Abstract
Cancer-associated fibroblasts (CAF), comprised of activated fibroblasts or myofibroblasts, are found in stroma surrounding solid tumors; these myofibroblasts promote invasion and metastasis of cancer cells. Activation of stromal fibroblasts into myofibroblasts is induced by expression of cystoskeleton protein, palladin, at early stages in tumorigenesis and increases with neoplastic progression. Expression of palladin in fibroblasts is triggered by paracrine signaling from adjacent k-ras-expressing epithelial cells. Three-dimensional co-cultures of palladin-expressing fibroblasts and pancreatic cancer cells reveals that the activated fibroblasts lead the invasion by creating tunnels through the extracellular matrix through which the cancer cells follow. Invasive tunneling occurs as a result of the development of invadopodia-like cellular protrusions in the palladin-activated fibroblasts and the addition of a wounding/inflammatory trigger. Abrogation of palladin reduces the invasive capacity of these cells. CAF also play a role in cancer resistance and immuno-privilege, making the targeting of activators of these cells of interest for oncologists.
The soil in which cancer grows has a profound effect on tumor destiny. Will an incipient cancer remain occult and indolent, or become aggressive and invasive? Work in the past decade has highlighted some of the essential ways in which the stroma fibroblasts can influence neoplastic progression. Pancreatic adenocarcinoma has frequently been used as a model tumor type because the cancer cells are embedded in a sea of activated myofibroblasts. Myofibroblasts, also referred to as cancer-associated fibroblasts (CAF), have smooth muscle cell-like contractile properties and positive α-smooth muscle actin (α-SMA) staining.Citation1
The mechanism by which myofibroblasts enhance tumorigenesis and metastases is complex and may involve the enhanced secretion of soluble growth factors, increased contractility and mechanostimulation of the cancer cells, and physical remodeling of the extracellular matrix to create metastasis-promoting channels.Citation2-Citation8 Myofibroblasts can have a critical influence on immune surveillance, as well as chemo and radio-resistance to tumors.Citation8-Citation11 Moreover, hypoxic conditions caused by the exuberant growth of CAF surrounding cancer may also contribute chemotherapy resistance through increased hydrostatic pressure and compression/loss of local vasculature.Citation7,Citation12 Recent breakthroughs shed light on the timing and mechanism of this important step in tumorigenesis: fibroblast activation in cancer.
Stromal Fibroblasts are Activated Early in Tumorigenesis
Stromal fibroblast activation occurs early prior to cancer development. In human pancreatic cancer and mouse models of pancreatic cancer, CAFs are present surrounding the high-grade dysplastic lesions in the pancreas and even to a lesser extent in the low-grade dysplastic lesions.Citation13,Citation14 Similar findings in hepatocellular carcinoma and oral squamous cell carcinoma and their dysplastic precursor lesions have been found.Citation15,Citation16 These data suggest that cancer is not necessary for the transformation of CAF, but rather myofibroblast activation occurs earlier in the process of neoplastic progression when dysplasia is present.
