Abstract
Bacillus thuringiensis (Bt) is a unique bacterium in that it shares a common place with a
number of chemical compounds which are used commercially to control insects important to
agriculture and public health. Although other bacteria, including B. popilliae and B. sphaericus,
are used as microbial insecticides, their spectrum of insecticidal activity is quite limited
compared to Bt. Importantly, Bt is safe for humans and is the most widely used environmentally
compatible biopesticide worldwide. Furthermore, insecticidal Bt genes have been incorporated
into several major crops, rendering them insect resistant, and thus providing a model for genetic
engineering in agriculture.
This review highlights what the authors consider the most relevant issues and topics
pertaining to the genomics and proteomics of Bt. At least one of the authors (LAB) has spent
most of his professional life studying different aspects of this bacterium with the goal in mind of
determining the mechanism(s) by which it kills insects. The other authors have a much shorter
experience with Bt but their intellect and personal insight have greatly enriched our
understanding of what makes Bt distinctive in the microbial world. Obviously, there is personal
interest and bias reflected in this article notwithstanding oversight of a number of published
studies. This review contains some material not published elsewhere although several ideas and
concepts were developed from a broad base of scientific literature up to 2010.
Acknowledgements
We are deeply grateful for the expert technical assistance of Shweta Biliya and Gayathri Raghupathy in preparing this manuscript.
Figures and Tables
Figure 1 Transmission electron micrograph of a sporulated cell of Bacillus thuringiensis subsp. morrisoni strain C18 (× 44,000). Strain C18 was isolated from dead cotton bollworm larvae. The strain is unique in that it harbors 15 cry genes. The host range of C18 includes three major insect orders: Lepidoptera, Diptera and Coleoptera as well as nematodes. The parasporal crystals of C18 vary in shape and size (arrows), and represent ∼50% of the cell dry weight.
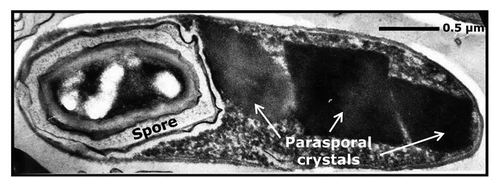
Figure 2 Bootstrapped neighbor-joining tree of 20 strains belonging to different species of the BC group. The tree was generated based on nucleotide sequence alignment of 16S rRNA. Bootstrap volumes are reported on the branches. The 20 strains are divided into two main groups. Group A includes all Ba, Bc and Bpm strains and Group B includes Bw strain KBAB4 and Bm strain SDANFMO448. There are two clusters within Group A, each marked with Roman numerals I and II. Cluster I includes Ba strains Sterne CDC684 and Ames Ancestor A0248; Bc strains AH187 and AH 820 and ATCC 10987 and 14579; Bpm strain CIP 5259; and Bt serovar konkukian and Bt Al-Hakm. Bpm strain CIP 5259 and Bc ATCC 14579 are the most divergent in this cluster. Cluster II includes Bt serovars tenebrionis, morrisoni, kurstaki, sotto, israelensis and berliner and Bc G9842. The horizontal bar represents 0.02% differences in nucleotide similarities.
Figure 3 Cadherin receptor and Cry1Ab toxin binding analysis. (A) SDS-PAGE. Lane 1, molecular markers; lane 2, Cry1Ab toxin; lane 3, proteinase K-digested Cry1Ab. (B) Immunoligand blot. Cry1Ab toxin and its proteinase K proteolytic fragments were trans-blotted to a PVDF membrane and tested for their ability to bind the soluble toxin-binding region of BT-R1 (TBR). Lane 1, Cry1AB toxin; lane 2, proteinase K digested Cry1Ab. The smallest binding fragment of Cry1Ab (∼22 kDa, double arrow) was subjected to N-terminal amino acid sequence analysis. The N-terminal amino acid sequence is 442NSSVSIIRAPMFSWIHR458.
