
Abstract
It would be hard to argue that live-cell imaging has not changed our view of biology. The past 10 years have seen an explosion of interest in imaging cellular processes, down to the molecular level. There are now many advanced techniques being applied to live cell imaging. However, cellular health is often under appreciated. For many researchers, if the cell at the end of the experiment has not gone into apoptosis or is blebbed beyond recognition, than all is well. This is simply incorrect. There are many factors that need to be considered when performing live-cell imaging in order to maintain cellular health such as: imaging modality, media, temperature, humidity, PH, osmolality, and photon dose. The wavelength of illuminating light, and the total photon dose that the cells are exposed to, comprise two of the most important and controllable parameters of live-cell imaging. The lowest photon dose that achieves a measureable metric for the experimental question should be used, not the dose that produces cover photo quality images. This is paramount to ensure that the cellular processes being investigated are in their in vitro state and not shifted to an alternate pathway due to environmental stress. The timing of the mitosis is an ideal canary in the gold mine, in that any stress induced from the imaging will result in the increased length of mitosis, thus providing a control model for the current imagining conditions.
Live-Cell Imaging—Why?
As long as there have been microscopes, there have been inquisitive researchers peering through them at specimens, many of which were alive. The microscope as well as live-cell imaging has come a long way since the days-of-old where the illumination source was the sun or a candle and images had to be hand-drawn.
The needs and benefits of documenting dynamic cellular and sub-cellular processes in real or near real-time have been understood for a long time. The limitations of population averaging,Citation1 the ability to obtain real-time measurements, and to obtain data from in vivo systems (complete cellular systems) has led to an increased use of live-cell imaging. The organisms or cell types along with the questions being investigated today are very broad. Some examples are, temperature dependency of drug-induced events in neurons,Citation2 fast imaging of zebra fish,Citation3 monitoring molecular interaction,Citation4 egg development in Drosophila,Citation5 how viral replication in cells occurs,Citation6 and, for even more examples, see reviews.Citation1,7,8
Fluorescent probes and proteins have forever changed the biological sciences and especially live-cell imaging. It is now possible to tag and image cellular structures and macromolecular complexes over a broad range of sizes. It is possible to capture very rapid cellular events such as signaling by the transcription factor nuclear factor kappa B (NFkappaB)Citation9 or imaging and tracking multiple fluorophores over time and depth.
While the benefits and need for live-cell imaging is well appreciated, the need to ensure cellular health is not. Imaging cells and their structures in three dimensions has challenges, which while not unique to imaging are often amplified, especially in multi-dimensional live cell imaging. The additional photon dose needed for multi-dimensional imaging requires balancing many of the imaging parameters to achieve the desired metric without compromising cellular health. The natural tendency while imaging is to adjust imaging parameters such that the resulting image is both biologically relevant and aesthetically pleasing. For the vast majority of experiments, this will compromise cellular health and is not necessary. The process of imaging a biological sample damages the sample to some extent. Cells are more sensitive to light than most researchers appreciate. But now quantum physicists are beginning to apply their theories to bio-imaging in the hope of reducing this damage.Citation10
Which Imaging Modality To Use?—What Do You Really Need To Detect?
If the experimental question(s) can be answered with transmitted light images, then use transmitted light (lower photon dose) rather than fluorescence (higher photon dose). The same holds true for the imaging modality, practice “simplest first.” If the specimen is a monolayer then widefield microscopy (WFM) is almost always a better choice than a confocal laser scanning microscope (CLSM). There are of course exceptions, but make the choice based on what metrics are needed to answer the experimental question. What follows are summaries of the different modalities; for more details, see reviews.Citation7,8
Widefield microscopy (WFM)
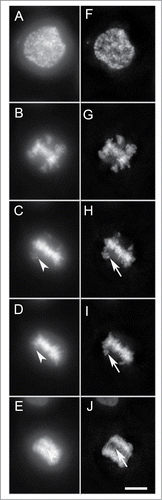
This is the simplest, least expensive, and oldest imaging modality used for live-cell imaging. It has the advantage of requiring the lowest photon dose, especially for transmitted light imaging, i.e., phase or differential interference contrast (DIC). While fluorescence has largely replaced transmitted light imaging, these techniques are still very useful in answering biological questions. The advancement in CCD cameras, now boasting quantum efficiencies >90%, has reduced the photon dose required for imaging in both transmitted and epi-fluorescence modes. This modality is ideally suited for monolayers of cells or thin <10 μM tissue slices. The low photon dose of this modality allows multi-dimensional data to be collected while still maintaining happy and healthy cells. The use of filter wheels allows for relatively rapid switching between fluorophores and piezoelectric objective drives allows for rapid collection of Z-series. Post-processing such as deconvolution can recover details obscured due to blurring of the specimen. Deconvolution redistributes the out of focus light by applying the point spread function (PSF) to the collected images.Citation11,12 See for an illustration of this approach. Combining WFM microscopy with deconvolution for thin specimens yields images that approach confocal “quality” at a fraction of the photon dose of a CLSM, which makes the cells being imaged healthier.
