the suppressor domain (a.a. 1-225) and the Ins(1,4,5)P3-binding core [Ins(1,4,5)P3BC; a.a. 226-604)] (Fig. S2A and S2B) using a GST-pull-down assay. The presence of the full-length GST-fusion proteins corresponding to the different Ins(1,4,5)P3R domains in the pull-down reactions () was confirmed by western blot analysis using a GST antibody (Fig. S2B). The purified GST-Ins(1,4,5)P3R fragments were able to pull down purified Beclin 1 through a direct interaction. For Ins(1,4,5)P3R1, the strongest Beclin 1 binding was observed to correspond to the suppressor domain, whereas for Ins(1,4,5)P3R3, Beclin 1 interacted more potently with the entire LBD (). All other domains of the Ins(1,4,5)P3R, encompassing the entire protein, except for the transmembrane domain, were also tested, but showed no specific interaction with Beclin 1 (data not shown). This suggests that Beclin 1 directly binds to both Ins(1,4,5)P3R isoforms through their respective suppressor domains and to a lesser extent through the Ins(1,4,5)P3-binding core, but the intramolecular determinants for Beclin 1 binding may be somewhat different for Ins(1,4,5)P3R1 and Ins(1,4,5)P3R3.
Because on the one hand Beclin 1 is able to bind to the N-terminal suppressor domain of the Ins(1,4,5)P3R, which controls Ins(1,4,5)P3R-channel gating by interacting with the C-terminal Ca2+-channel domain,Citation47 and on the other hand the Beclin 1-Ins(1,4,5)P3R interaction increases during starvation, we tested whether Beclin 1 was responsible for the Ins(1,4,5)P3R sensitization observed during starvation. Therefore, we used two independent experimental approaches: (1) knockdown of BECN1 using siRNA and (2) investigating the direct effect of recombinantly expressed and purified Beclin 1 on Ins(1,4,5)P3-induced Ca2+-release using the unidirectional 45Ca2+-flux assay in permeabilized HeLa cells.
First, we performed siRNA-mediated knockdown of Beclin 1 using two independent siRNAs directed against two different regions of the BECN1 mRNA. A control siRNA (siCtrl) was developed to assess non-specific effects. shows a western blot analysis of lysates obtained from siRNA-treated HeLa cells, monitoring the expression of Ins(1,4,5)P3Rs, the Atg12–Atg5 complex, Beclin 1 and the loading control, GAPDH. The results indicate an efficient knockdown of Beclin 1 by siBECN-1 1 and siBECN-1 2, while siCtrl did not reduce Beclin 1 protein levels. The Ins(1,4,5)P3R and Atg12–Atg5 were not significantly affected.
Figure 5. siRNA-mediated knockdown of Beclin 1, but not of Atg5, abolishes starvation-induced sensitization of Ins(1,4,5)P3R. (A) Western blot analysis of indicated proteins in HeLa cells treated with indicated siRNA duplexes. (B) Unidirectional 45Ca2+-flux experiments in siRNA-treated permeabilized cells pretreated with (3 h) or without (0 h) HBSS. During the experiment cells were exposed to 0.7 µM Ins(1,4,5)P3 (circles) or 10 µM A23187 (triangles). The arrows indicate the addition of Ins(1,4,5)P3 or A23187. Representative traces are shown of fractional 45Ca2+ release (%/2 min) as a function of time in cells treated with siRNA against BECN1 siBECN-1 1. (C) Unidirectional 45Ca2+-flux experiments in control siRNA (siCtrl)-treated cells. (D) Unidirectional 45Ca2+-flux experiments in siRNA-treated cells against ATG5 (siAtg5). (E) Quantitative analysis of the Ins(1,4,5)P3-induced 45Ca2+ release relative to the A23187-induced 45Ca2+ release in cells pretreated with (3 h) or without (0 h) HBSS (n = 4). *p < 0.05; **p < 0.01; N.S. not significant (paired t-test).
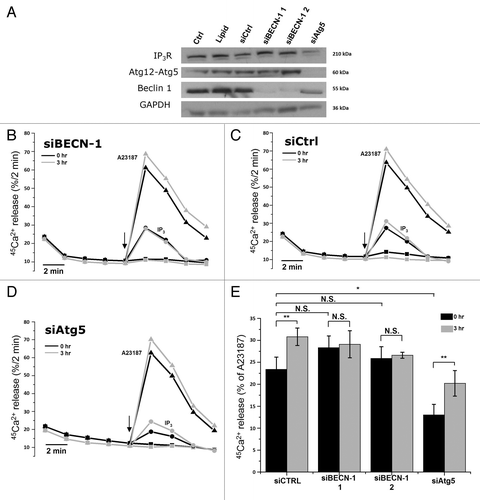
Importantly, as demonstrated by unidirectional 45Ca2+-flux assays in permeabilized HeLa cells treated with different siRNA probes, the starvation-induced sensitization of the Ins(1,4,5)P3R was abolished when Beclin 1 was knocked down using either siBECN-1 1 or siBECN-1 2 (). In contrast, siCtrl did not affect the sensitization of the Ins(1,4,5)P3R (). The sensitization of the Ins(1,4,5)P3R was again calculated as the amount of Ins(1,4,5)P3-induced Ca2+ release, relative to the Ca2+ ionophore-induced Ca2+ release. Importantly, in nonstarved cells, Beclin 1 knockdown had no significant effect on Ins(1,4,5)P3-induced Ca2+ release, suggesting a specific action of cellular Beclin 1 on the Ins(1,4,5)P3R during starvation-induced autophagy.
Since Beclin 1 knockdown also leads to a deficiency in the stimulation of autophagy,Citation9 we tested whether the absence of Ins(1,4,5)P3R sensitization was due to the absence of Beclin 1 or to autophagic deficiency. Therefore, we performed identical experiments in siAtg5-treated cells. Atg5 is another essential key player in autophagy, but one step more downstream in the autophagic pathway. Hence, like Beclin 1, knockdown of Atg5 will prevent autophagy stimulation. However, Beclin 1 still dissociates from the Bcl-2-family proteins in response to starvation. Interestingly, in siAtg5-treated cells, the starvation-induced sensitization of the Ins(1,4,5)P3R was still observed (), indicating that Ins(1,4,5)P3R sensitization did not result from a downstream event during autophagy but was dependent on the presence of Beclin 1. The decreased Ins(1,4,5)P3-induced Ca2+ release in siAtg5-treated cells in control conditions () is likely due to the decline in Ins(1,4,5)P3R-expression level, which was observed in western blots (). However, the reason for this is not clear.
These experiments point toward a direct action of Beclin 1 on the Ins(1,4,5)P3R. Therefore, we expressed and purified recombinant Beclin 1 as a GST-fusion protein in BL21 (DE3) E. coli and removed the N-terminal GST tag using PreScission protease (Fig. S2C). After its dialysis against efflux medium, we examined its direct effect on Ins(1,4,5)P3R-mediated Ca2+ flux using the unidirectional 45Ca2+-flux assay in permeabilized HeLa cells. Since Beclin 1 is a relatively large protein (~55 kDa), its yield was relatively low (~2.5 µM), though unidirectional 45Ca2+-flux assays require a high amount of protein for adequate testing.Citation48,Citation49 A contaminant obtained with this procedure consists of the E. coli Hsp70 chaperone DnaK, which copurifies during bacterial GST-fusion protein purifications.Citation50 We added recombinant Beclin 1 at this maximal concentration of 2.5 µM, 2 min before Ins(1,4,5)P3 addition and during Ins(1,4,5)P3 addition. We used a submaximal [Ins(1,4,5)P3] (0.7 µM, i.e., just below the EC50 value) and a supramaximal [Ins(1,4,5)P3] (30 µΜ). As an additional control, we also used 2.5 µM GST. GST at 2.5 µM did not significantly alter the Ins(1,4,5)P3-induced Ca2+ release (Fig. S2D). Beclin 1 at 2.5 µM potentiated Ins(1,4,5)P3R-mediated Ca2+ release in response to 0.7 µM Ins(1,4,5)P3 (+13 ± 3%), but not to 30 µM Ins(1,4,5)P3 (-6 ± 4%) (). These results indicate that purified Beclin 1 is able to acutely and directly sensitize Ins(1,4,5)P3Rs.