The Mechanism of Fibroblast Activation Implicates Two Factors: Palladin Expression and Inflammation
Palladin appears to play a key role in fibroblast transformation in some cancers, including pancreatic cancer and breast cancer.Citation14,Citation17 Palladin is an embryonic protein that plays a key role in cellular migration. It is a cytoskeletal protein that acts as a scaffold and serves to crosslink the components of stress fibers, actin bundles, Z discs, focal adhesions and other subcellular structures.Citation18,Citation19 Palladin is upregulated in the leading edge of wounds and in the cancer-associated fibroblasts of metastatic cancers.Citation14,Citation17,Citation20 Interestingly, palladin has also been detected in expression screens for invasion-specific genes in pancreatic and breast cancer.Citation21,Citation22
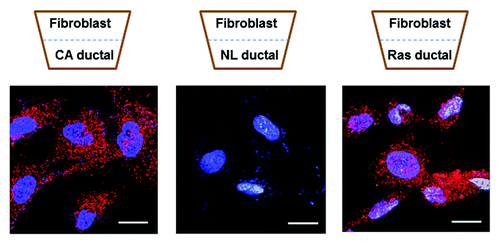
Palladin is upregulated in the CAF of pancreatic cancer early during tumorigenesis—it is overexpressed in the stromal fibroblasts immediately surrounding low and high-grade dysplasia. The expression of palladin closely correlates with the expression of α-SMA in these pre-cancerous lesions.Citation13 Our recent studies to unravel the role of palladin in fibroblast activation in cancer reveal that simply co-culturing a normal human fibroblast next to a pancreatic cancer cell is sufficient to impart myofibroblast properties to the fibroblast; this process occurs in a palladin-dependent fashion (). Because myofibroblasts can be detected early in tumorigenesis, we tested whether the initiating event in pancreatic ductal adenocarcinoma, e.g., a k-ras mutation in an epithelial cell, was sufficient to cause transformation of an adjacent resting fibroblast into a myofibroblast. Transwell experiments involving normal fibroblasts co-cultured with normal epithelial cell expressing wild-type or mutated k-ras were performed: activated k-ras (wild-type or mutated) paracrine signaling is sufficient to induce the adjacent, but non-touching, quiescent fibroblasts to become myofibroblasts. Abrogation of palladin, using siRNA, causes loss of the myofibroblast phenotype, including loss of common myofibroblast markers such as α-SMA, and loss of myofibroblast function, such as migration and invasion.Citation13
Figure 1. Normal human dermal fibroblasts were grown adjacent to pancreatic ductal cells in a transwell plate. Left: fibroblasts grown adjacent to pancreatic cancer ductal cells caused upregulation of palladin (stained red) in the fibroblasts. Middle: fibroblasts growing adjacent to normal pancreatic ductal cells do not express palladin. Right: k-ras expression (either wild type or mutated k-ras) in a normal pancreatic ductal cell was sufficient to upregulate palladin in the adjacent normal fibroblast. Once fibroblasts express palladin they develop the myofibroblast phenotype. Nuclei are stained blue with DAPI. Scale bars indicate 20 μm.
Curiously, palladin-expressing fibroblasts appear to be primed but not activated. The palladin-expressing fibroblasts have the phenotype of a myofibroblast: an elongated shape and expression of α-SMA and vimentin; however, an inflammatory or wounding signal is required for the myofibroblasts to become migratory and invasive. In absence of the inflammatory signal, the palladin-expressing myofibroblasts remain dormant, with diminished capability for migration or invasion.Citation13 This finding might be one reason for the underlying inactivity of some indolent tumors, if inflammation is absent the fibroblast-led invasion cannot not occur. In summary, while a palladin-expressing fibroblast is primed, expressing all of the proteins one would expect in a myofibroblast, it does not yet act as a leading partner for cancer cell invasion without the inflammatory signal. In the clinical setting, such inflammation could be driven from environmental factors such as smoking, infections or inflammatory cytokines associated with obesity.
Stromal-Assisted Cancer Invasion and Metastases
Myofibroblasts can produce tracks within the extracellular matrix, which in effect, create tunnels for the carcinoma cells to follow.Citation23 More recently, we have identified the mechanism of how this fibroblast-led cancer invasion occurs. Upregulation of palladin causes the fibroblast to develop a fusiform/mesenchymal shape with apparent invadopodia—feet that contain proteolytic enzymes (). To identify the contents of the “feet” of palladin-activated myofibroblasts, we ensnared the myofibroblasts in the act of invasion in a sieve that was large enough to let the feet through, but too small for a whole cell to pass through. Proteomic analysis performed on the ensnared and isolated “feet” revealed the overexpression of proteolytic enzymes such as metalloproteinases and cathepsin, invadopodia proteins and proteins associated with poor prognosis in cancer. Functional studies demonstrated that, in the setting of an inflammatory or wounding signal, the palladin-activated fibroblasts can both rip and destroy the extracellular matrix literally creating tunnels through which the cancer cells follow.Citation13 Fibroblasts without palladin expression have markedly diminished capacity to create tunnels and cancer cells do not follow them (). Remarkably, once the activated myofibroblasts escort the cancer cells through tunnels in the organ of origin, labeling studies have shown that the cancer cell and myofibroblasts invade together through blood vessels and implant in metastatic sites.Citation24,Citation25 In support of these studies, we found palladin-expressing fibroblasts adjacent to cancer cells in lymph node and liver metastases.Citation14
Figure 2. Normal human fibroblasts transfected with an empty vector (EV) remained boxy in appearance and had no effect on collagen or matrigel when exposed to wounding media (A and B). In contrast, fibroblasts transfected with wild-type palladin (WT) became elongated with mesenchymal features (C), caused destruction of the collagen matrix (D) with apparent clumping of the collagen edges. Additionally, palladin-expressing fibroblasts created tunnels in matrigel (stained red) when exposed to wounding media in 3D invasion cultures (E). Fibroblasts, stained white in (F), became quite elongated when tunneling. Tunnel is delineated by yellow arrowheads. Scale bars indicate 20 μm.