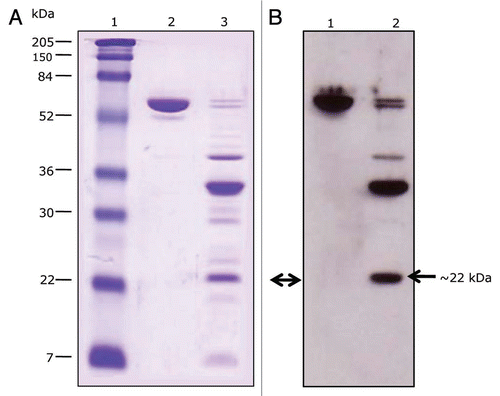
Figure 4 Proposed secondary structure and three-dimensional model for the putative receptor-binding regions of Cry1Ab, Cry3A and Cry4B. (A) Multiple sequence alignments and secondary structure prediction of the amino acid residues spanning β10, loop 3, β11–β16 (end of Domain II and beginning of Domain III) of Cry1Ab (residues 422–498); Cry3A (residues 466–537) and Cry4B (residues 437–506), using PfamCitation181 and SASCitation182 databases. The sequence in red represents solvent-exposed residues. Underlined, bold residues in the three Cry toxins constitute loop 3. The symbols (*), (:) and (.) beneath the alignments denote identical, similar and less similar amino acid residues, respectively. (B) Three-dimensional structure in ribbon format for the putative receptor-binding regions on Cry1Ab, Cry3A and Cry4B. The Cry3A and Cry4B structures are from crystal structures (PDB codes 1DLC and 1W99, respectively). The Cry1Ab structure was generated as a model by the SWISS-MODEL protein modeling serverCitation173 using the crystal structure of the Cry1Aa toxin (88% sequence identity; PDB code 1CIY) as a template. Blue, red and golden shaded areas highlight predicted β-strands and loops.
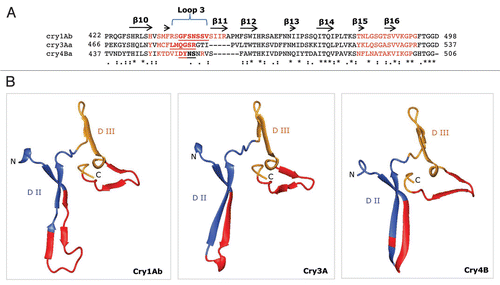
Figure 5 Domain structure and putative Cry toxin-binding regions in cadherin-like proteins from mosquito (red), moth (green) and beetle (golden). EC, ectodomain; MPED, membrane proximal extracellular domain; TM, transmembrane domain; CYTO, cytoplasmic domain, TBR, toxin binding region.
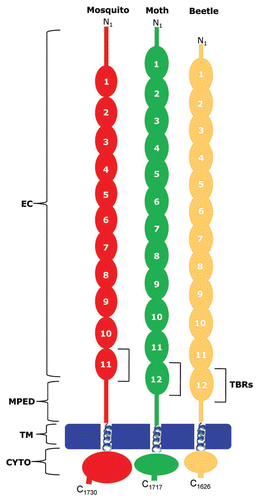
Figure 6 Predicted secondary structure and three-dimensional models of EC modules containing the TBRs for moth, mosquito and beetle. (A) Multiple sequence alignments and secondary structure prediction of the amino acid residues spanning the TBRs of BT-R1 (residues 1349–1460), BT-R3 (residues 1339–1451) and BT-R4 (residues 1292–1405). The structures were generated using the PfamCitation181 and SASCitation182 databases. The two black boxes contain the conserved residues in the upstream and downstream regions of the Cry toxin-binding motifs. The designations beneath the alignments are as described in the legend to . (B) Three-dimensional models of the TBRs of BT-R1, BT-R3 and BT-R4 generated from crystal structures of homologous cadherin domains from a mouse E-cadherin (PDB code ledh), a mouse N-cadherin (PDB code 1ncj), and a frog C-cadherin (PDB code 1l3w). The three TBRs were templated onto the superimposed crystal structures by sequence alignment using the DeepView (Swiss-PdbViewer) computer program.Citation183 The alignment was guided by secondary structure prediction by the JPred 3 serverCitation184 to match predicted β-strands in the receptor sequences to β-strands in the cadherin crystal structures. The templated models were then refined on the SWISS-MODEL protein modeling server.Citation173 Golden arrows and gray lines highlight predicted β-strands and loops, respectively. Blue arrows and blue lines represent sequences that flank the TBRs and are required for binding.
