Abstract
In recent years, the process of selective autophagy has received much attention with respect to the clearance of protein aggregates, damaged mitochondria, and bacteria. However, until recently, there have been virtually no studies on the selective autophagy of viruses, although they are perhaps one of the most ubiquitous unwanted constituents in human cells. Recently, we have shown that the ability of neuronal Atg5 to protect against lethal Sindbis virus central nervous system (CNS) infection in mice is associated with impaired viral capsid clearance, increased p62 accumulation, and increased neuronal cell death. In vitro, we showed that p62 interacts with the Sindbis capsid protein and targets it for degradation in autophagosomes. Herein, we review these findings and broadly speculate about potential roles of selective viral autophagy in the regulation of host immunity and viral pathogenesis.
Acknowledgements
The work in the authors' laboratory was supported by the Ellison Medical Foundation Senior Scholars Award in Infectious Diseases (B.L.) and NIH RO1 AI151367 (B.L.). We thank Angela Diehl for expert scientific illustration.
Figures and Tables
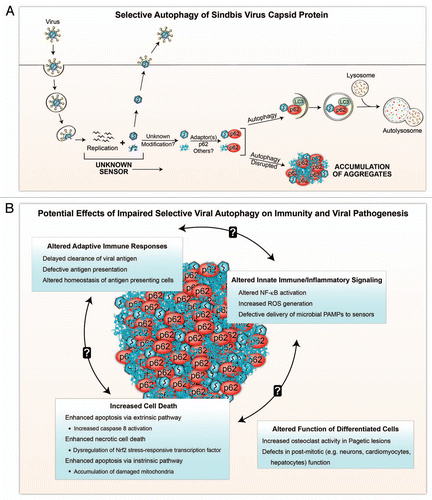
Figure 1 Schematic model of selective viral autophagy (A) and the potential biological consequences of viral protein-p62 aggregate accumulation during impaired selective autophagy (B). (A) After entry into the host cell and uncoating, Sindbis virus (SIN) replicates in the cytosol, generating newly synthesized viral nucleic acids and proteins. Autophagy is triggered by an unknown sensor(s) during viral replication, and SIN capsid protein is targeted to the autophagic machinery in a process that requires the selective autophagy adaptor protein, p62. It is unknown whether p62 recognizes free capsid (monomeric or aggregated) or assembled capsid (containing viral or other nucleic acids), whether the targeted capsid proteins undergo modification prior to recognition by p62, or whether interactions with additional adaptor proteins are required for selective autophagy of SIN capsid. Disruption of autophagy results in the accumulation of aggregates of viral proteins and p62 within the host cell. (B) In the absence of selective autophagy, aggregates containing viral capsid and p62 may perturb innate and adaptive immune response, and result in increased cell death and disruption of cellular functions. The processes listed are speculations based on extrapolations from the literature on p62 functions (see text for more detailed explanations). Further studies are required to test these speculations in models of viral infection.