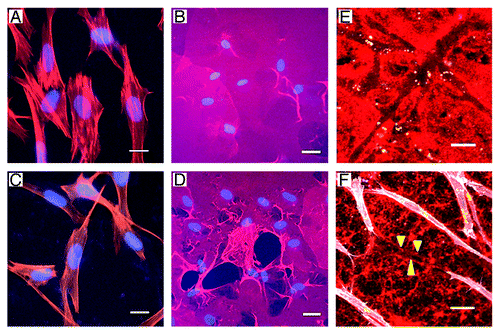
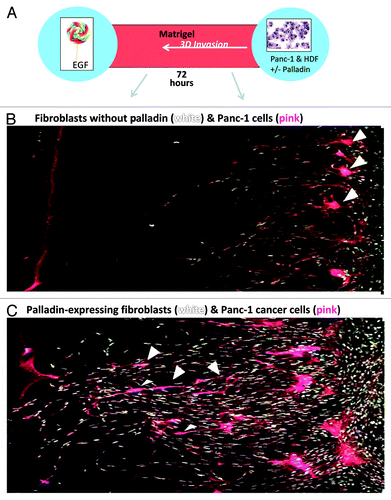
Figure 3. The cartoon in (A) depicts the 3D invasion culture chamber: two wells, one containing fibroblasts and cancer cells (right) and the other filled with chemoattractant EGF (left) are separated by a chamber filled with matrigel. Fibroblasts without palladin (B) and with palladin (C) were co-cultured with pancreatic cancer ductal cell line, Panc-1, over a period of 72 h. Fibroblasts were stained with white Q-dots and cancer cells were stained pink. Note in (C), the palladin-expressing fibroblasts tunneled through the matrigel and were followed by the pink cancer cells (arrow heads) as the cells moved toward the EGF. In (B), pancreatic cancer cells remained at the baseline and did not invade in the 3D cultures when the fibroblasts did not express palladin. Wounding media was provided in all of the 3D invasion cultures.
Metastasis Can Occur before Cancer Formation
Elegant studies by Rhim et al., using lineage tracing in a pancreatic cancer engineered mouse model, revealed that mutant ductal cells undergo epithelial mesenchymal transformation (EMT), invade into the blood stream, and lodge into metastatic sites such as the liver prior to histologic evidence of cancer.Citation26 The invasion of these mutant epithelial cells occurs in 2.7% of all PanIN 2 (low-grade dysplasia) and 6.8% of PanIN 3 (high-grade dysplasia) lesions, but never in the setting of PanIN 1 (hyperplasia). Inflammation is required for the dissemination of PanIN 2 and 3 cells to occur. Not surprisingly, COX-2, an inflammatory mediator, is increasingly overexpressed between PanIN lesions and malignant pancreatic tissues.Citation27 In the studies by Rhim, if dexamethasone was added as an anti-inflammatory agent, the dissemination of mutated circulated pancreatic epithelial cells was abolished. Even more amazing was the loss of PanIN lesions and associated myofibroblasts within the pancreatic parenchyma in the setting of dexamethasone taken on a daily basis: the pancreata return to a normal appearance, while the control mice proceed to get pancreatic adenocarcinoma. Taken as a whole, this work implicates the invasion of mutated cells earlier than originally thought in cancer and would help explain the very lethal nature of some cancers, such as pancreatic, even when the tumors are quite small. The early activation of fibroblasts into tunneling myofibroblasts by k-ras mutated epithelial cells fits in mechanistically with the model of earlier invasion of epithelial cells prior to cancer formation. Abolition of inflammation reverses the invasion process.
