Abstract
The special architecture of neurons in the peripheral nervous system, with axons extending for long distances, represents a major challenge for the intracellular transport system. Two recent studies show that mutations in the small heat shock protein HSPB1, which cause an axonal type of Charcot-Marie-Tooth (CMT) neuropathy, affect microtubule dynamics and impede axonal transport. Intriguingly, while at presymptomatic age the neurons in the mutant HSPB1 mouse show a hyperstable microtubule network, at postsymptomatic age, the microtubule network completely lost its stability as reflected by a marked decrease in tubulin acetylation levels. We here propose a model explaining the role of microtubule stabilization and tubulin acetylation in the pathogenesis of HSPB1 mutations.
The peripheral nervous system is responsible for exchanging information between the central nervous system and the rest of our body. To do so, peripheral neurons project their axons throughout the body over distances that can range from a few millimeters up to one meter, in the case of nerves connecting the spinal cord with our hands and feet. This particular anatomical architecture poses a significant challenge on these neurons and requires an efficient transport of proteins, RNA, vesicles and organelles between the cell body and the axon tip. This transport, generally called axonal transport, is mediated by the motor proteins dynein and kinesin and a highly polarized microtubule network, in which the microtubule-minus end is pointed toward the cell body and the microtubule-plus end points toward the axon tip. Microtubules are cytoskeletal structures composed of heterodimers of α- and β-tubulin; they extend in all directions throughout the cell forming a dynamic network that continuously grows, retracts, bends and breaks. Therefore, rather than providing cellular rigidity, microtubules are important for enabling dynamic processes such as intracellular transport or mitotic spindle formation that heavily depend on their ability to be polymerized, depolymerized and severed.Citation1 The tight regulation of their dynamics is pivotal to ensure efficient transport of cargoes along the axons.Citation2,Citation3
While all neuronal cell types depend on an efficient axonal transport for their function, peripheral neurons seem to be particularly susceptible to disturbances in axonal transport, as evidenced by the large number of cellular transport related genesCitation4-Citation6 in which mutations specifically lead to peripheral nerve degeneration. Furthermore, several chemotherapeutic drugs that target the microtubule network cause peripheral neurodegeneration, which is their major dose limiting side-effect.Citation7,Citation8
Missense mutations in the small heat shock protein HSPB1 (also known as HSP27) cause two types of peripheral neuropathy: Charcot-Marie-Tooth disease (CMT) type 2F and distal hereditary motor neuropathy (distal HMN).Citation9 Both diseases are very similar and clinically characterized by a length-dependent degeneration of peripheral nerves, resulting in progressive weakness in the limbs and wasting of foot and hand muscles. In contrast to most other chaperonopathies in which mutations generally lead to a loss in chaperone activity, a subset of HSPB1 mutations led to an increase in HSPB1 chaperone activity, which was associated with an enhanced binding to their client proteins.Citation10 In a recent study we found that the main targets of hyperactive HSPB1 mutants appeared to be tubulin and microtubules.Citation11 This anomalous binding resulted in an increased stability of the microtubule network in cells expressing the hyperactive mutants,Citation11 reminiscent of the activity of classical microtubule-associated proteins (MAP).Citation12 Importantly, we were able to confirm the enhanced binding to tubulin and increased microtubule stability in dorsal root ganglia (DRG) neurons isolated from 3 month-old (pre-symptomatic) mice expressing the hyperactive HSPB1-S135F mutantCitation13 (see also further). Intriguingly, the stabilization caused by the hyperactive HSPB1 mutants was not reflected by an increase in tubulin acetylation,Citation11 a post-translational modification commonly associated with increased microtubule stability.Citation14-Citation16 Furthermore despite being more in the pause phase, microtubules from cells expressing mutant HSPB1 depolymerize at a much faster speed than wild type microtubules, once they do. Therefore, we hypothesized that both phenomena (the absence of acetylation and the higher depolymerization speed) reflect the fact that the enhanced stability is not the result of a proper stabilization event, controlled by appropriate cellular signals, but rather the result of an incomplete or aberrant microtubule stabilization event due to the presence of a mutated chaperone with strongly increased binding properties.Citation11
In another recent study, d’Ydewalle et al.Citation13 describe that the mouse model expressing the hyperactive HSPB1 (S135F) mutation present at 8 months of age, when symptoms are clear and axonal loss became apparent, markedly decreased tubulin acetylation levels, which is an indication of loss of microtubule stability. Moreover, chemical inhibition of HDAC6, an α-tubulin deacetylating enzyme,Citation17 was able to increase the acetylation levels of microtubules in peripheral neurons and rescue the CMT phenotype.Citation13
Importantly, as mentioned above, in the same mouse model but at an earlier age (presymptomatic state at three months of age), the opposite results could be observed and an enhanced stability of the microtubule network was detected.Citation11 This suggests that the pathology of this CMT type is composed of two temporally separated and opposing microtubule states, a presymptomatic hyperstable state followed by a postsymptomatic unstable state, and that the transition between these states might trigger the appearance of symptoms.