Figure 6. Purified Beclin 1 directly sensitizes the Ins(1,4,5)P3R toward Ins(1,4,5)P3, independently of Bcl-2. (A) Unidirectional 45Ca2+-flux experiments in permeabilized HeLa cells with (gray; Beclin 1) or without (black; Ctrl) the addition of 2.5 µM purified Beclin 1. Mean fractional 45Ca2+ release (%/2 min) as a function of time is shown in the absence or presence of 2.5 µM purified Beclin 1, added for 4 min, starting 2 min prior to the addition of 0.7 (circles) or 30 µM Ins(1,4,5)P3 (triangles). The arrow indicates the addition of Ins(1,4,5)P3. (B) Quantification of the 45Ca2+ release triggered by the indicated concentration of Ins(1,4,5)P3, normalized to control conditions (n = 4). (C) Mean traces of unidirectional 45Ca2+-flux experiments in permeabilized HeLa cells without (black; Ctrl) or with 10 µM purified Beclin 1 domains (N-BH3: red; CCD: green; ECD-C: blue), added for 4 min, starting 2 min prior to the addition of 0.7 µM Ins(1,4,5)P3. The arrow indicates the addition of Ins(1,4,5)P3. (D) Quantification of the 45Ca2+ release triggered by 0.7 µM Ins(1,4,5)P3, normalized to control conditions, after addition of the indicated Beclin 1 domains (n = 4). (E) Representative example of pull-down experiments with GST-fused domains of Ins(1,4,5)P3R1 and purified Beclin 1-F123A. The GST-fusion proteins were visualized using SYPRO Orange, while Beclin 1 was detected using anti-Beclin 1. IN = input. The asterisks indicate the different domains: ligand-binding domain (1–604); suppressor domain (1–225); Ins(1,4,5)P3-binding core (226–604). (F) Representative unidirectional 45Ca2+-flux experiment in permeabilized cells with (gray; Beclin 1-F123A) or without (black; Ctrl) 2.5 µM purified Beclin 1-F123A added for 4 min, starting 2 min prior to the addition of 0.7 µM Ins(1,4,5)P3 (circles). The arrow indicates the addition of Ins(1,4,5)P3. (G) Representative co-immunoprecipitation experiment of Beclin 1-F123A with Ins(1,4,5)P3R1 from lysates of HeLa cells transfected with Beclin 1-F123A and pretreated with (3 h) or without (0 h) HBSS. IgG = negative control; IN = Input. (H) Representative co-immunoprecipitation experiment of Beclin 1 with Bcl-2 from lysates of HeLa cells transfected with Beclin 1 and pretreated with (3 or 5 h) or without (0 h) HBSS. IgG = negative control; IN = Input. *p < 0.05; **p < 0.01; N.S. not significant (paired t-test).
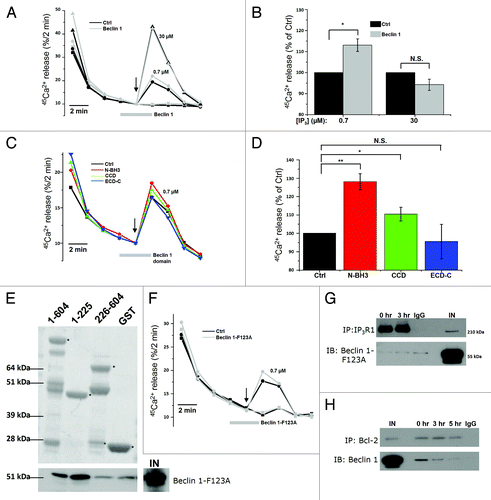
To further substantiate these findings, we examined three functional domains of Beclin 1 expressed as recombinant proteins: the N-terminal domain with the BH3 domain (N-BH3; a.a. 1-150), the coiled-coil domain (CCD; a.a. 151-244) and the evolutionarily conserved domain together with the C-terminal tail (ECD-C; a.a. 244-450) (Fig. S3A). Similarly to full-length Beclin 1, we expressed and purified these domains as GST-fused proteins and removed the GST tag. Since these proteins are relatively small, much higher yields were obtained, and they could be diluted to a final concentration of 10 μM. These recombinant Beclin 1 domains were found to be of high purity and stable (Fig. S3B). Their identity was confirmed using site-specific Beclin 1 antibodies (Fig. S3C). Similar to the full-length Beclin 1, these domains were applied in the unidirectional 45Ca2+-flux assays to assess their effect on the Ins(1,4,5)P3R (). We found that both N-BH3 and CCD stimulated Ins(1,4,5)P3-induced Ca2+ release, of which N-BH3 was the most potent. In contrast, ECD-C did not alter Ins(1,4,5)P3R function. Strikingly, this stimulatory effect of N-BH3 on the Ins(1,4,5)P3R was even larger than for full-length Beclin 1, probably reflecting the higher concentrations used for these shorter domains (10 μM vs. 2.5 μM). These experiments therefore underpin our findings concerning full-length Beclin 1 and point toward a role for both N-BH3 and CCD in Ins(1,4,5)P3R regulation. Moreover, these experiments rule out potential artifacts due to the contaminating bacterial DnaK present in our GST-fusion protein preparations, given the (very) low level of DnaK in the active N-BH3 and CCD preparations.
Finally, because Beclin 1 binds to Bcl-2, and Bcl-2 can suppress Ins(1,4,5)P3-induced Ca2+ release,Citation34,Citation51,Citation52 we assessed whether the effect of Beclin 1 on Ins(1,4,5)P3Rs could be due to its binding of Bcl-2. Therefore, we recombinantly expressed and purified Beclin 1-F123A, a Beclin 1 mutant that is unable to bind Bcl-2 (Fig. S2C).Citation53 Beclin 1-F123A’s deficiency in binding Bcl-2 was confirmed by a pull-down analysis (Fig. S2E). Similar to wild-type Beclin 1, Beclin 1-F123A still interacted with the complete LBD as well as with the suppressor domain of the Ins(1,4,5)P3R in GST-pull-down assays () and potentiated the Ins(1,4,5)P3-induced Ca2+ release in permeabilized HeLa cells in unidirectional 45Ca2+-flux assays (). This indicates that Beclin 1-mediated Ins(1,4,5)P3R sensitization is independent of its interaction with Bcl-2. However, co-immunoprecipitation studies in lysates of HeLa cells overexpressing Beclin 1-F123A revealed that in a cellular context this mutant did not interact with Ins(1,4,5)P3R1, neither at 0 h nor after 3 h starvation (), indicating an indirect role of Bcl-2 at the ER in the Beclin 1-Ins(1,4,5)P3R association. This is supported by co-immunoprecipitation experiments indicating that Beclin 1 was released from Bcl-2 during starvation (), as was previously described.Citation12,Citation53 Thus, these data suggest that Beclin 1 at the ER shuttles from Bcl-2 to Ins(1,4,5)P3Rs, thereby enhancing its Ca2+-release activity.
Cytosolic Ca2+ and Ins(1,4,5)P3Rs are required for starvation-induced autophagy
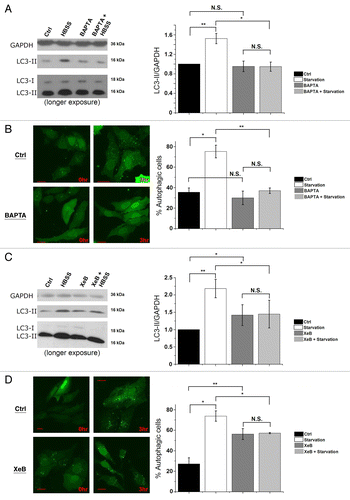
Using the cell-permeable Ca2+-chelating agent BAPTA-AM or the Ins(1,4,5)P3R inhibitor xestospongin B (XeB),Citation54 we analyzed whether Ins(1,4,5)P3-induced Ca2+ signaling is critical for the autophagy response toward starvation. One hour before collecting the cells, we added 100 nM bafilomycin A1 (Baf A1), a blocker of lysosomal H+-ATPase, in order to measure the formation of LC3-II during the last hour, independently of its degradation in autolysosomes. Using the methods proposed to properly analyze autophagy,Citation36 we calculated the LC3-II/GAPDH ratio. Interestingly, loading the cells with 10 µM BAPTA-AM significantly diminished the increase of LC3-II levels () and of GFP-LC3 puncta () after 3 h of starvation. Addition of 2 µM XeB also blunted the increased lipidation of LC3 () and GFP-LC3-puncta formation () after 3 h of starvation. It should be noted that, as previously decribed,Citation23,Citation31 XeB increased basal autophagy (), albeit less strongly than starvation. Similar results for BAPTA and XeB treatment were obtained in mouse L15 cells (Fig. S1F). These results strongly suggest that proper cytosolic Ca2+ signaling through Ins(1,4,5)P3Rs is a requisite for autophagy induction by starvation.