Targeting the Stromal Fibroblasts
Because of the interdependent behavior of cancer cells and stromal fibroblasts, the latter have become a target of interest for oncologists.Citation28 Pancreatic cancer cells have increased resistance to gemcitabine, in part due to direct activation of the Hedgehog pathway resulting from cross-talk between myofibroblasts and adjacent cancer cells.Citation29,Citation30 Recent chemotherapy using a Hedgehog inhibitor results in a significant loss of the tumor-associated fibroblasts in pancreatic cancer and prolonged survival in mouse models.Citation7 However, in the latter trial the mice relapse when the myofibroblasts repopulate. This finding, combined with the negative outcome of a recent human phase III clinical trial testing the efficacy of chemotherapy and hedgehog pathway inhibition, suggests that compensatory pathways may exist if only one pathway in the targeting of CAF is abrogated. This is particularly of issue because there are usually myofibroblasts remaining at the surgically resected pancreatic cancer margins and the sources for tumor stromal fibroblasts may be derived from both local and potentially non-local sources.Citation31
Other methods of directly targeting the CAF have included use of monoclonal antibodies, drugs, and vaccines. A novel monoclonal antibody targeting fibroblast activation protein (FAP), a cell surface protease of activated tumor fibroblasts, has been shown to induce long-lasting inhibition of tumor growth and complete regression in xenograft models of lung, pancreas, and head and neck cancers.Citation32 Vaccination against stromal fibroblasts targeting FAP has also shown some promise in mouse models.Citation33 While no current therapy targets palladin expression in fibroblasts, we have performed preliminary studies suggesting that reagents that target anti-SMA and regulate hepatic stellate cell activation, (such as PPARγ agonists and metformin) are useful when used in combination to block palladin expression in myofibroblasts and human CAF. These combined palladin-targeting therapies concomitantly block α-SMA expression and the myofibroblast phenotype (unpublished data). Further investigation using small molecular and high-throughput drug screening is required to identify which drugs are most effective in blocking palladin and whether these drugs are effective in the early and the late stages of cancer in mouse models.
Therapies to decrease the inflammatory component of carcinogenesis have included use of aspirin,Citation34 COX2 inhibitors such as celcoxib,Citation35,Citation36 NFκB inhibitors such as curcuminCitation37,Citation38 and PPARγ agonists such as troglitazone.Citation39,Citation40 Some of these anti-inflammatory drugs have been used in human phase II trials with mixed success,Citation35,Citation36,Citation41,Citation42 where many of the patients had later stage tumors. With our current knowledge of the role of inflammation in driving forth the early dissemination of myofibroblast-aided cancer cells, it is possible that the inflammation needs to be treated earlier in the neoplastic progression—before the cancer cells have escaped. In keeping with this concept, the effective use of anti-inflammatory drugs has been reported in chemoprevention trialsCitation43,Citation44.
Cross-talk between fibroblasts and cancer cells is essential to invasion and potentially chemotherapeutic agents that disrupt this process could be effective. Inflammatory cytokines and signaling molecules including TNFα, IL-6, IL-1α/β, NFκB and TGF-β play key roles in paracrine signaling between tumor cells and fibroblasts, as outlined in an excellent review by Bhomick and Moses.Citation45-Citation49 Therapeutics designed to modulate these molecules are described elsewhere; in general most of these therapies are relatively new and thus trials of some of these agents are just being undertaken in humans.Citation40,Citation50
summarizes steps and mechanism of myofibroblast activation and the partnership these cells play in cancer invasion. Although difficult, targeting the stromal fibroblasts remains an attractive strategy in the fight of aggressive cancers because of the interdependence of the CAF and the cancer cells. In addition, it may be valuable to determine when during the neoplastic process targeting the CAF is most effective: in the initiating stages of tumorigenesis or whether the strategy can be effective in an established, even metastatic cancer.