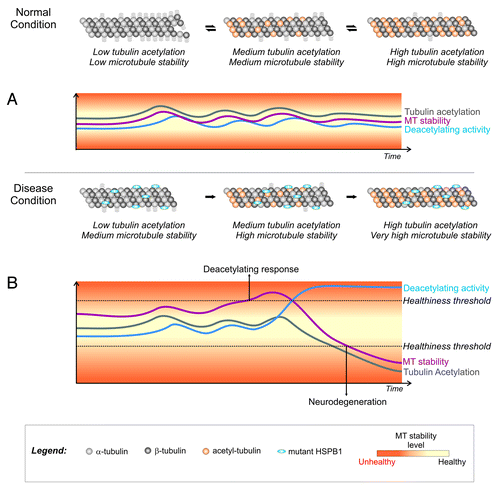
How the transition occurs from an overstable microtubule network to a deacetylated and unstable microtubule network is still unknown. Here we propose a simplified model where, under normal conditions, the dynamicity of the microtubule network in neurons is kept at an optimal level by the controlled action of tubulin acetylating (Elp3 and MEC-17)Citation18-Citation20 and deacetylating (HDAC6 and SIRT2)Citation17,Citation21 enzymes (). In disease conditions, when neurons express the hyperactive HSPB1 mutants, there is a discrepancy between the dynamicity of the microtubule network and tubulin acetylation levels (). After an extended period of increased microtubule stability the cell triggers a tubulin deacetylating response by increasing the recruitment of HDAC6 (or other deacetylating enzymes) to microtubules in an attempt to restore normal microtubule dynamics. This response results in an excessive loss of tubulin acetylation and, consequently, an extensive destabilization of the microtubule network (). Moreover, the excessively low levels of tubulin acetylation also reduce the recruitment of motor proteins to the microtubules.Citation22 With a severely destabilized microtubule network and a disturbed axonal transport system, these neurons are not able to function properly and degenerate.
Figure 1. Model for the role of tubulin acetylation in the stability of the neuronal microtubule network under normal and disease conditions (*). (A) Under normal conditions, there is a correlation between tubulin acetylation levels and microtubule stability. Acetylating and deacetylating enzymes are able to control acetylation levels and maintain microtubule stability at healthy levels. (B) In the presence of the microtubule-stabilizing CMT-causing HSPB1 mutants, tubulin acetylation levels do not reflect the degree of stability of the microtubule network. An increase in microtubule stability would eventually trigger a cellular deacetylating response as an attempt to restore normal stability levels. Due to the discrepancy between acetylation and microtubule stability levels, this cellular response results in an excessive loss of acetylation that reduces integrity of the microtubule network, impairs axonal transport and causes neurodegeneration. *To simplify the model we highlighted only the tubulin acetylation levels in the figure. We recognize that a complete model for microtubule stability control must also include the role of MAPs and other post-translational modifications.