Figure 1. Selected maximum intensity projections (6 Z slices) from a time-lapse series of HT1080 H2B-GFP transgenic cells progressing through mitosis, imaged with wide-field microscopy (A–E). Deconvolved (F–J) maximum intensity projections (6 Z slices) of the same time points in A–E. There is a clear improvement in both the over-all signal-to-noise as well as the resolution, compare arrow heads to arrows. Bar = 10 μm.
Confocal laser scanning microscopy (CLSM)
This type of microscopy is the workhorse of the imaging lab; however, it is less than ideal for keeping cells healthy and functioning normally. The photon dose required for imaging in this modality is high due to the amount of light being rejected by the pin holes (what makes the image “deblurred”) and the low quantum efficiency (10–20% standard, 40% new GaAsP13) of the photomultipliers used to generate the image. This is especially true for 4D microscopy. There are however, certain exceptions, such as photo activation or uncaging, which require specific lasers commonly found on CLSM, which may not be available on wide-field scopes.
Two-photon or multi-photon microscopy (MPM)
There are several factors that account for these instruments being a healthy choice for live-cell imaging. With MPM, fluorescent excitation of the specimen only occurs within a diffraction-limited region formed by the objective lens, which is significantly smaller than other modalities. shows the beam profiles of WFM, CLSM, and MPM. MPM is nonlinear in nature and the fluorescence excitation is achieved with longer wavelengths. These longer wavelengths combined with generating fluorescence only at the region being imaged allows for much deeper penetration (maximum of ∼1 mm). Since the fluorescence is only generated within the imaging region, there is no need for de-scanning the image typical of CLSM, this allows special non-descanned detectors to be placed in close proximity to the objective lens, which in turn, allows more signal to be collected. Counter intuitively, photobleaching rates with two-photon excitation increases faster with excitation intensity than with single-photon excitation.14 Even more importantly, phototoxicity observed with two-photon excitation may be higher than single-photon excitation in thin specimens (<10 μm).Citation15 For thick specimens, such as in vivo imagingCitation16 or where UV excitation is required (uncaging, excitation, etc.), MPM is an excellent and often only choice. This is especially true for live-organism imaging.Citation13 Additionally, with MPM, it is possible to image label-free samples such as collagen fibers or other non- centrosymmetric samples with second harmonic generated microscopy.Citation17
Figure 2. Distribution of fluorescein fluorescence (green) as viewed in the x-z plane, on inverted microscope, focused with 20 X objective lens 0.75NA. (A) Wide-field microscopy (WFM) mercury source and filter cube excitation. (B) Confocal scanning microcopy (CLSM), 488 nm laser excitation, and (C) two-photon (2P) excitation using femtosecond pulses of 850-nm light.

Spinning disk confocal microscopy (SDCM)
There are a number of implementations of the confocal principle where a pinhole rejects the out-of-focus light but maintains the ability to generate wide-field images, thereby overcoming the speed limitations and phototoxicity of CLSM. These include slit scanning and pinhole multiplexing methods, including swept-field and spinning disk confocal. The most commonly used is SDCM. This technique uses a pair of rotating disks with thousands of pinholes in a spiral. The Yokogawa scanner adds microlenses to the second disk, which increases the light efficiency of the system.Citation18 Because of the high scan speed (up to 360 frames per second) and the parallel collection of images with a high-QE CCD camera, there is a dramatic (10–15-fold) decreased photobleaching and phototoxicity compared with point scanning.Citation19 While it does not have as low a photon dose as WFM, it does offer the advantage of confocality at a reasonable photon dose, thereby making it well suited to high magnification (due to single pinhole size) live-cell imaging, especially in 4D. It should be noted that for thicker specimens the light from a single point can easily diverge (depends on the scattering within a given sample) and enters an adjacent pinhole “pinhole crosstalk,” thereby degrading the image.
Light sheet fluorescence microscopy (LSM)
In this technique the specimen is illuminated with a thin sheet of light from the side of the specimen, thereby illuminating a single XY plane with fluorescence detection occurring in the direction perpendicular to the excitation. This optical system has major advantages compared with CLSM. Speed is greatly increased relative to point-scanning methods, as well as the entire field of view being captured in parallel on high QE, low noise CCD camera. The excitation is confined to the focal plane without the use of a pinhole, thereby increasing the light throughput and reducing the photon dose. Bessel beam and structured illumination have been used to improve the axial resolution of these systems; however, it is still lower than MPM. Additionally, the spatial resolution is lower than other techniques, therefore most studies have been of whole embryos or tissue slicesCitation3,20,21 rather than at the single cell level. LSFM has allowed remarkable observations of vertebrate embryonic development due to its speed and sensitivityCitation3,20 as well as having the ability to image over long time intervals.Citation22 One caveat is that there are no commercial systems available to date, although there will be in the very near future.