Figure 7. Intracellular Ca2+ and Ins(1,4,5)P3Rs are requisite for starvation-induced autophagy. (A) LC3 western blotting in HeLa cells treated without (Ctrl) or with HBSS for 3 h, 10 µM BAPTA-AM or both (BAPTA + HBSS). One hour before collecting cells, 100 nM Baf A1 was added. Cells were given BAPTA-AM for 30 min, then washed with complete cell culture medium (DMEM) as the control or HBSS and incubated for 3 h before collecting. Left: Representative blots. Right: Quantification of LC3-II/GAPDH ratio, normalized to control conditions (n = 4). (B) GFP-LC3-puncta formation in HeLa cells transfected with GFP-LC3 constructs. Cells were treated without (0 h) or with HBSS (3 h), and without (Ctrl) or with 10 µM BAPTA-AM (BAPTA). Left: Representative pictures. The scale bar represents 20 µm. Right: Quantification of autophagic cells. Only cells displaying more than 10 puncta were considered autophagic (n = 3). Similar results were obtained when the number of GFP-LC3 puncta were quantified. (C) LC3 western blotting in cells treated without (Ctrl) or with HBSS for 3 h, 2 µM XeB or both (XeB + HBSS). One hour before collecting cells, 100 nM Baf A1 was added. Left: Representative blots. Right: Quantification of LC3-II/GAPDH ratio, normalized to control conditions (n = 5). (D) GFP-LC3-puncta formation in HeLa cells transfected with GFP-LC3 constructs. Cells were treated without (0 h) or with HBSS (3 h), and without (Ctrl) or with 2 µM XeB. Left: Representative pictures. The scale bar represents 20 µm. Right: Quantification of autophagic cells. Only cells displaying more than 10 puncta were considered autophagic (n = 3). *p < 0.05; **p < 0.01; N.S. not significant (paired t-test).
Discussion
The key finding of the present study is a bidirectional feedback regulation between intracellular Ca2+ signaling and autophagy initiation, making both processes interdependent: Ins(1,4,5)P3-mediated Ca2+ signaling is transiently sensitized during autophagy stimulation by starvation, while proper autophagy initiation requires Ins(1,4,5)P3-mediated Ca2+ signaling. This finding is supported by different observations: Ca2+ measurements in starved cells displayed an increased ER Ca2+-store content as well as a sensitization of the Ins(1,4,5)P3R. Both events resulted in an increased Ca2+ release. We identified several parameters underlying these events: intralumenal CaBPs (Grp78/BiP and CRT) were upregulated, the ER Ca2+-leak rate was decreased and Beclin 1 association to the Ins(1,4,5)P3R was enhanced. Beclin 1 bound to the N-terminal Ins(1,4,5)P3R suppressor domain, a region important for Ins(1,4,5)P3R-channel gating and sensitized the Ins(1,4,5)P3R to low/medium [Ins(1,4,5)P3]. These findings elucidate Beclin 1 as a novel Ins(1,4,5)P3R-regulating protein that is dynamically regulated during autophagy stimulation. Finally, by dampening intracellular Ca2+ signals using BAPTA-AM or XeB, we identified cytosolic Ca2+ and Ins(1,4,5)P3Rs as critical factors in starvation-induced autophagy.
Stimulation of autophagy induces a sensitization of the Ca2+ machinery
Our results demonstrate that starvation induced a molecular remodeling process leading to a transient sensitization of the Ca2+-signaling machinery. Interestingly, the time course of this sensitization correlated well with the stimulation of autophagy (as evidenced by the LC3-II levels and GFP-LC3 puncta formation), while ER stress and apoptosis were not yet initiated. This indicates that a sensitization of Ca2+ signaling is a proximal response during the first 3 h of starvation. After 5 h of starvation, the increase in ER Ca2+-store content, in agonist-induced Ca2+ signaling and in LC3 lipidation were abolished in HeLa cells (). Also in other cell types, MEF and L15 cells, the transient changes in Ca2+ signaling correlated well with the time course of LC3 lipidation (Fig. S1A–D).
We verified several key players that regulate ER Ca2+ homeostasis, like SERCA2b, intralumenal CaBPs, and ER Ca2+ leak by monitoring their levels/activity during this proximal phase of starvation. Interestingly, SERCA2b levels were significantly reduced by ~50% even as early as after 1 h of starvation. After 3–5 h, these levels slowly increased again, but they were still lower than in control conditions. The mechanism underpinning the decline in SERCA2b levels remains elusive, but may involve reactive oxygen species (ROS)-dependent damage to the SERCA Ca2+ pumps. Indeed, ROS accumulate within 30 min of starvation.Citation55 Importantly, ROS (and in particular 1O2) was already shown to rapidly lead to damage to SERCA Ca2+ pumps, at least during photodynamic therapy.Citation56 In contrast to SERCA, CaBPs were upregulated during starvation. While the exact underlying mechanism remains unknown, it is likely that CaBPs are upregulated at the transcriptional level, since amino acid depletion has been shown to augment the amount of stress proteins via this mechanism.Citation57 The augmented CaBP levels correlated with a declined unidirectional ER Ca2+-leak rate in starved cells. Indeed, increasing the Ca2+-buffering capacity by upregulation of the intralumenal CaBPs may decrease the lumenal free Ca2+ available for the ER Ca2+-leak channels.Citation58 Hence, the proximal responses toward starvation consist of an upregulation of CaBPs, resulting in a reduced Ca2+-leak rate and an increased ER Ca2+-store content. This inevitably enhances Ca2+ release from the ER to the cytosol, e.g., in response to basal levels of Ins(1,4,5)P3 or in response to agonists that induce Ins(1,4,5)P3 production, which is in line with previous publications concerning the role of intracellular Ca2+ stores in autophagy.Citation59,Citation60 Besides the increased ER Ca2+-store content, enhanced ER Ca2+ signaling was also due to Ins(1,4,5)P3R sensitization toward low [Ins(1,4,5)P3].
Beclin 1 is essential for Ins(1,4,5)P3R sensitization during autophagy stimulation
Using co-immunoprecipitation and GST-pull-down experiments, we identified an interaction of Beclin 1 with the LBD (a.a. 1-604) of the Ins(1,4,5)P3R. This interaction was enhanced during autophagy stimulation in response to starvation. Moreover, depleting cells of Beclin 1 using siRNA abolished the sensitization of the Ins(1,4,5)P3R after starvation. The effect of Beclin 1 represents a direct modulation of the Ins(1,4,5)P3R for two reasons: (1) depleting cells of another essential autophagy protein, Atg5, which acts downstream of Beclin 1, did not affect Ins(1,4,5)P3R sensitization during starvation, and (2) bacterially expressed and purified recombinant Beclin 1 was sufficient to directly elicit Ins(1,4,5)P3R sensitization in the absence of starvation. The in vitro binding and stimulation of the Ins(1,4,5)P3R by Beclin 1 was also established for Beclin 1-F123A, a mutant that cannot bind Bcl-2. This excludes that the effects of Beclin 1 are mediated indirectly by counteracting the binding of Bcl-2 to the Ins(1,4,5)P3R, which too would result in an increased Ins(1,4,5)P3R-mediated Ca2+ flux.Citation34,Citation52 However, in a cellular context, Beclin 1-F123A neither formed endogenous protein complexes with the Ins(1,4,5)P3Rs nor displayed increased binding to Ins(1,4,5)P3Rs upon starvation. These data correlate well with the findings observed by Vicencio et al. who showed that Bcl-2 knockdown prevented the binding of Beclin 1 to the Ins(1,4,5)P3R in a cellular context.Citation31 Moreover, Beclin 1 is released from Bcl-2 during starvation ().Citation12,Citation53 Collectively, these data suggest an indirect role of Bcl-2 in this process, in which Bcl-2 at the ER may be needed to tether Beclin 1 in close proximity of its target, the Ins(1,4,5)P3R, at the ER during autophagy (). This is in line with the observations that overexpression of ER-targeted, but not mitochondrial, Bcl-2 is able to affect autophagy.Citation27,Citation53
Figure 8. Model representing changes in the Ca2+ signalosome during starvation. In the non-starved state (left) Ins(1,4,5)P3Rs inhibit autophagy through a Ca2+ signal in the ER-mitochondria microdomains to fuel mitochondrial energetics, thereby inhibiting AMPK and autophagy.Citation26 In this situation, Beclin 1 likely is kept at the ER in the proximity of the Ins(1,4,5)P3R through ER-localized Bcl-2. Alternatively, there may be a scaffolding role for Ins(1,4,5)P3R/Bcl-2 complexes capturing Beclin 1.Citation31 During starvation (right), however, an upregulation of Ca2+-binding proteins (CaBP) together with the direct binding of Beclin 1 to Ins(1,4,5)P3R’s ligand-binding domain, underpins a sensitized Ca2+ signaling through Ins(1,4,5)P3Rs, leading to autophagy stimulation. The target of this Ca2+ signal is probably cytosolic. While Bcl-2 is essential for facilitating Ins(1,4,5)P3R-Beclin 1 complex formation in a cellular context, the mechanism by which Beclin 1 sensitizes Ins(1,4,5)P3R activity is direct and does not involve Bcl-2. We propose that upon starvation Beclin 1 at the ER shuttles from Bcl-2 to the Ins(1,4,5)P3R.