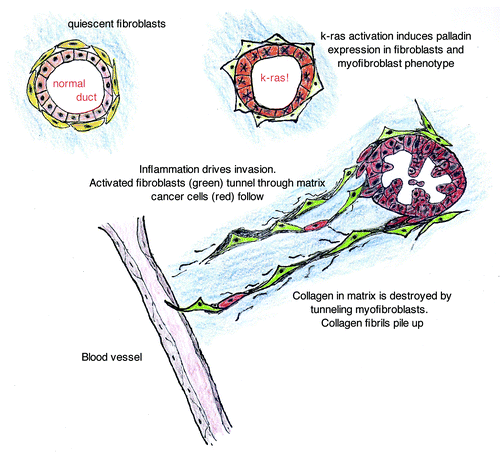
Figure 4. Stromal fibroblasts in normal pancreas are quiescent and without palladin staining. K-ras activation in ductal cells leads to paracrine signaling that is sufficient to induce palladin-associated myofibroblast transformation of the adjacent fibroblasts. This event occurs early in tumorigenesis, when epithelial cells are dysplastic, and increases with neoplastic progression. In the setting of a wounding signal, the palladin-activated fibroblasts develop cellular protrusions (feet) that express invadopodia proteins, proteases and enhance the capacity for invasion. The palladin-activated fibroblasts create tunnels through the matrix, assisting the escape of cancer cells into the neo-vasculature. The activated fibroblasts appear to accompany the cancer cells to their metastatic niche in breast and pancreatic cancer models.
Summary
The arousal of the stroma is a key and transformative event in the invasive stages of tumorigenesis in many solid tumors. Stromal fibroblasts can be transformed through paracrine signaling of adjacent k-ras overexpressing epithelial cells. The fibroblast then undergoes phenotypic change into a myofibroblast that is mediated through palladin, a cytoskeletal protein essential in cell motility. However, this change is insufficient to cause fibroblast-assisted cancer cell invasion and migration. For the latter events to occur, an additional wounding or inflammatory signal is required. The presence of these three events (overexpression of k-ras, palladin-expression in the fibroblasts and inflammatory signal) instigates the dynamic relationship between the stroma and the mutated epithelial cell. These three events are sufficient for the activated myofibroblast to tunnel through the extracellular matrix and provide avenues for the dysplastic and cancerous epithelial cells to follow. Abrogation of palladin or the inflammatory signal is sufficient to shut down the process.Citation13 Future studies will help elucidate the role of epithelial-mesenchymal transition to enhance the migration of cancer cells through the fibroblast-created tunnels and the potential for chemotherapeutic targeting of the initiating events in cancer invasion.
Related Research Data
References
- Räsänen K, Vaheri A. Activation of fibroblasts in cancer stroma. Exp Cell Res 2010; 316:2713 - 22; http://dx.doi.org/10.1016/j.yexcr.2010.04.032; PMID: 20451516
- Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature 2004; 432:332 - 7; http://dx.doi.org/10.1038/nature03096; PMID: 15549095
- Chu GC, Kimmelman AC, Hezel AF, DePinho RA. Stromal biology of pancreatic cancer. J Cell Biochem 2007; 101:887 - 907; http://dx.doi.org/10.1002/jcb.21209; PMID: 17266048
- De Wever O, Demetter P, Mareel M, Bracke M. Stromal myofibroblasts are drivers of invasive cancer growth. Int J Cancer 2008; 123:2229 - 38; http://dx.doi.org/10.1002/ijc.23925; PMID: 18777559
- Olumi AF, Grossfeld GD, Hayward SW, Carroll PR, Tlsty TD, Cunha GR. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res 1999; 59:5002 - 11; PMID: 10519415
- Ostman A, Augsten M. Cancer-associated fibroblasts and tumor growth--bystanders turning into key players. Curr Opin Genet Dev 2009; 19:67 - 73; http://dx.doi.org/10.1016/j.gde.2009.01.003; PMID: 19211240
- Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009; 324:1457 - 61; http://dx.doi.org/10.1126/science.1171362; PMID: 19460966