At the bleeding edge
In the past 20 y or so, the simple recording of cellular processes has been replaced by the following advanced techniques, such as fluorescence resonance energy transfer (FRET), fluorescence correlation spectroscopy (FCS), fluorescence recovery after photobleaching (FRAP), fluorescence-lifetime imaging (FLIM), and others. In the very near future, although some would argue it is possible today, many of the sub-resolution techniques will be applicable to live-cell imaging. Nanoscopy will allow new areas of research at detection levels previously unattainable and frankly unimaginable 10 y ago.
Super-resolution (nanoscopy)
One of the most significant challenges in 3D imaging is the resolution limit especially the ∼3X lower axial resolution. To circumvent the limits imposed by diffraction and the anisotropic PSF, several of the super-resolution techniques have been able to push the resolution limits below 100 nm. However, to achieve the improved resolution, either hundreds (Structured Illumination Microscopy -SIM) to thousands of images (photoactivated localization microscopy PALM or stochastic optical reconstruction microscopy STORM and their variants) need to be acquired or two relatively high-powered lasers must be used (stimulated emission-depletion microscopy STED). However, with the use of a pulsed laser in STED, live-cell imaging is possible, even with two colors.Citation23-27 While the SIM has been used in the exploration of chromatin structure in 3D,Citation28 the technique is limited by requiring multiple images, thereby increasing the photon dose and decreasing speed significantly. While the pointillist approaches (STORM and PALM) offering resolution down to 20 nm, they require even longer collection cycles than SIM and rely on iterative cycles of activation, excitation, photobleaching, and image collection. These techniques have been used in 3D; however, the use is limited and not amiable to all samples.Citation29
Live-Cell Imaging Considerations
Illumination-sun glasses and sunscreen should not be required
The illumination wavelength and photon dose are two of the most important imaging parameters to consider when performing live-cell imaging. There should be stringent control of both the wavelength(s) and the total photon dose delivered to the sample. Typically, photon dose is calculated from the illumination power (usually mW) at the sample and the focused or spot size of the objective lens and expressed in photons/μm2. For fluorescence, there is not a 1 to 1 relationship of photons produced to photons captured.Citation30 The efficiency of the detection system is equally important, in such that the lower the detection efficiency, the more illuminating photons will be required to produce an image. For example, based on the same illumination dose, a wide-field microscope will produce an image that is ∼3 times more intense than a spinning disk confocal and 10–15 times that of a point scanning confocal.Citation31 It should be noted that photon dose, in virtually any amount, is damaging to cells.Citation10,32,33 Photobleaching and phototoxicity are a result of high energy electrons from fluorescent excitation not being released as photons, rather reacting with dissolved oxygen, thus, bleaching the fluorophore and producing reactive oxygen species (ROS), which cause phototoxic effects. While ROS are the major contributors to phototoxicity, other non-ROS photodamage may also occur.Citation34 This does not translate to: all imaging produces artifacts or all live-cell imaging data are invalid. However, there needs to be careful consideration to experimental design, including the use of appropriate controls to detect artifacts or invalid data. One of the main advantages of live-cell imaging is the direct coupling of structural and dynamic information. However, this information is only relevant if obtained without perturbing the physiology of the process. It cannot be stressed too much that the imaging paradigm should be such that the images/image analysis addresses the hypotheses being tested and not the generation of a journal cover image. This includes the imaging modality, the number of Z sections, number of channels, and the timing. The timing of mitotic stages is an excellent phototoxicity control.Citation35 While other environmental stresses (see section on temperature, for example) can cause a dilation of the timing or aberrant mitosis, these stresses are more easily determined and eliminated. For example, measuring the temperature at the cell level or measuring the PH.