Tubulin acetylation was considered for many years a passive player in the regulation of microtubule dynamics.Citation23 However, the finding that acetylation is able to control the binding of kinesin to microtubules,Citation22,Citation24 the discovery that the lack of the tubulin acetylase Mec-17 renders microtubules unstableCitation23 and the results from Almeida-SouzaCitation11 and d’YdewalleCitation13 that we describe above, place tubulin acetylation into the spotlight as an essential player in the function of the neuronal microtubule cytoskeleton, especially in the peripheral nervous system.
Despite the enormous advances on the understanding of the role of tubulin acetylation on the microtubule cytoskeleton,Citation25 a lot of work remains to be done to build a full picture on how this post-translational modification is controlled in neurons in vivo and how it interacts with other microtubule dynamics regulators. The modulation of tubulin acetylation levels has shown to be a promising therapeutic strategy for neurodegenerative diseases.Citation13,Citation24 For that reason, a detailed knowledge of this process is essential to enable the development of efficient and safe drugs for peripheral neuropathies and possibly a multitude of neurodegenerative diseases.
Acknowledgments
This work was supported by the Methusalem program of the University of Antwerp, the Fund for Scientific Research (FWO-Flanders), the Medical Foundation Queen Elisabeth, the “Association Belge contre les Maladies Neuromusculaires,” and the American Muscular Dystrophy Association.
References
- Baas PW, Karabay A, Qiang L. Microtubules cut and run. Trends Cell Biol 2005; 15:518 - 24; http://dx.doi.org/10.1016/j.tcb.2005.08.004; PMID: 16126385
- Gundersen GG. Evolutionary conservation of microtubule-capture mechanisms. Nat Rev Mol Cell Biol 2002; 3:296 - 304; http://dx.doi.org/10.1038/nrm777; PMID: 11994749
- Conde C, Ćceres A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat Rev Neurosci 2009; 10:319 - 32; http://dx.doi.org/10.1038/nrn2631; PMID: 19377501
- Verhoeven K, De Jonghe P, Coen K, Verpoorten N, Auer-Grumbach M, Kwon JM, et al. Mutations in the small GTP-ase late endosomal protein RAB7 cause Charcot-Marie-Tooth type 2B neuropathy. Am J Hum Genet 2003; 72:722 - 7; http://dx.doi.org/10.1086/367847; PMID: 12545426
- Züchner S, Noureddine M, Kennerson M, Verhoeven K, Claeys K, De Jonghe P, et al. Mutations in the pleckstrin homology domain of dynamin 2 cause dominant intermediate Charcot-Marie-Tooth disease. Nat Genet 2005; 37:289 - 94; http://dx.doi.org/10.1038/ng1514; PMID: 15731758
- Rivière JB, Ramalingam S, Lavastre V, Shekarabi M, Holbert S, Lafontaine J, et al. KIF1A, an axonal transporter of synaptic vesicles, is mutated in hereditary sensory and autonomic neuropathy type 2. Am J Hum Genet 2011; 89:219 - 30; http://dx.doi.org/10.1016/j.ajhg.2011.06.013; PMID: 21820098
- Wolf S, Barton D, Kottschade L, Grothey A, Loprinzi C. Chemotherapy-induced peripheral neuropathy: prevention and treatment strategies. Eur J Cancer 2008; 44:1507 - 15; http://dx.doi.org/10.1016/j.ejca.2008.04.018; PMID: 18571399
- Lee JJ, Swain SM. Peripheral neuropathy induced by microtubule-stabilizing agents. J Clin Oncol 2006; 24:1633 - 42; http://dx.doi.org/10.1200/JCO.2005.04.0543; PMID: 16575015
- Evgrafov OV, Mersiyanova I, Irobi J, Van Den Bosch L, Dierick I, Leung CL, et al. Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat Genet 2004; 36:602 - 6; http://dx.doi.org/10.1038/ng1354; PMID: 15122254