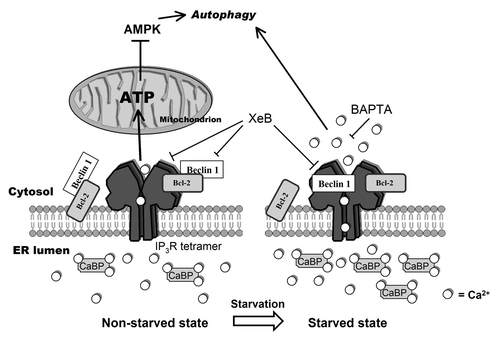
While the interaction between Beclin 1 and the Ins(1,4,5)P3R has previously been reported by Kroemer and coworkers,Citation31 our data provide a novel role for the Ins(1,4,5)P3R-Beclin 1 complexes during autophagy stimulation. The former report focuses on Beclin 1-Ins(1,4,5)P3R interaction in basal conditions and hypothesizes that Ins(1,4,5)P3Rs act as scaffold proteins that bind Beclin 1 and Bcl-2 separately, thereby promoting Bcl-2’s interaction with and inhibition of Beclin 1. In their study,Citation31 knockdown of Beclin 1 did not affect intracellular Ca2+ homeostasis. Collectively, our results presented here and the report by Vicencio et al.Citation31 indicate that the state of the cell, starved or nonstarved, may determine the role of Beclin 1 on the Ins(1,4,5)P3R (). In nonstarved cells, Beclin 1 could interact with the Ins(1,4,5)P3R, but indirectly through Bcl-2, thereby not affecting Ins(1,4,5)P3R function. In this situation, knockdown of Bcl-2 would reduce the interaction of Beclin 1 with the Ins(1,4,5)P3R,Citation31 but overexpression or knockdown of Beclin 1 would have no effect on Ca2+ homeostasis ().Citation16,Citation31 It should be noted that in the nonstarved situation we did not consistently find Beclin 1 binding to Ins(1,4,5)P3Rs, which may be due to a low affinity of the binding under our experimental conditions. In the starved state, however, Beclin 1 is released from Bcl-2, and is thereby able to interact directly with the LBD and so to sensitize Ins(1,4,5)P3Rs (). This is supported by our experiments using recombinantly expressed wild-type Beclin 1 and Beclin 1-F123A, which is unable to bind Bcl-2 (). These data indicate that Beclin 1 can sensitize the Ins(1,4,5)P3R independently of its ability to bind Bcl-2 and is thus an inherent property of Beclin 1. In this situation, knockdown of Beclin 1 abolished the effect of starvation on Ins(1,4,5)P3R’s Ca2+-flux properties ().
Vicencio et al. observed a decline in the Beclin 1-Ins(1,4,5)P3R interaction after 3–6 h of starvation.Citation31 In agreement with this, we also found a decline after 5 h (). However, in the early phase of autophagy induction (1–3 h) we found a clear increase of the interaction, correlating with the increased LC3-II levels (). The exact reason for these different findings is not clear and may reflect differences in the experimental conditions, as the enhanced Beclin 1 association to Ins(1,4,5)P3Rs that we observed was very transient. The interaction of Beclin 1 with the Ins(1,4,5)P3R closely correlated with LC3 lipidation, suggesting a tight link between Beclin 1-Ins(1,4,5)P3R interaction and events occurring during the initial steps of autophagy induction. We observed this tight interplay between increased autophagy and enhanced Ca2+ signaling in three independent cell lines. Importantly, it should be noted that the timing at which autophagy was induced and at which Ca2+ signaling was enhanced, differed among the three different cell lines, but always correlated with each other within a particular cell type. Furthermore, we also observed a decrease in Beclin 1 binding to Ins(1,4,5)P3Rs at later stages (), correlating with the decline in LC3-II levels and GFP-LC3-puncta formation.
Beclin 1 binds to both Ins(1,4,5)P3R1 and Ins(1,4,5)P3R3, but the molecular determinants are slightly different
Vicencio et al. already pointed to the Ins(1,4,5)P3BC (a.a. 224–604) as the binding site for Beclin 1.Citation31 By performing a detailed analysis of the N-terminal domain of the Ins(1,4,5)P3R, we confirmed the binding of Beclin 1 to the Ins(1,4,5)P3BC, particularly for Ins(1,4,5)P3R3, but we found that the suppressor domain (a.a. 1-225) showed the most prominent interaction with Beclin 1 (). The presence of different interaction domains is also compatible with our finding that two separate Beclin 1 fragments (N-BH3 and CCD) were able to enhance Ins(1,4,5)P3R activity, albeit with different potencies (). Strikingly, the binding of Beclin 1 to the suppressor domain of Ins(1,4,5)P3R1 was much stronger than the binding to the complete LBD. Hence, the Ins(1,4,5)P3BC might fine-tune the Beclin 1-Ins(1,4,5)P3R1 interaction, as deletion of this Ins(1,4,5)P3BC increased the binding of Beclin 1 to the suppressor domain of Ins(1,4,5)P3R1. Interestingly, this did not occur for Ins(1,4,5)P3R3, suggesting a different mode of interaction for Ins(1,4,5)P3R1 and Ins(1,4,5)P3R3. This is in agreement with the observations that properties and regulation of the LBD in Ins(1,4,5)P3R1 and -3 are not identical.Citation35,Citation47 Intramolecular interactions between the suppressor domain and the Ins(1,4,5)P3BC have been described,Citation61 and it is known that isoform-specific differences between Ins(1,4,5)P3R1 and Ins(1,4,5)P3R3 exist with respect to these intramolecular interactions.Citation35 In addition, thiol-reactive agents, like thimerosal, regulate these interactions. Hence, the redox state of the Ins(1,4,5)P3R may affect its ability to interact with Beclin 1, which may be very important, since ROS arise very early during starvation.Citation55 ROS may therefore affect Ins(1,4,5)P3R function by altering the accessibility of the Beclin 1-binding site of the Ins(1,4,5)P3R.