Sources
Probably the most common illumination source for fluorescent imaging in WFM is the mercury arc-lamp. These are being replaced quickly by light emitting diodes (LED) sources, which have several key advantages.Citation36 LEDs require less filtering (peaks more closely match excitation sources), no mechanical shutter is required (reduces vibration), the illumination can be easily pulsed (reducing photon dose), and lower cost of ownership. All the major microscope manufacturers as well many other companies (Lumen Dynamics Group Inc., Lumencor, Inc., Thorlabs Inc.) offer LED illuminators both for transmitted and Epi-fluorescence. There are also a large selection of lab-built LED illuminators.Citation36-39 Pulsing the illumination has been shown to reduce photo-toxicityCitation38, Citation40-42 while maintaining image quality. Compared with continuous wave (CW) illumination, shorter pulse widths reduced illumination and the time lag between two pulses allows all excited molecules to non-radiatively relax to the ground states via non-radiative and intermolecular (i.e., conduction and convection) as well as radiative (i.e., flourescence and phosphorecence) de-excitation processes. With pulse illumination there is also reduced heating of the sample.Citation43 demonstrates the lower photo-toxicity of pulsed LED illumination as compared with traditional mercury illumination. The same numbers of images with the same average brightness were collected with both illumination conditions. The companies listed above (LED illuminators) offer models that can be pulsed, alternatively, is a simple system that allows pulsing of most LEDs. Most of the other imaging modalities require laser illumination, such as confocal and light sheet. LEDs have not achieved high enough brightness levels, and therefore, are not a suitable substitute. Solid state lasers have reduced the operating cost and filled in some of the spectral gaps of more traditional lasers (Argon-Ion and Helium-Neon).
Table 1. Author please provide title
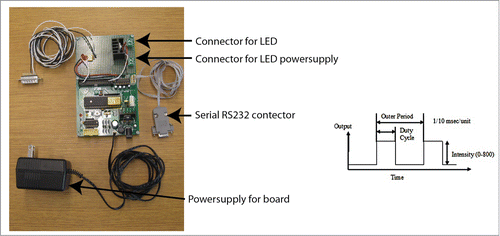
Figure 3. This device provides computer controlled pulse generation for high-power LEDs via simple serial commands. It operates as a high-speed switch generating square waves to modulate the light output. It turns the LED off when the camera is not exposing, limiting the total light load on the cells. The driver consists of three elements: the microcontroller with support electronics, an opto-isolated input for the camera signal, and a MOSFET transistor to switch the LED. The microcontroller is an ATMega16 device on a Futurlec development board. The optoisolator is a standard H11L1 isolator with support components. (The small indicator light is on when the camera is not exposing.) A 26 pin connector is wired to the optoisolator in the correct configuration for the camera outputs. The switch transistor is a BUZ80 n-type MOSFET with a maximum 3.4 A at 800 V. Schematic diagram available upon request. The LED pulse output is designed to generate square wave pulses that will excite fluorescent proteins. It generates a relatively low frequency square wave, called the “outer” pulse, and a 10 kHz PWM high-frequency pulse that is used to control the amplitude. The period and duty cycle of the outer pulse is highly variable. The units of the outer pulse is in 1/10 000 s/unit, and the PWM intensity varies between 0–800.
Temperature-happy and homeostatic cells
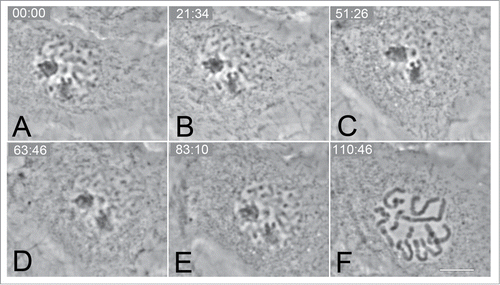
While maintaining homeostatic temperatures during imaging may seem like a no-brainer, the actual temperature of the cells being imaged is often lower than anticipated. So does a couple of degrees really matter? Yes, many cells types will stall and even reverse the cell cycle at 20–22°C,Citation44 see . At temperatures between 23 and 35, the timing of mitosis is dilated indicating a substantial stress on the cells. One of the main reasons for lower temperatures than expected is the use of immersion objective lenses without the use of an objective lens heater. The microscope itself acts as a massive heat sink, causing as much as a 10°C temperature differential. Even with dry objectives there can be a significant temperature differential between the edges of the specimen holder and where the cells are being imaged. It is therefore imperative that the temperature where the cells are growing be measured not just the chamber holding them. This can be accomplished with an inexpensive non-contact thermometer equipped w/laser sighting, such as OS-PAL (∼$55.00 from OMEGA Engineering, INC.). When using immersion objectives or where there is not a line of sight to the imaging area (e.g., inverted microscopes), a small thermocouple probe can be directly inserted at the focal point of the microscope. This can be safely done by attaching a small probe to the coverslip (where the cells would normally be attached) and assembling the chamber normally, including filling with media or water. Depending on the chamber type, a small hole may need to be drilled to feed the probe wire through.
Figure 4. Selected frames from a time-lapse series of CFPAC-1 cell showing a reversal of mitosis (A–C) in response to mild hypothermia (20°C). The chromatin can be clearly seen decondensing to an “interphase” appearance (C). Normal progression of mitosis can be seen (D–F) including nuclear envelope break (F) down once the temperature was returned to 37°C. Bar = 10 μm.