- Hwang RF, Moore T, Arumugam T, Ramachandran V, Amos KD, Rivera A, et al. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res 2008; 68:918 - 26; http://dx.doi.org/10.1158/0008-5472.CAN-07-5714; PMID: 18245495
- Singh S, Ross SR, Acena M, Rowley DA, Schreiber H. Stroma is critical for preventing or permitting immunological destruction of antigenic cancer cells. J Exp Med 1992; 175:139 - 46; http://dx.doi.org/10.1084/jem.175.1.139; PMID: 1309851
- Kim EJ, Simeone DM. Advances in pancreatic cancer. Curr Opin Gastroenterol 2011; 27:460 - 6; http://dx.doi.org/10.1097/MOG.0b013e328349e31f; PMID: 21778878
- Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, et al. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science 2010; 330:827 - 30; http://dx.doi.org/10.1126/science.1195300; PMID: 21051638
- Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012; 21:418 - 29; http://dx.doi.org/10.1016/j.ccr.2012.01.007; PMID: 22439937
- Brentnall TA, Lai LA, Coleman J, Bronner MP, Pan S, Chen R. Arousal of cancer-associated stroma: overexpression of palladin activates fibroblasts to promote tumor invasion. PLoS One 2012; 7:e30219; http://dx.doi.org/10.1371/journal.pone.0030219; PMID: 22291919
- Goicoechea SM, Bednarski B, Stack C, Cowan DW, Volmar K, Thorne L, et al. Isoform-specific upregulation of palladin in human and murine pancreas tumors. PLoS One 2010; 5:e10347; http://dx.doi.org/10.1371/journal.pone.0010347; PMID: 20436683
- Chaudhary M, Gadbail AR, Vidhale G, Mankar Gadbail MP, Gondivkar SM, Gawande M, et al. Comparison of Myofibroblasts Expression in Oral Squamous Cell Carcinoma, Verrucous Carcinoma, High Risk Epithelial Dysplasia, Low Risk Epithelial Dysplasia and Normal Oral Mucosa. Head Neck Pathol 2012; 6:305 - 13; http://dx.doi.org/10.1007/s12105-012-0335-x; PMID: 22392407
- Nhieu JT, Brochériou I, Préaux AM, Mallat A, Cherqui D, Zafrani ES, et al. Myofibroblasts and hepatocellular carcinoma: an in vivo and in vitro study. J Hepatol 1998; 29:120 - 8; http://dx.doi.org/10.1016/S0168-8278(98)80186-4; PMID: 9696500
- Goicoechea SM, Bednarski B, García-Mata R, Prentice-Dunn H, Kim HJ, Otey CA. Palladin contributes to invasive motility in human breast cancer cells. Oncogene 2009; 28:587 - 98; http://dx.doi.org/10.1038/onc.2008.408; PMID: 18978809
- Parast MM, Otey CA. Characterization of palladin, a novel protein localized to stress fibers and cell adhesions. J Cell Biol 2000; 150:643 - 56; http://dx.doi.org/10.1083/jcb.150.3.643; PMID: 10931874
- Goicoechea SM, Arneman D, Otey CA. The role of palladin in actin organization and cell motility. Eur J Cell Biol 2008; 87:517 - 25; http://dx.doi.org/10.1016/j.ejcb.2008.01.010; PMID: 18342394
- Gupta V, Bassi DE, Simons JD, Devarajan K, Al-Saleem T, Uzzo RG, et al. Elevated expression of stromal palladin predicts poor clinical outcome in renal cell carcinoma. PLoS One 2011; 6:e21494; http://dx.doi.org/10.1371/journal.pone.0021494; PMID: 21738681
- Ryu B, Jones J, Hollingsworth MA, Hruban RH, Kern SE. Invasion-specific genes in malignancy: serial analysis of gene expression comparisons of primary and passaged cancers. Cancer Res 2001; 61:1833 - 8; PMID: 11280733
- Wang W, Goswami S, Lapidus K, Wells AL, Wyckoff JB, Sahai E, et al. Identification and testing of a gene expression signature of invasive carcinoma cells within primary mammary tumors. Cancer Res 2004; 64:8585 - 94; http://dx.doi.org/10.1158/0008-5472.CAN-04-1136; PMID: 15574765