- Almeida-Souza L, Goethals S, de Winter V, Dierick I, Gallardo R, Van Durme J, et al. Increased monomerization of mutant HSPB1 leads to protein hyperactivity in Charcot-Marie-Tooth neuropathy. J Biol Chem 2010; 285:12778 - 86; http://dx.doi.org/10.1074/jbc.M109.082644; PMID: 20178975
- Almeida-Souza L, Asselbergh B, d’Ydewalle C, Moonens K, Goethals S, de Winter V, et al. Small heat-shock protein HSPB1 mutants stabilize microtubules in Charcot-Marie-Tooth neuropathy. J Neurosci 2011; 31:15320 - 8; http://dx.doi.org/10.1523/JNEUROSCI.3266-11.2011; PMID: 22031878
- Faller EM, Brown DL. Modulation of microtubule dynamics by the microtubule-associated protein 1a. J Neurosci Res 2009; 87:1080 - 9; http://dx.doi.org/10.1002/jnr.21920; PMID: 18951470
- d’Ydewalle C, Krishnan J, Chiheb DM, Van Damme P, Irobi J, Kozikowski AP, et al. HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat Med 2011; 17:968 - 74; http://dx.doi.org/10.1038/nm.2396; PMID: 21785432
- Takemura R, Okabe S, Umeyama T, Kanai Y, Cowan NJ, Hirokawa N. Increased microtubule stability and alpha tubulin acetylation in cells transfected with microtubule-associated proteins MAP1B, MAP2 or tau. J Cell Sci 1992; 103:953 - 64; PMID: 1487506
- Westermann S, Weber K. Post-translational modifications regulate microtubule function. Nat Rev Mol Cell Biol 2003; 4:938 - 47; http://dx.doi.org/10.1038/nrm1260; PMID: 14685172
- Tanabe K, Takei K. Dynamic instability of microtubules requires dynamin 2 and is impaired in a Charcot-Marie-Tooth mutant. J Cell Biol 2009; 185:939 - 48; http://dx.doi.org/10.1083/jcb.200803153; PMID: 19528294
- Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, et al. HDAC6 is a microtubule-associated deacetylase. Nature 2002; 417:455 - 8; http://dx.doi.org/10.1038/417455a; PMID: 12024216
- Creppe C, Malinouskaya L, Volvert ML, Gillard M, Close P, Malaise O, et al. Elongator controls the migration and differentiation of cortical neurons through acetylation of alpha-tubulin. Cell 2009; 136:551 - 64; http://dx.doi.org/10.1016/j.cell.2008.11.043; PMID: 19185337
- Akella JS, Wloga D, Kim J, Starostina NG, Lyons-Abbott S, Morrissette NS, et al. MEC-17 is an alpha-tubulin acetyltransferase. Nature 2010; 467:218 - 22; http://dx.doi.org/10.1038/nature09324; PMID: 20829795
- Shida T, Cueva JG, Xu Z, Goodman MB, Nachury MV. The major alpha-tubulin K40 acetyltransferase alphaTAT1 promotes rapid ciliogenesis and efficient mechanosensation. Proc Natl Acad Sci U S A 2010; 107:21517 - 22; http://dx.doi.org/10.1073/pnas.1013728107; PMID: 21068373
- North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell 2003; 11:437 - 44; http://dx.doi.org/10.1016/S1097-2765(03)00038-8; PMID: 12620231
- Reed NA, Cai D, Blasius TL, Jih GT, Meyhofer E, Gaertig J, et al. Microtubule acetylation promotes kinesin-1 binding and transport. Curr Biol 2006; 16:2166 - 72; http://dx.doi.org/10.1016/j.cub.2006.09.014; PMID: 17084703
- Palazzo A, Ackerman B, Gundersen GG. Cell biology: Tubulin acetylation and cell motility. Nature 2003; 421:230; http://dx.doi.org/10.1038/421230a; PMID: 12529632
- Dompierre JP, Godin JD, Charrin BC, Cordelières FP, King SJ, Humbert S, et al. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington’s disease by increasing tubulin acetylation. J Neurosci 2007; 27:3571 - 83; http://dx.doi.org/10.1523/JNEUROSCI.0037-07.2007; PMID: 17392473
- Janke C, Bulinski JC. Post-translational regulation of the microtubule cytoskeleton: mechanisms and functions. Nat Rev Mol Cell Biol 2011; 12:773 - 86; http://dx.doi.org/10.1038/nrm3227; PMID: 22086369