One side-effect of heating above ambient temperature is stage drift. Microscopes are constructed mostly of metal with little or no engineered thermal breaks. This is especially problematic with stage type heaters but can also affect other types. To minimize the effect of temperature, allow the entire system to equilibrate (∼45–60 min), including a mock specimen chamber and focused objective lens (if immersion). While this will help, it will not be enough to maintain the same focal plane for long-term filming. The good news is that all the microscope manufacturers offer some sort of focal correction that will maintain the same focal plane indefinitely.
How to keep the cells warm and happy
There are two approaches, a heater that holds the specimen and attaches to the stage of the microscope or enclosing the entire microscope in a heated box. There are commercial (just a few examples: CellAsci, Harvard Apparatus, Bioptechs Inc., Physitemp Instruments, Inc., In Vivo Scientific) and Lab-built versionsCitation2, Citation45-48 of both of these approaches. They range from a simple cardboard box heated by a hair dryer to a complicated system utilizing external temperature probes and PPD temperature controllers to prevent temperature over-shooting. The main advantages of the “stage” type heater are that it allows for access to the cells for electrophysiology, and has lower cost. However, if an immersion objective is going to be used, an additional objective heater is required. Gas exchange control is more challenging, but not impossible with stage type chambers, see . The enclosed microscope type tends to have more stable temperature regulation and does not require objective lens heaters. It is also easier to control gas exchange, C02 for PH control, or other gases for specific experimental conditions. The C02 levels can be controlled by either flowing premixed C02/air mixture or by mixing it at the enclosure. While the premixed gas is easier, it is significantly more expensive per tank and limited to a single ratio.
Figure 5. Environmental chamber designed for imaging glass bottom 35 mm culture dishes (MatTek or similar). The chamber is designed to fit into a Ludl motorized stage (Ludl Electronic Products Ltd.), although small modification would allow adaptation to other stage manufactures. The top panel shows the heated (PID controlled) chamber. A portion of the aluminum plate is machined out and resistive heaters, temperature probes, and a sand/glue mixture is added to form-fit the culture dish and provide the source of heat. The bottom panel shows the humidified gas chamber on top of the stage (opacity is reduced to allow the juxtaposition with stage to be seen). The machined hole (coverslip covered) in the top allows transmitted light imaging. The two small resistive heaters on top of the humidified gas chamber are used to keep the coverslip fog “free.” The humidity inside the chamber is controlled by adding cotton saturated with sterile water into the chamber. The C02 balance is maintained by flowing at a low rate, premixed gases (5% C02) into the chamber. Electromechanical drawings would be happily provided upon request.

Visualization chambers—Home away from home
Similar to temperature control, there are two basic designs, open and closed. As the names imply, they allow gas exchange or not. Additionally, the open systems allow for manipulation of the cells, for example, micro-injection. However, with open styles, humidity control is necessary. The substitution of liquids is an option in both types of chambers, but is easier in the closed systems, which allow for laminar flow. Again, there are a large selection of both commercial (most of the companies listed above) and lab-built chambers.Citation2,4,45, Citation47-49 The simplest open style is a petri dish with a glass coverslip inserted into the bottom, which improves image quality (MatTek Corp.), while the simplest closed is a slide-coverslip sandwich sealed with VALAP 1:1:1 (Vaseline: lanolin: paraffin).Citation50 Most home-built chambers are built to accomplish a specific set of experiments; therefore, a complete list here would be ephemeral.
Media—Environment and food (yum)
The media not only provides the nutrients needed by the cells, but it is also their environment. There are various media formulations which are specifically designed to match the growth requirements for specific cell types. For the purpose of live-cell imaging, there are either C02-dependent or -independent styles of media. The dependent style requires a specific concentration (usually 5%) of C02 in the atmosphere to buffer and maintain the PH of the media. For the C02-independent style, HEPES (usually 25 nm) is used to maintain the PH balance. It is possible to grow cells in a C02-dependent media and then transition them to a C02-independent media for filming. This can be accomplished by switching to a C02-independent media, Leibovitz L-15 media (Life Technologies), or custom designed growth media.Citation51 Alternatively, HEPES can be added to the cells preferred growth media. For some cell lines, the addition of HEPES needs to be gradual and done over period of time (up to 24 h). All media used for filming in fluorescence should be phenol red “free.”
For some cell lines, the rapid transition to “new non-conditioned” media can cause significant cell cycle delays.Citation52 Conditioned media is simply media which cells have been growing in for a period of time. Cells in their normal existence “condition” the media by releasing/secreting proteins, cytokines, chemicals, etc., into it.Citation53,54 Mixing conditioned and “new” media (1:3) usually alleviates the “stress” of new media and allows filming immediately. Most cell types will completely recover when placed into “new” media in approximately 3 h. It should be mentioned here that whichever media type is being used, the media should be at the optimal temperature before usage.