- Gaggioli C, Hooper S, Hidalgo-Carcedo C, Grosse R, Marshall JF, Harrington K, et al. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol 2007; 9:1392 - 400; http://dx.doi.org/10.1038/ncb1658; PMID: 18037882
- Xu Z, Vonlaufen A, Phillips PA, Fiala-Beer E, Zhang X, Yang L, et al. Role of pancreatic stellate cells in pancreatic cancer metastasis. Am J Pathol 2010; 177:2585 - 96; http://dx.doi.org/10.2353/ajpath.2010.090899; PMID: 20934972
- Duda DG, Duyverman AM, Kohno M, Snuderl M, Steller EJ, Fukumura D, et al. Malignant cells facilitate lung metastasis by bringing their own soil. Proc Natl Acad Sci U S A 2010; 107:21677 - 82; http://dx.doi.org/10.1073/pnas.1016234107; PMID: 21098274
- Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012; 148:349 - 61; http://dx.doi.org/10.1016/j.cell.2011.11.025; PMID: 22265420
- Hermanova M, Trna J, Nenutil R, Dite P, Kala Z. Expression of COX-2 is associated with accumulation of p53 in pancreatic cancer: analysis of COX-2 and p53 expression in premalignant and malignant ductal pancreatic lesions. Eur J Gastroenterol Hepatol 2008; 20:732 - 9; http://dx.doi.org/10.1097/MEG.0b013e3282f945fb; PMID: 18617777
- Li X, Ma Q, Xu Q, Duan W, Lei J, Wu E. Targeting the cancer-stroma interaction: a potential approach for pancreatic cancer treatment. Curr Pharm Des 2012; 18:2404 - 15; PMID: 22372501
- Onozuka H, Tsuchihara K, Esumi H. Hypoglycemic/hypoxic condition in vitro mimicking the tumor microenvironment markedly reduced the efficacy of anticancer drugs. Cancer Sci 2011; 102:975 - 82; http://dx.doi.org/10.1111/j.1349-7006.2011.01880.x; PMID: 21255190
- Onishi H, Kai M, Odate S, Iwasaki H, Morifuji Y, Ogino T, et al. Hypoxia activates the hedgehog signaling pathway in a ligand-independent manner by upregulation of Smo transcription in pancreatic cancer. Cancer Sci 2011; 102:1144 - 50; http://dx.doi.org/10.1111/j.1349-7006.2011.01912.x; PMID: 21338440
- Engels B, Rowley DA, Schreiber H. Targeting stroma to treat cancers. Semin Cancer Biol 2012; 22:41 - 9; http://dx.doi.org/10.1016/j.semcancer.2011.12.008; PMID: 22212863
- Ostermann E, Garin-Chesa P, Heider KH, Kalat M, Lamche H, Puri C, et al. Effective immunoconjugate therapy in cancer models targeting a serine protease of tumor fibroblasts. Clin Cancer Res 2008; 14:4584 - 92; http://dx.doi.org/10.1158/1078-0432.CCR-07-5211; PMID: 18628473
- Wen Y, Wang CT, Ma TT, Li ZY, Zhou LN, Mu B, et al. Immunotherapy targeting fibroblast activation protein inhibits tumor growth and increases survival in a murine colon cancer model. Cancer Sci 2010; 101:2325 - 32; http://dx.doi.org/10.1111/j.1349-7006.2010.01695.x; PMID: 20804499
- Rothwell PM, Fowkes FG, Belch JF, Ogawa H, Warlow CP, Meade TW. Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet 2011; 377:31 - 41; http://dx.doi.org/10.1016/S0140-6736(10)62110-1; PMID: 21144578
- Dragovich T, Burris H 3rd, Loehrer P, Von Hoff DD, Chow S, Stratton S, et al. Gemcitabine plus celecoxib in patients with advanced or metastatic pancreatic adenocarcinoma: results of a phase II trial. Am J Clin Oncol 2008; 31:157 - 62; http://dx.doi.org/10.1097/COC.0b013e31815878c9; PMID: 18391600
- Lipton A, Campbell-Baird C, Witters L, Harvey H, Ali S. Phase II trial of gemcitabine, irinotecan, and celecoxib in patients with advanced pancreatic cancer. J Clin Gastroenterol 2010; 44:286 - 8; http://dx.doi.org/10.1097/MCG.0b013e3181cda097; PMID: 20216081