The Ca2+-signaling machinery is required for proper autophagy stimulation by starvation
Previous findings already reported that cytosolic Ca2+ may stimulate autophagy, but these studies predominantly involved Ca2+-mobilizing triggers such as ionomycin, TG or Cd2+.Citation27-Citation30 These treatments led to disturbed intracellular Ca2+ homeostasis, and subsequent stimulation of autophagy through a Ca2+-dependent pathway. The present study, however, reveals that autophagy induction is also a Ca2+-dependent process in response to a non-Ca2+-mobilizing trigger, starvation, since chelating the cytosolic Ca2+ with BAPTA-AM during starvation blunted autophagy stimulation. In addition, there is an important role for Ins(1,4,5)P3Rs in this process, since blocking Ins(1,4,5)P3Rs with XeB also abolished LC3 lipidation. This finding therefore suggests that autophagy stimulation depends on proper Ins(1,4,5)P3-mediated Ca2+ signaling, and is not only provoked by aberrant Ca2+ homeostasis. Moreover, this study reveals the Ins(1,4,5)P3R as a prominent key player in starvation-induced autophagy, since its sensitivity toward Ins(1,4,5)P3 was directly regulated by Beclin 1 during this process. In this regard, a role for Ins(1,4,5)P3R-mediated Ca2+ release has also been described for Cd2+-induced autophagy.Citation29
The exact mechanism by which Ca2+ and Ins(1,4,5)P3Rs mediate autophagy remains unclear, although several pathways have already been proposed. Increasing [Ca2+]cyt induces autophagy by activation of AMPK via calmodulin kinase kinase-β (CaMKK-β),Citation27 although other reports indicate that this effect could occur independently of AMPK activation.Citation28 In this respect, other groups proposed Ca2+ dependence for protein kinase Cθ (PKCθ) in ER stress-induced autophagy,Citation30 or for extracellular signal regulated kinase (ERK) in Cd2+-induced autophagy.Citation29
Two different Ca2+ signals regulate autophagy in opposite ways
The findings presented in this work provide novel insights in the molecular mechanism underpinning autophagy regulation by Ca2+, which has been reported to both inhibit,Citation23-Citation25,Citation62 as well as to stimulate autophagy.Citation27-Citation30 These differences might reflect different spatiotemporal Ca2+ signals in unstressed vs. stressed conditions.Citation63 In unstressed cells a constitutive Ca2+ release from ER to mitochondria is required for mitochondrial ATP production. A high ATP/AMP ratio inhibits AMPK and subsequently autophagy.Citation26 Consequently, unstressed cells display an autophagy-inhibiting Ca2+ signal, specifically targeted to mitochondria and specific for the typical ER-mitochondria microdomains. Reducing basal Ins(1,4,5)P3 levels,Citation25 knocking down Ins(1,4,5)P3Rs,Citation23,Citation24 or XeB () leads therefore to a stimulation of the autophagic pathway. It is important to note that in contrast to XeB, treating the cells with BAPTA-AM was not sufficient to induce autophagy (). This underpins an important role for Ins(1,4,5)P3R-mediated Ca2+ signals in specialized microdomains in non-starved conditions. However, the enhancement of autophagy during starvation was abolished with both BAPTA-AM and XeB, thereby suggesting that the Ca2+ signal that is necessary for autophagy induction and its downstream target may be cytosolic and not restricted to a specialized microdomain (). Also stimulation of autophagy via the inhibition of L-type Ca2+ channels in the plasma membrane can be explained in this way,Citation62 since Ca2+ is a co-activator of Ins(1,4,5)P3Rs.Citation17 Independently of Ca2+, the Ins(1,4,5)P3R can still act as a scaffold protein in this situation, thereby promoting the Bcl-2-mediated inhibition of Beclin 1.Citation31
In stressed cells (e.g., during starvation), however, the intracellular Ca2+ machinery becomes sensitized, resulting in increased Ca2+ release from the ER into the cytosol. This Ca2+ signal can lead to a stimulation of autophagy, in accordance with previous observations.Citation27-Citation30 In this manner, cells can switch from an “unstressed” autophagy-inhibiting and mitochondrial Ca2+ signal to a “stressed” autophagy-activating and cytosolic Ca2+ signal (). This dual role for Ca2+ is supported by our experiments using XeB: addition of this Ins(1,4,5)P3R inhibitor in “unstressed” conditions increased LC3-II formation (),Citation23,Citation31 albeit not as high as for starvation, while the extra enhancement of LC3-II was abolished in the “stressed” situation (XeB + HBSS) (). The molecular “switch” between both Ca2+ signals may lie in the differential targeting of Beclin 1 to the Ins(1,4,5)P3R in both conditions.
Materials and Methods
Cell culture
Wild-type human cervix carcinoma HeLa cells, mouse embryonic fibroblasts (MEF) and mouse L fibroblast L15 cells (a kind gift from Dr. K. Mikoshiba, Brain Science Institute, Wakō, Saitama, Japan)Citation33 were grown in Glutamax-containing Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated fetal calf serum (FCS) and 10 mM HEPES buffer. The cells were grown in the presence of 85 IU ml−1 penicillin and 85 µg ml−1 streptomycin at 37°C and 5% CO2. For starvation, cells were transferred to Hank’s Balanced Salt Solution (HBSS). All materials were purchased from Gibco, Invitrogen.
DNA construction, siRNA and transfection
pCMV6-XL5-Beclin 1 (OriGene Technologies) and pCR3.1-Flag-Beclin 1-F123A, a kind gift from Dr. B. Levine (University of Texas Southwestern Medical Center, TX, USA),Citation53 were used for transfection or for subcloning in pGEX-6P-2 (GE Healthcare) for GST-fusion with 5′-AGT AGT GGA TCC ATG GAA GGG TCT AAG ACG TCC as forward and 5′-AGT AGT GAA TTC AGG CAA ACC TCC CCC TAA GGA as reverse primer. These primers were also used for subcloning of the Beclin 1 domains in combination with 5′-AGG AGC GAA TTC TCA AGT GTC CAG CTG GTC TAA AAG as reverse primer for N-BH3 construction or 5′-AGG CGT GGA TCC GAT GAT GAG CTG AAG AGT GTT G as forward primer for ECD-C construction. For the CCD domain, 5′-AGT CGT GGA TCC ACT CAG CTC AAC GTC ACT G was used as forward and 5′-AGT CGT GAA TTC TCA ATC CAG CTC CAG CTG CTG TG as reverse primer. pcDNA3.1(-)-GFP-LC3 was cloned from pBABEpuro-GFP-LC3 (a kind gift from Dr. J. Debnath, University of California, San Francisco)Citation64 using 5′-AGT CGT GCG GCC GCA TGG TGA GCA AGG GCG A as forward and 5′-AGT AGT GAA TTC TTA CAC TGA CAA TTT CAT CCC as reverse primer. siRNA-duplex oligonucleotides against human BECN1 and ATG5 were designed and purchased from Eurogentec. Two siRNA duplexes were made for BECN1 (sense siBECN-1 1: 5′-UGA GUG UCA GAA CUA CAA AdTdT; sense siBECN-1 2: 5′-CUC ACA GCU CCA UUA CUU AdTdT), one siRNA duplex for ATG5 (sense siAtg5: 5′-GAA GUU UGU CCU UCU GCU AdTdT) and one control siRNA duplex (sense siCtrl: 5′-GGU AAA CGG AAC GAG AAG AdTdT). DNA transfection was achieved with jetPRIME™ from Polyplus Transfection (114-75), and siRNA transfection with HiPerfect (Qiagen, 301704). Twenty-four h after transfection, the medium was changed and 48 h later, the cells were treated, collected or measured.
Antibodies and reagents
For immunoblot, the following antibodies were used: anti-GAPDH (Sigma-Aldrich NV, G8795), anti-BiP (Sigma-Aldrich NV, G8918), anti-LC3 (nanoTools Antikörpertechnik GmbH and Co., 0231-100), N-terminal and C-terminal anti-Beclin 1 (Santa Cruz Biotechnology, Inc., sc-48341 and sc-10087, respectively), central anti-Beclin 1 (BD Biosciences, 612112) anti-calreticulin (anti-CRT) (Affinity Bioreagents, PA1-903), anti-Atg12 (Cell Signaling Technologies, 2010), anti-caspase 3 (Calbiochem, 235412), anti-GST (Zymed, 13-6700) and anti-Ins(1,4,5)P3R3 (BD Biosciences, 610313). Anti-Ins(1,4,5)P3R1 and anti-pan-Ins(1,4,5)P3R are Rbt03 and Rbt475,Citation35,Citation65,Citation66 respectively; SERCA2b antibody was a kind gift from Dr. P. Vangheluwe and Dr. F. Wuytack (K.U. Leuven, Belgium).Citation67 For immunoprecipitation experiments, antibodies against Ins(1,4,5)P3R1 (Santa Cruz Biotechnologies, Inc., sc-6093) or Ins(1,4,5)P3R3 (Santa Cruz Biotechnologies, Inc., sc-7277) were used. Other chemicals used are the following: Ca2+ ionophore A23187 (Sigma-Aldrich NV, C7522), tunicamycin (TM) (Sigma-Aldrich NV, T7765), Ins(1,4,5)P3 (Sigma-Aldrich NV, I7012), isopropyl-β-D-thiogalactoside (IPTG) (Sigma-Aldrich NV, I5502), EGTA (Acros Organics, 409910250), thapsigargin (Enzo Life Sciences BVBA, ALX-350-004-M010), ionomycin (LC Laboratories, I-6800), staurosporine (LC laboratories, S-9300), Baf A1 (LC Laboratories, B-1080), ATP (Roche Applied Science, 10127531001), 45Ca2+ (PerkinElmer, NEZ-013), Fura-2-AM (Biotium, 50033), and BAPTA-AM (Molecular Probes, Invitrogen, B6769). Xestospongin BCitation68 was purified from Xestospongia exigua as previously described.Citation54,Citation68
Fluorescent Ca2+ measurements in intact cells
For the [Ca2+]cyt measurements in intact cells, HeLa cells were seeded in 96-well plates (Greiner) at a density of approximately 1.2 × 104 cells cm−2 and investigated two days after seeding. The cells were loaded for 30 min with 5 µM Fura-2-AM at 25°C in modified Krebs solution containing 135 mM NaCl, 5.9 mM KCl, 1.2 mM MgCl2, 11.6 mM HEPES (pH 7.3), 11.5 mM glucose and 1.5 mM Ca2+. They were then incubated for at least 30 min in the absence of Fura-2-AM. Fluorescence was monitored on a FlexStation 3 microplate reader (Molecular Devices) by alternately exciting the Ca2+ indicator at 340 and 380 nm and collecting emission fluorescence at 510 nm. [Ca2+]cyt was derived after in situ calibration according to the following equation: Kd is the dissociation constant of Fura-2 for Ca2+ at room temperature (220 nM), q is the fluorescence ratio of the emission intensity in the absence of Ca2+, to that in the presence of saturating Ca2+, R is the fluorescence ratio, Rmin and Rmax are the minimal and maximal fluorescence ratios, respectively. Rmin was measured by perfusion with 10 mM EGTA in Ca2+-free modified Krebs solution and Rmax was obtained by perfusion with 10 µM ionomycin and 5 mM CaCl2.