Osmolality is another very important parameter and often goes unchecked. All “homemade” buffers should be checked to assure that they are within an appropriate range for the cell type being imaged. It is important that there is not significant evaporation of the media during the experiment because this will cause a change in the osmolality, sending the cells into “shock.”
Vitamins, specifically riboflavin and pyridoxal, have been found to decrease photostability of Enhanced Green Fluorescent Protein (EGFP) present in imaging media. Conversely, flavonoid rutin (antioxidants) greatly enhances photostability of EGFP during live cell microscopy. The removal of the two vitamins and the addition flavonoid rutin further suppress EGFP photobleaching allowing a higher photon dose without any detectable side-effects.Citation55,56
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the New York State Department of Health and Wadsworth Center's Advanced Light Microscopy and Image Analysis Core Facility.
References
- Sung MH, McNally JG. Live cell imaging and systems biology. Wiley Interdiscip Rev Syst Biol Med 2011; 3:167-82; PMID:20730797; http://dx.doi.org/10.1002/wsbm.108
- Cheng WY, Hsu WL, Cheng HH, Huang ZH, Chang YC. An observation chamber for studying temperature-dependent and drug-induced events in live neurons using fluorescence microscopy. Anal Biochem 2009; 386:105-12; PMID:19111514; http://dx.doi.org/10.1016/j.ab.2008.12.004
- Keller PJ, Schmidt AD, Santella A, Khairy K, Bao Z, Wittbrodt J, Stelzer EH. Fast, high-contrast imaging of animal development with scanned light sheet-based structured-illumination microscopy. Nat Methods 2010; 7:637-42; PMID:20601950; http://dx.doi.org/10.1038/nmeth.1476
- Day RN, Schaufele F. Imaging molecular interactions in living cells. Mol Endocrinol 2005; 19:1675-86; PMID:15761028; http://dx.doi.org/10.1210/me.2005-0028
- He L, Wang X, Montell DJ. Shining light on Drosophila oogenesis: live imaging of egg development. Curr Opin Genet Dev 2011; 21:612-9; PMID:21930372; http://dx.doi.org/10.1016/j.gde.2011.08.011
- Campbell EM, Hope TJ. Live cell imaging of the HIV-1 life cycle. Trends Microbiol 2008; 16:580-7; PMID:18977142; http://dx.doi.org/10.1016/j.tim.2008.09.006
- Stephens DJ, Allan VJ. Light microscopy techniques for live cell imaging. Science 2003; 300:82-6; PMID:12677057; http://dx.doi.org/10.1126/science.1082160
- Fischer RS, Wu Y, Kanchanawong P, Shroff H, Waterman CM. Microscopy in 3D: a biologist's toolbox. Trends Cell Biol 2011; 21:682-91; PMID:22047760; http://dx.doi.org/10.1016/j.tcb.2011.09.008
- Nelson DE, Ihekwaba AE, Elliott M, Johnson JR, Gibney CA, Foreman BE, Nelson G, See V, Horton CA, Spiller DG, et al. Oscillations in NF-kappaB signaling control the dynamics of gene expression. Science 2004; 306:704-8; PMID:15499023; http://dx.doi.org/10.1126/science.1099962
- Taylor MA, Janousek J, Daria V, Knittel J, Hage B, Bachor HA, Bowen WP. Biological measurement beyond the quantum limit. Nat Photonics 2013; 7:229-33; http://dx.doi.org/10.1038/nphoton.2012.346
- Swedlow JR. Quantitative fluorescence microscopy and image deconvolution. Methods Cell Biol 2007; 81:447-65; PMID:17519179; http://dx.doi.org/10.1016/S0091-679X(06)81021-6
- Swedlow JR, Platani M. Live cell imaging using wide-field microscopy and deconvolution. Cell Struct Funct 2002; 27:335-41; PMID:12502887; http://dx.doi.org/10.1247/csf.27.335
- Benninger RK, Piston DW. Two-photon excitation microscopy for the study of living cells and tissues. Current Protocols in Cell Biology 2013; Chapter 4:Unit 4.11.