Figure 7 Proposed model for Cry toxin action. The univalent binding of Cry toxin monomer to BT-R initiates the progression of cell death by transmitting a death signal into the cell. A signal transduction pathway, involving G protein (Gα) adenylyl cyclase (AC) and protein kinase A (PKA), is activated. Activation of the signaling pathway mediates exocytosis of the BT-R receptor from intracellular vesicles to the cell membrane. The resulting enhanced display of BT-R on the cell surface facilitates recruitment of additional toxin molecules which, in turn, amplifies the original signal in a cascade-like fashion. The signaling kinase PKA modifies downstream molecules that promote the biochemical activities that destroy the cell. Toxin oligomers incorporated into the plasma membrane of living cells do not form lytic pores and are not toxic.
Figure 8 Morphological changes associated with BT-R-transfected High-Five cells treated with mosquito and moth toxins. High-Five cells transfected with BT-R3 cDNA (M5 cells, upper photos) and BT-R1 cDNA (S5 cells, lower photos) were treated with Cry4Ba and Cry1Ab, respectively. The sequence of cytological changes during the course of toxin-induced cell death was captured by phase contrast microscopy. The long arrow beneath the photographs indicates the stages of cell death: toxin binding, membrane blebbing and cellular swelling. Cytotoxicity is Mg2+-dependent and involves exocytosis of the cadherin receptors (BT-R).
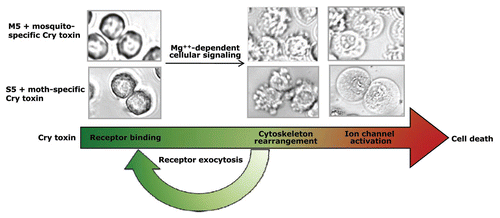
Figure 9 A proposed model showing docking of the Cry1Ab toxin to BT-R1. (A) Ribbon structures showing docking of the putative receptor-binding sequence of the Cry1Ab toxin (residues 422–498) to the TBR (residues 1349–1460) in BT-R1. The structure for the TBR was generated from the X-ray crystal structures of homologous cadherin domains as described in the legend to . Note that in the interface between the Cry1Ab toxin and the TBR of BT-R1 there are potential hydrophobic interactions involving surface-exposed Phe440 in Cry1Ab and Leu1358, Tyr1453 and Val1455 in the TBR of BT-R1. Also, there is probable electrostatic interaction between the positively charged Arg437 in Cry1Ab and the negatively charged Glu1357 in the TBR. Glu1357, Leu1358, Tyr1453 and Val1455 lie either within or immediately adjacent to the signature sequences in the TBR of BT-R1 ( and blue arrows). Ribbon (B) and space-filling (C) representations showing docking of Cry1Ab toxin to TBR. Domains I, II and III of the toxin are colored green, blue and gold, respectively. The TBR is colored pink. Surface-exposed residues 432–449 and 480–493 in Cry1Ab are colored red, and residues 1349–1354 and 1451–1460 (signature sequences) in the TBR are colored purple.
Figure 10 Possible mechanisms of insect resistance to Bt. Once Bt parasporal crystals are ingested by an insect larva, the crystals are dissociated, followed by proteolytic activation of protoxin and conversion to toxin. The activated toxin binds to the receptor, effecting a cascade of signal transduction events that lead to eventual larval death. There are three ways by which the insect larva could become resistant: (A) an enhanced immune response that inhibits dissociation and accessibility of protoxin; (B) erroneous protease production or defective protease activity that interferes with protoxin/toxin activation; (C) reduced number of binding sites or mutated receptors that retard or prevent appropriate toxin-receptor interaction.
Figure 11 A genomics/proteomics platform for new insecticide discovery. There are three pathways to discovery, identification and validation of insecticide targets. (A) Bioinformatics pipeline to identify target proteins and to design specific primers or probes for cDNA screening. The process of target selection starts with data mining of a list of protein sequences from available genomic databases. The proteins are selected and categorized based on their potential to mediate insecticidal action. (B) Construction of cDNA libraries from tissue or cell samples, cloning and transfection of selected genes in insect cells and formatting of transfected cells for gene expression. (C) Construction of engineered toxins and target screening for evaluation and validation.