45Ca2+ measurements in permeabilized cells
Unidirectional 45Ca2+-flux experiments were basically performed as previously described.Citation43,Citation69,Citation70 After permeabilization of HeLa cells with 20 µg ml−1 saponin, the non-mitochondrial Ca2+ stores were loaded for 45 min in 120 mM KCl, 30 mM imidazole-HCl (pH 6.8), 5 mM MgCl2, 5 mM ATP, 0.44 mM EGTA, 10 mM NaN3 and 150 nM free 45Ca2+ (28 μCi ml−1). Efflux medium containing 120 mM KCl, 30 mM imidazole-HCl (pH 6.8) and 1 mM EGTA was subsequently added and replaced every 2 min. The indicated [Ins(1,4,5)P3] or 10 µM of the Ca2+ ionophore A23187 were added for 2 min after 10 min of efflux. Eight min later, the 45Ca2+ remaining in the stores was released by incubation with sodium dodecyl sulfate solution for 30 min. The amount of 45Ca2+ present in each sample was measured using the Liquid Scintillation Analyzer (Packard BioScience, PerkinElmer).
Immunoblots and co-immunoprecipitation
HeLa cells were scraped into ice-cold phosphate-buffered saline and lysed in a modified RIPA buffer containing 10 mM sodium phosphate (pH 7.5), 150 mM NaCl, 1.5 mM MgCl2, 0.5 mM DTT, 1% Triton X-100, 10% glycerol and complete EDTA-free protease inhibitor tablets (Roche Applied Science, 04 693 132 001). After 30 min of incubation on ice, the lysates were cleared via centrifugation. Protein concentrations were determined by the Bradford procedure. For sample separation we used commercial Tris-glycine or Bis-Tris SDS-PAGE gels (Invitrogen). For co-immunoprecipitation of Ins(1,4,5)P3R3, lysates were first incubated with anti-Ins(1,4,5)P3R3 antibody overnight at 4°C, then one additional hour with Protein A/G PLUS-agarose beads (Santa Cruz Biotechnology, Inc., sc-2003), before washing the beads four times with modified RIPA buffer. For co-immunoprecipitation of Ins(1,4,5)P3R1, we used the Pierce co-immunoprecipitation kit (Thermo Scientific, 26149). Negative controls were prepared with goat IgG (Santa Cruz Biotechnology, Inc., sc-2028) instead of the antibody. Both 0 h and 3 h samples were used as negative controls but showed no differences between them. The beads were then boiled in SDS with β-mercaptoethanol and the supernatant was collected and loaded on an 8% Tris-glycine gel. After transfer to a PVDF membrane (Immobilon®-P, Millipore, IPVH00010) the membranes were blocked with Tris-buffered saline containing 0.1% (v/v) Tween-20 and 5% (w/v) non-fat dry milk powder. Subsequently the membranes were incubated with primary antibody and horseradish peroxidase-conjugated secondary antibody. For immunoblot of full-length Beclin 1, we used the N-terminal anti-Beclin 1 described above. For the co-immunoprecipitation experiments, we used TrueBlot™ ULTRA horseradish peroxidase-conjugated secondary antibody from eBioscience (18-8817-313). The immunoreactive bands were visualized with ECL substrate and exposed to CL-XPosure™ film (Thermo Scientific). The film was developed using a Kodak X-Omat 1000 (Kodak). Quantification was done with ImageJ software (rsbweb.nih.gov/ij/).
GFP-LC3 measurements
HeLa cells transfected with pcDNA3.1(-)-GFP-LC3 were fixed in 4% paraformaldehyde 48 h after transfection. Cells were then observed under a Zeiss LSM510 confocal microscope using a 63× lens. The number of puncta per cell was determined using the WatershedCounting3D plug-in for ImageJ.Citation71 Cells were considered autophagic if they displayed more than 10 puncta.
Purification of GST-fusion proteins
pGEX-6P-2 constructs were transformed into BL21 (DE3) E. coli. Colonies were grown overnight in 50 ml of dYT medium (16 g/l peptone, 10 g/l yeast extract, 5 g/l NaCl, pH 7.4) at 37°C. dYT medium (~400 ml) was added to this preculture, and bacteria were further grown at 28°C until the A600 reached 0.8–1. Protein expression was induced by adding 0.1 mM IPTG and bacteria were further grown at 28°C for 4 h. Bacterial cells were harvested and lysed by sonication (9 x 10 sec, 12 kHz). Lysates were cleared by centrifugation (30 min, 15,000xg). The soluble fractions were then incubated during 2 h with glutathione-Sepharose™ 4B beads (GE Healthcare, 27-4574-01) at 4°C. After washing the beads, fusion proteins were eluted with 10 mM glutathione or cut with PreScission Protease. Purified proteins were dialyzed overnight against PBS or efflux medium, using Slide-A-Lyzer® dialysis cassettes with a 20 kDa cut-off (Thermo Fisher Scientific, 66003).
GST-pull-down assays
Purified and dialyzed GST-fusion proteins or parental GST (control) were incubated with purified Beclin 1 or Beclin 1-F123A in pull-down buffer (0.25% Triton X-100 and 1 mM DTT in Tris-buffered saline solution) and immobilized on glutathione-Sepharose™ 4B beads via rotation in a head-over-head rotator for 2 h at 4°C. The beads were washed 4 times and complexed GST-fusion proteins were eluted by boiling in LDS (Invitrogen, NP0007). Eluates were further analyzed using SDS-PAGE and western blotting.
XBP-1-mRNA splicing
RNA was extracted from HeLa cells treated with HBSS or TM using the RNeasy RNA-extraction kit (Qiagen, 74104). Reverse transcription was done using the Cloned AMV First-Strand cDNA-Synthesis Kit (Invitrogen, 12328032). XBP-1-splicing products were assessed by PCR using 5′-AAC TTT TGC TAG AAA ATC AGC as forward primer and 5′-CCA TGG GGA GAT GTT CTG GAG G as reverse primer. PCR products were loaded and run on a 1% agarose gel in Tris-borate-EDTA buffer and finally visualized with Gene Flash (Westburg).
XTT-assay
The XTT-assay was performed according to the manufacturer’s protocol (Biotium, 30007). Cell viability was determined in a 96-well plate by measuring the absorbance at 490 nm (A490) corrected for the absorbance at 630 nm (A630) as the reference wavelength.
Statistical analysis
Results are expressed as means ± SEM, and n refers to the number of independent experiments. For statistical analyses, significance was determined using one-tailed paired Student’s t-test. Differences were considered significant at p < 0.05.