- Patterson GH, Piston DW. Photobleaching in two-photon excitation microscopy. Biophys J 2000; 78:2159-62; PMID:10733993; http://dx.doi.org/10.1016/S0006-3495(00)76762-2
- Hopt A, Neher E. Highly nonlinear photodamage in two-photon fluorescence microscopy. Biophys J 2001; 80:2029-36; PMID:11259316; http://dx.doi.org/10.1016/S0006-3495(01)76173-5
- Herron BJ, Smith J, Cole RW. New method to quantify angiogenesis in vivo using multi-photon imaging. Microscopy Today 2009; 17:24-7; http://dx.doi.org/10.1017/S1551929509000339
- Ilina O, Bakker GJ, Vasaturo A, Hofmann RM, Friedl P. Two-photon laser-generated microtracks in 3D collagen lattices: principles of MMP-dependent and -independent collective cancer cell invasion. [Erratum appears in Phys Biol. 2011 Apr;8(2):029501]. Phys Biol 2011; 8:015010; PMID:21301056; http://dx.doi.org/10.1088/1478-3975/8/1/015010
- Gräf R, Rietdorf J, Zimmermann T. Live cell spinning disk microscopy. Adv Biochem Eng Biotechnol 2005; 95:57-75; PMID:16080265; http://dx.doi.org/10.1007/b102210
- Wang E, Babbey CM, Dunn KW. Performance comparison between the high-speed Yokogawa spinning disc confocal system and single-point scanning confocal systems. J Microsc 2005; 218:148-59; PMID:15857376; http://dx.doi.org/10.1111/j.1365-2818.2005.01473.x
- Huisken J, Swoger J, Del Bene F, Wittbrodt J, Stelzer EH. Optical sectioning deep inside live embryos by selective plane illumination microscopy. Science 2004; 305:1007-9; PMID:15310904; http://dx.doi.org/10.1126/science.1100035
- Holekamp TF, Turaga D, Holy TE. Fast three-dimensional fluorescence imaging of activity in neural populations by objective-coupled planar illumination microscopy. Neuron 2008; 57:661-72; PMID:18341987; http://dx.doi.org/10.1016/j.neuron.2008.01.011
- Verveer PJ, Swoger J, Pampaloni F, Greger K, Marcello M, Stelzer EH. High-resolution three-dimensional imaging of large specimens with light sheet-based microscopy. Nat Methods 2007; 4:311-3; PMID:17339847
- Eggeling C, Willig KI, Barrantes FJ. STED microscopy of living cells–new frontiers in membrane and neurobiology. J Neurochem 2013; 126:203-12; PMID:23506404; http://dx.doi.org/10.1111/jnc.12243
- Takasaki KT, Ding JB, Sabatini BL. Live-cell superresolution imaging by pulsed STED two-photon excitation microscopy. Biophys J 2013; 104:770-7; PMID:23442955; http://dx.doi.org/10.1016/j.bpj.2012.12.053
- Wagner E, Lauterbach MA, Kohl T, Westphal V, Williams GS, Steinbrecher JH, Streich JH, Korff B, Tuan HT, Hagen B, et al. Stimulated emission depletion live-cell super-resolution imaging shows proliferative remodeling of T-tubule membrane structures after myocardial infarction. Circ Res 2012; 111:402-14; PMID:22723297; http://dx.doi.org/10.1161/CIRCRESAHA.112.274530
- Willig KI, Nagerl UV. Stimulated emission depletion (STED) imaging of dendritic spines in living hippocampal slices. Cold Spring Harb Protoc 2012; 5.
- Willig KI, Stiel AC, Brakemann T, Jakobs S, Hell SW. Dual-label STED nanoscopy of living cells using photochromism. Nano Lett 2011; 11:3970-3; PMID:21786833; http://dx.doi.org/10.1021/nl202290w
- Schermelleh L, Carlton PM, Haase S, Shao L, Winoto L, Kner P, Burke B, Cardoso MC, Agard DA, Gustafsson MG, et al. Subdiffraction multicolor imaging of the nuclear periphery with 3D structured illumination microscopy. Science 2008; 320:1332-6; PMID:18535242; http://dx.doi.org/10.1126/science.1156947
- York AG, Ghitani A, Vaziri A, Davidson MW, Shroff H. Confined activation and subdiffractive localization enables whole-cell PALM with genetically expressed probes. Nat Methods 2011; 8:327-33; PMID:21317909; http://dx.doi.org/10.1038/nmeth.1571
- Waters JC. Accuracy and precision in quantitative fluorescence microscopy. J Cell Biol 2009; 185:1135-48; PMID:19564400; http://dx.doi.org/10.1083/jcb.200903097
- Murray JM, Appleton PL, Swedlow JR, Waters JC. Evaluating performance in three-dimensional fluorescence microscopy. J Microsc 2007; 228:390-405; PMID:18045334; http://dx.doi.org/10.1111/j.1365-2818.2007.01861.x
- Rieder CL, Cole R. Microscopy-induced radiation damage, microtubules, and progression through the terminal stage of G2 (prophase) in vertebrate somatic cells. Cold Spring Harb Symp Quant Biol 2000; 65:369-76; PMID:12760052; http://dx.doi.org/10.1101/sqb.2000.65.369