- Dhillon N, Aggarwal BB, Newman RA, Wolff RA, Kunnumakkara AB, Abbruzzese JL, et al. Phase II trial of curcumin in patients with advanced pancreatic cancer. Clin Cancer Res 2008; 14:4491 - 9; http://dx.doi.org/10.1158/1078-0432.CCR-08-0024; PMID: 18628464
- Epelbaum R, Schaffer M, Vizel B, Badmaev V, Bar-Sela G. Curcumin and gemcitabine in patients with advanced pancreatic cancer. Nutr Cancer 2010; 62:1137 - 41; http://dx.doi.org/10.1080/01635581.2010.513802; PMID: 21058202
- Burstein HJ, Demetri GD, Mueller E, Sarraf P, Spiegelman BM, Winer EP. Use of the peroxisome proliferator-activated receptor (PPAR) gamma ligand troglitazone as treatment for refractory breast cancer: a phase II study. Breast Cancer Res Treat 2003; 79:391 - 7; http://dx.doi.org/10.1023/A:1024038127156; PMID: 12846423
- Uomo I, Miraglia S, Pastorello M. Inflammation and pancreatic ductal adenocarcinoma: a potential scenario for novel drug targets. JOP 2010; 11:199 - 202; PMID: 20442512
- Pino MS, Milella M, Gelibter A, Sperduti I, De Marco S, Nuzzo C, et al. Capecitabine and celecoxib as second-line treatment of advanced pancreatic and biliary tract cancers. Oncology 2009; 76:254 - 61; http://dx.doi.org/10.1159/000205388; PMID: 19246950
- Pruthi RS, Derksen JE, Moore D, Carson CC, Grigson G, Watkins C, et al. Phase II trial of celecoxib in prostate-specific antigen recurrent prostate cancer after definitive radiation therapy or radical prostatectomy. Clin Cancer Res 2006; 12:2172 - 7; http://dx.doi.org/10.1158/1078-0432.CCR-05-2067; PMID: 16609031
- Harris RE, Beebe-Donk J, Doss H, Burr Doss D. Aspirin, ibuprofen, and other non-steroidal anti-inflammatory drugs in cancer prevention: a critical review of non-selective COX-2 blockade (review). [review] Oncol Rep 2005; 13:559 - 83; PMID: 15756426
- Cheng AL, Hsu CH, Lin JK, Hsu MM, Ho YF, Shen TS, et al. Phase I clinical trial of curcumin, a chemopreventive agent, in patients with high-risk or pre-malignant lesions. Anticancer Res 2001; 21:4B 2895 - 900; PMID: 11712783
- Bhowmick NA, Moses HL. Tumor-stroma interactions. Curr Opin Genet Dev 2005; 15:97 - 101; http://dx.doi.org/10.1016/j.gde.2004.12.003; PMID: 15661539
- Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-kappaB-dependent manner. Cancer Cell 2010; 17:135 - 47; http://dx.doi.org/10.1016/j.ccr.2009.12.041; PMID: 20138012
- Stuelten CH, DaCosta Byfield S, Arany PR, Karpova TS, Stetler-Stevenson WG, Roberts AB. Breast cancer cells induce stromal fibroblasts to express MMP-9 via secretion of TNF-alpha and TGF-beta. J Cell Sci 2005; 118:2143 - 53; http://dx.doi.org/10.1242/jcs.02334; PMID: 15855236
- Tjomsland V, Spångeus A, Välilä J, Sandström P, Borch K, Druid H, et al. Interleukin 1α sustains the expression of inflammatory factors in human pancreatic cancer microenvironment by targeting cancer-associated fibroblasts. Neoplasia 2011; 13:664 - 75; PMID: 21847358
- Guo Y, Xu F, Lu T, Duan Z, Zhang Z. Interleukin-6 signaling pathway in targeted therapy for cancer. Cancer Treat Rev 2012; 38:904 - 10; http://dx.doi.org/10.1016/j.ctrv.2012.04.007; PMID: 22651903
- Cox AD, Olive KP. Silencing the killers: paracrine immune suppression in pancreatic cancer. Cancer Cell 2012; 21:715 - 6; http://dx.doi.org/10.1016/j.ccr.2012.05.029; PMID: 22698396