Additional material
Download Zip (7.6 MB)Acknowledgments
We thank Marina Crabbé and Anja Florizoone for their technical assistance. This work was supported by Grant GOA/09/12 and OT START1/10/044 from the Research Council of the K.U. Leuven, by grant G073109N from the Research Foundation Flanders (FWO), and by the Interuniversity Poles of Attraction Programme-Belgian State, Prime Minister’s Office, Federal Office for Scientific, Technical, and Cultural Affairs, IUAP P6/28. JPD is recipient of a Ph.D. Fellowship from the Agency for Innovation by Science and Technology (IWT). The authors are very grateful to Dr. B. Levine (University of Texas Southwestern Medical Center, TX USA) for providing the Beclin 1-F123A construct, Dr. J. Debnath (University of California, San Francisco, CA USA) for the GFP-LC3 construct, Dr. K. Mikoshiba (Brain Science Institute, Wakō, Saitama, Japan) for the L15 cell line and Dr. P. Vangheluwe and Dr. F. Wuytack (K.U. Leuven, Belgium) for providing the SERCA2b antibody.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 2010; 90:1383 - 435; http://dx.doi.org/10.1152/physrev.00030.2009; PMID: 20959619
- Lee JY, Yao TP. Quality control autophagy: A joint effort of ubiquitin, protein deacetylase and actin cytoskeleton. Autophagy 2010; 6:555 - 7; http://dx.doi.org/10.4161/auto.6.4.11812; PMID: 20404488
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27 - 42; http://dx.doi.org/10.1016/j.cell.2007.12.018; PMID: 18191218
- Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol 2010; 12:823 - 30; http://dx.doi.org/10.1038/ncb0910-823; PMID: 20811354
- Cao Y, Klionsky DJ. Physiological functions of Atg6/Beclin 1: a unique autophagy-related protein. Cell Res 2007; 17:839 - 49; http://dx.doi.org/10.1038/cr.2007.78; PMID: 17893711
- Noble CG, Dong JM, Manser E, Song H. Bcl-xL and UVRAG cause a monomer-dimer switch in Beclin1. J Biol Chem 2008; 283:26274 - 82; http://dx.doi.org/10.1074/jbc.M804723200; PMID: 18641390
- He C, Levine B. The Beclin 1 interactome. Curr Opin Cell Biol 2010; 22:140 - 9; http://dx.doi.org/10.1016/j.ceb.2010.01.001; PMID: 20097051
- Burman C, Ktistakis NT. Regulation of autophagy by phosphatidylinositol 3-phosphate. FEBS Lett 2010; 584:1302 - 12; http://dx.doi.org/10.1016/j.febslet.2010.01.011; PMID: 20074568
- Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 2004; 6:463 - 77; http://dx.doi.org/10.1016/S1534-5807(04)00099-1; PMID: 15068787
- Ciechomska IA, Goemans GC, Skepper JN, Tolkovsky AM. Bcl-2 complexed with Beclin-1 maintains full anti-apoptotic function. Oncogene 2009; 28:2128 - 41; http://dx.doi.org/10.1038/onc.2009.60; PMID: 19347031
- Erlich S, Mizrachy L, Segev O, Lindenboim L, Zmira O, Adi-Harel S, et al. Differential interactions between Beclin 1 and Bcl-2 family members. Autophagy 2007; 3:561 - 8; PMID: 17643073
- Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell 2008; 30:678 - 88; http://dx.doi.org/10.1016/j.molcel.2008.06.001; PMID: 18570871
- Zalckvar E, Berissi H, Eisenstein M, Kimchi A. Phosphorylation of Beclin 1 by DAP-kinase promotes autophagy by weakening its interactions with Bcl-2 and Bcl-XL. Autophagy 2009; 5:720 - 2; http://dx.doi.org/10.4161/auto.5.5.8625; PMID: 19395874
- Tang D, Kang R, Livesey KM, Cheh CW, Farkas A, Loughran P, et al. Endogenous HMGB1 regulates autophagy. J Cell Biol 2010; 190:881 - 92; http://dx.doi.org/10.1083/jcb.200911078; PMID: 20819940
- Chang NC, Nguyen M, Germain M, Shore GC. Antagonism of Beclin 1-dependent autophagy by BCL-2 at the endoplasmic reticulum requires NAF-1. EMBO J 2010; 29:606 - 18; http://dx.doi.org/10.1038/emboj.2009.369; PMID: 20010695
- Vicencio JM, Lavandero S, Szabadkai G. Ca2+, autophagy and protein degradation: thrown off balance in neurodegenerative disease. Cell Calcium 2010; 47:112 - 21; http://dx.doi.org/10.1016/j.ceca.2009.12.013; PMID: 20097418
- Foskett JK, White C, Cheung KH, Mak DO. Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev 2007; 87:593 - 658; http://dx.doi.org/10.1152/physrev.00035.2006; PMID: 17429043
- Harr MW, Distelhorst CW. Apoptosis and autophagy: decoding calcium signals that mediate life or death. Cold Spring Harb Perspect Biol 2010; 2:a005579; http://dx.doi.org/10.1101/cshperspect.a005579; PMID: 20826549
- Sammels E, Parys JB, Missiaen L, De Smedt H, Bultynck G. Intracellular Ca2+ storage in health and disease: a dynamic equilibrium. Cell Calcium 2010; 47:297 - 314; http://dx.doi.org/10.1016/j.ceca.2010.02.001; PMID: 20189643
- Decuypere JP, Monaco G, Bultynck G, Missiaen L, De Smedt H, Parys JB. IP3 receptor-mitochondria connection in apoptosis and autophagy. Biochim Biophys Acta 2011; 1813:1003-13.
- Decuypere JP, Monaco G, Missiaen L, De Smedt H, Parys JB, Bultynck G. IP3 receptors, mitochondria, and Ca2+ signaling: Implications for aging. J Aging Res 2011; 2011:920178.
- Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol 2000; 1:11 - 21; http://dx.doi.org/10.1038/35036035; PMID: 11413485
- Criollo A, Maiuri MC, Tasdemir E, Vitale I, Fiebig AA, Andrews D, et al. Regulation of autophagy by the inositol trisphosphate receptor. Cell Death Differ 2007; 14:1029 - 39; PMID: 17256008
- Khan MT, Joseph SK. Role of inositol trisphosphate receptors in autophagy in DT40 cells. J Biol Chem 2010; 285:16912 - 20; http://dx.doi.org/10.1074/jbc.M110.114207; PMID: 20308071
- Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M, et al. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol 2005; 170:1101 - 11; http://dx.doi.org/10.1083/jcb.200504035; PMID: 16186256
- Cárdenas C, Miller RA, Smith I, Bui T, Molgo J, Muller M, et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010; 142:270 - 83; http://dx.doi.org/10.1016/j.cell.2010.06.007; PMID: 20655468
- Høyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell 2007; 25:193 - 205; http://dx.doi.org/10.1016/j.molcel.2006.12.009; PMID: 17244528
- Grotemeier A, Alers S, Pfisterer SG, Paasch F, Daubrawa M, Dieterle A, et al. AMPK-independent induction of autophagy by cytosolic Ca2+ increase. Cell Signal 2010; 22:914 - 25; http://dx.doi.org/10.1016/j.cellsig.2010.01.015; PMID: 20114074
- Wang SH, Shih YL, Ko WC, Wei YH, Shih CM. Cadmium-induced autophagy and apoptosis are mediated by a calcium signaling pathway. Cell Mol Life Sci 2008; 65:3640 - 52; http://dx.doi.org/10.1007/s00018-008-8383-9; PMID: 18850067
- Sakaki K, Wu J, Kaufman RJ. Protein kinase Ctheta is required for autophagy in response to stress in the endoplasmic reticulum. J Biol Chem 2008; 283:15370 - 80; http://dx.doi.org/10.1074/jbc.M710209200; PMID: 18356160
- Vicencio JM, Ortiz C, Criollo A, Jones AW, Kepp O, Galluzzi L, et al. The inositol 1,4,5-trisphosphate receptor regulates autophagy through its interaction with Beclin 1. Cell Death Differ 2009; 16:1006 - 17; http://dx.doi.org/10.1038/cdd.2009.34; PMID: 19325567
- Lytton J, Westlin M, Hanley MR. Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. J Biol Chem 1991; 266:17067 - 71; PMID: 1832668
- Miyawaki A, Furuichi T, Maeda N, Mikoshiba K. Expressed cerebellar-type inositol 1,4,5-trisphosphate receptor, P400, has calcium release activity in a fibroblast L cell line. Neuron 1990; 5:11 - 8; http://dx.doi.org/10.1016/0896-6273(90)90029-F; PMID: 2164403
- Rong YP, Aromolaran AS, Bultynck G, Zhong F, Li X, McColl K, et al. Targeting Bcl-2-IP3 receptor interaction to reverse Bcl-2's inhibition of apoptotic calcium signals. Mol Cell 2008; 31:255 - 65; http://dx.doi.org/10.1016/j.molcel.2008.06.014; PMID: 18657507
- Bultynck G, Szlufcik K, Kasri NN, Assefa Z, Callewaert G, Missiaen L, et al. Thimerosal stimulates Ca2+ flux through inositol 1,4,5-trisphosphate receptor type 1, but not type 3, via modulation of an isoform-specific Ca2+-dependent intramolecular interaction. Biochem J 2004; 381:87 - 96; http://dx.doi.org/10.1042/BJ20040072; PMID: 15015936
- Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy 2007; 3:542 - 5; PMID: 17611390
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008; 4:151 - 75; PMID: 18188003
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313 - 26; http://dx.doi.org/10.1016/j.cell.2010.01.028; PMID: 20144757
- Klionsky DJ, Cuervo AM, Seglen PO. Methods for monitoring autophagy from yeast to human. Autophagy 2007; 3:181 - 206; PMID: 17224625
- Szeto J, Kaniuk NA, Canadien V, Nisman R, Mizushima N, Yoshimori T, et al. ALIS are stress-induced protein storage compartments for substrates of the proteasome and autophagy. Autophagy 2006; 2:189 - 99; PMID: 16874109
- Sriburi R, Jackowski S, Mori K, Brewer JW. XBP1: a link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J Cell Biol 2004; 167:35 - 41; http://dx.doi.org/10.1083/jcb.200406136; PMID: 15466483
- Missiaen L, De Smedt H, Parys JB, Raeymaekers L, Droogmans G, Van Den Bosch L, et al. Kinetics of the non-specific calcium leak from non-mitochondrial calcium stores in permeabilized A7r5 cells. Biochem J 1996; 317:849 - 53; PMID: 8760372
- Missiaen L, De Smedt H, Droogmans G, Casteels R. Ca2+ release induced by inositol 1,4,5-trisphosphate is a steady-state phenomenon controlled by luminal Ca2+ in permeabilized cells. Nature 1992; 357:599 - 602; http://dx.doi.org/10.1038/357599a0; PMID: 1608471
- Missiaen L, De Smedt H, Parys JB, Casteels R. Co-activation of inositol trisphosphate-induced Ca2+ release by cytosolic Ca2+ is loading-dependent. J Biol Chem 1994; 269:7238 - 42; PMID: 8125936
- Parys JB, Missiaen L, De Smedt H, Casteels R. Loading dependence of inositol 1,4,5-trisphosphate-induced Ca2+ release in the clonal cell line A7r5. Implications for the mechanism of quantal Ca2+ release. J Biol Chem 1993; 268:25206 - 12; PMID: 8227085
- Missiaen L, Van Acker K, Van Baelen K, Raeymaekers L, Wuytack F, Parys JB, et al. Calcium release from the Golgi apparatus and the endoplasmic reticulum in HeLa cells stably expressing targeted aequorin to these compartments. Cell Calcium 2004; 36:479 - 87; http://dx.doi.org/10.1016/j.ceca.2004.04.007; PMID: 15488597
- Chan J, Yamazaki H, Ishiyama N, Seo MD, Mal TK, Michikawa T, et al. Structural studies of inositol 1,4,5-trisphosphate receptor: coupling ligand binding to channel gating. J Biol Chem 2010; 285:36092 - 9; http://dx.doi.org/10.1074/jbc.M110.140160; PMID: 20843799
- Missiaen L, Parys JB, Weidema AF, Sipma H, Vanlingen S, De Smet P, et al. The bell-shaped Ca2+ dependence of the inositol 1,4,5-trisphosphate-induced Ca2+ release is modulated by Ca2+/calmodulin. J Biol Chem 1999; 274:13748 - 51; http://dx.doi.org/10.1074/jbc.274.20.13748; PMID: 10318777
- Rong YP, Bultynck G, Aromolaran AS, Zhong F, Parys JB, De Smedt H, et al. The BH4 domain of Bcl-2 inhibits ER calcium release and apoptosis by binding the regulatory and coupling domain of the IP3 receptor. Proc Natl Acad Sci USA 2009; 106:14397 - 402; http://dx.doi.org/10.1073/pnas.0907555106; PMID: 19706527
- Rial DV, Ceccarelli EA. Removal of DnaK contamination during fusion protein purifications. Protein Expr Purif 2002; 25:503 - 7; http://dx.doi.org/10.1016/S1046-5928(02)00024-4; PMID: 12182832
- Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, et al. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol 1998; 72:8586 - 96; PMID: 9765397
- Chen R, Valencia I, Zhong F, McColl KS, Roderick HL, Bootman MD, et al. Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. J Cell Biol 2004; 166:193 - 203; http://dx.doi.org/10.1083/jcb.200309146; PMID: 15263017
- Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005; 122:927 - 39; http://dx.doi.org/10.1016/j.cell.2005.07.002; PMID: 16179260
- Jaimovich E, Mattei C, Liberona JL, Cardenas C, Estrada M, Barbier J, et al. Xestospongin B, a competitive inhibitor of IP3-mediated Ca2+ signalling in cultured rat myotubes, isolated myonuclei, and neuroblastoma (NG108-15) cells. FEBS Lett 2005; 579:2051 - 7; http://dx.doi.org/10.1016/j.febslet.2005.02.053; PMID: 15811317
- Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 2007; 26:1749 - 60; http://dx.doi.org/10.1038/sj.emboj.7601623; PMID: 17347651
- Buytaert E, Callewaert G, Hendrickx N, Scorrano L, Hartmann D, Missiaen L, et al. Role of endoplasmic reticulum depletion and multidomain proapoptotic BAX and BAK proteins in shaping cell death after hypericin-mediated photodynamic therapy. FASEB J 2006; 20:756 - 8; PMID: 16455754
- Hinnebusch AG. Translational regulation of GCN4 and the general amino acid control of yeast. Annu Rev Microbiol 2005; 59:407 - 50; http://dx.doi.org/10.1146/annurev.micro.59.031805.133833; PMID: 16153175
- Guerrero-Hernandez A, Dagnino-Acosta A, Verkhratsky A. An intelligent sarco-endoplasmic reticulum Ca2+ store: Release and leak channels have differential access to a concealed Ca2+ pool. Cell Calcium 2010; 48:143 - 9; http://dx.doi.org/10.1016/j.ceca.2010.08.001; PMID: 20817294
- Gordon PB, Holen I, Fosse M, Rotnes JS, Seglen PO. Dependence of hepatocytic autophagy on intracellularly sequestered calcium. J Biol Chem 1993; 268:26107 - 12; PMID: 8253727
- Brady NR, Hamacher-Brady A, Yuan H, Gottlieb RA. The autophagic response to nutrient deprivation in the hl-1 cardiac myocyte is modulated by Bcl-2 and sarco/endoplasmic reticulum calcium stores. FEBS J 2007; 274:3184 - 97; http://dx.doi.org/10.1111/j.1742-4658.2007.05849.x; PMID: 17540004
- Chan J, Whitten AE, Jeffries CM, Bosanac I, Mal TK, Ito J, et al. Ligand-induced conformational changes via flexible linkers in the amino-terminal region of the inositol 1,4,5-trisphosphate receptor. J Mol Biol 2007; 373:1269 - 80; http://dx.doi.org/10.1016/j.jmb.2007.08.057; PMID: 17915250
- Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, et al. Novel targets for Huntington's disease in an mTOR-independent autophagy pathway. Nat Chem Biol 2008; 4:295 - 305; http://dx.doi.org/10.1038/nchembio.79; PMID: 18391949
- Decuypere JP, Bultynck G, Parys JB. A dual role for Ca2+ in autophagy regulation. Cell Calcium 2011; In press http://dx.doi.org/10.1016/j.ceca.2011.04.001; PMID: 21571367
- Fung C, Lock R, Gao S, Salas E, Debnath J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol Biol Cell 2008; 19:797 - 806; http://dx.doi.org/10.1091/mbc.E07-10-1092; PMID: 18094039
- Parys JB, De Smedt H, Missiaen L, Bootman MD, Sienaert I, Casteels R. Rat basophilic leukemia cells as model system for inositol 1,4,5-trisphosphate receptor IV, a receptor of the type II family: functional comparison and immunological detection. Cell Calcium 1995; 17:239 - 49; http://dx.doi.org/10.1016/0143-4160(95)90070-5; PMID: 7664312
- Ma HT, Venkatachalam K, Parys JB, Gill DL. Modification of store-operated channel coupling and inositol trisphosphate receptor function by 2-aminoethoxydiphenyl borate in DT40 lymphocytes. J Biol Chem 2002; 277:6915 - 22; http://dx.doi.org/10.1074/jbc.M107755200; PMID: 11741932
- Wuytack F, Eggermont JA, Raeymaekers L, Plessers L, Casteels R. Antibodies against the non-muscle isoform of the endoplasmic reticulum Ca2+-transport ATPase. Biochem J 1989; 264:765 - 9; PMID: 2482734
- Quirion JC, Sevenet T, Husson HP, Weniger B, Debitus C. Two new alkaloids from Xestospongia sp., a New Caledonian sponge. J Nat Prod 1992; 55:1505 - 8; http://dx.doi.org/10.1021/np50088a017; PMID: 1453184
- Missiaen L, Declerck I, Droogmans G, Plessers L, De Smedt H, Raeymaekers L, et al. Agonist-dependent Ca2+ and Mn2+ entry dependent on state of filling of Ca2+ stores in aortic smooth muscle cells of the rat. J Physiol 1990; 427:171 - 86; PMID: 2213595
- Decuypere JP, Monaco G, Kiviluoto S, Oh-hora M, Luyten T, De Smedt H, et al. STIM1, but not STIM2, is required for proper agonist-induced Ca2+ signaling. Cell Calcium 2010; 48:161 - 7; http://dx.doi.org/10.1016/j.ceca.2010.08.003; PMID: 20801505
- Gniadek TJ, Warren G. WatershedCounting3D: a new method for segmenting and counting punctate structures from confocal image data. Traffic 2007; 8:339 - 46; http://dx.doi.org/10.1111/j.1600-0854.2007.00538.x; PMID: 17319897