- Dewitt S, Hallett MB. Optical complexities of living cytoplasm–implications for live cell imaging and photo-micromanipulation techniques. J Microsc 2011; 241:221-4; PMID:21118242; http://dx.doi.org/10.1111/j.1365-2818.2010.03451.x
- Hoebe RA, Van der Voort HT, Stap J, Van Noorden CJ, Manders EM. Quantitative determination of the reduction of phototoxicity and photobleaching by controlled light exposure microscopy. J Microsc 2008; 231:9-20; PMID:18638185; http://dx.doi.org/10.1111/j.1365-2818.2008.02009.x
- Mora-Bermúdez F, Ellenberg J. Measuring structural dynamics of chromosomes in living cells by fluorescence microscopy. Methods 2007; 41:158-67; PMID:17189858; http://dx.doi.org/10.1016/j.ymeth.2006.07.035
- Cole RW, Turner JN. Light-emitting diodes are better illumination sources for biological microscopy than conventional sources. Microsc Microanal 2008; 14:243-50; PMID:18312724; http://dx.doi.org/10.1017/S1431927608080288
- Ahn HH, Kim SN, Kye YC. Fluorescence digital photography of acne using a light-emitting diode illuminator. Skin Res Technol 2006; 12:289-91; PMID:17026661; http://dx.doi.org/10.1111/j.0909-752X.2006.00166.x
- Nishigaki T, Wood CD, Shiba K, Baba SA, Darszon A. Stroboscopic illumination using light-emitting diodes reduces phototoxicity in fluorescence cell imaging. Biotechniques 2006; 41:191-7; PMID:16925021; http://dx.doi.org/10.2144/000112220
- Sun S, Francis J, Sapsford KE, Kostov Y, Rasooly A. Multi-wavelength Spatial LED illumination based detector for in vitro detection of Botulinum Neurotoxin A Activity. Sens Actuators B Chem 2010; 146:297-306; PMID:20498728; http://dx.doi.org/10.1016/j.snb.2010.02.009
- Borlinghaus RT. MRT letter: high speed scanning has the potential to increase fluorescence yield and to reduce photobleaching. Microsc Res Tech 2006; 69:689-92; PMID:16878313; http://dx.doi.org/10.1002/jemt.20363
- De AK, Goswami D. A systematic study on fluorescence enhancement under single-photon pulsed illumination. J Fluoresc 2009; 19:931-7; PMID:19507011; http://dx.doi.org/10.1007/s10895-009-0489-4
- Penjweini R, Loew HG, Hamblin MR, Kratky KW. Long-term monitoring of live cell proliferation in presence of PVP-Hypericin: a new strategy using ms pulses of LED and the fluorescent dye CFSE. J Microsc 2012; 245:100-8; PMID:21974829; http://dx.doi.org/10.1111/j.1365-2818.2011.03555.x
- Lakowicz J. Pulsed Xenon Lamps. Principles of Fluorescence Spectroscopy. New York: Springer; 2006.31-4
- Rieder CL, Cole RW. Cold-shock and the Mammalian cell cycle. Cell Cycle 2002; 1:169-75; PMID:12429927; http://dx.doi.org/10.4161/cc.1.3.119
- Lin PC, Cheng PC, Yu H. An engineered microenvironment for multidimensional microscopy of live cells. Scanning 2005; 27:284-92; PMID:16370396; http://dx.doi.org/10.1002/sca.4950270603
- Voiculescu O, Stern CD. Assembly of imaging chambers and high-resolution imaging of early chick embryos. Cold Spring Harb Protoc 2012; 12.
- Rieder CL, Cole RW. Perfusion chambers for high-resolution video light microscopic studies of vertebrate cell monolayers: some considerations and a design. Methods Cell Biol 1998; 56:253-75; PMID:9500142; http://dx.doi.org/10.1016/S0091-679X(08)60430-6
- Buchner O, Lütz C, Holzinger A. Design and construction of a new temperature-controlled chamber for light and confocal microscopy under monitored conditions: biological application for plant samples. J Microsc 2007; 225:183-91; PMID:17359253; http://dx.doi.org/10.1111/j.1365-2818.2007.01730.x
- Rose G. A separable and multipurpose tissue culture chamber. Tex Rep Biol Med 1954; 12:1074-83; PMID:13238275
- Fischer AH, Jacobson KA, Rose J, Zeller R. Mounting live cells attached to coverslips for microscopy. CSH Protoc 2008; 2008: t4927; PMID:21356764
- Vistica DT, Scudiero D, Skehan P, Monks A, Boyd MR. New carbon dioxide-independent basal growth medium for culture of diverse tumor and nontumor cells of human and nonhuman origin. J Natl Cancer Inst 1990; 82:1055-61; PMID:2112200; http://dx.doi.org/10.1093/jnci/82.12.1055
- Mikhailov A, Shinohara M, Rieder CL. Topoisomerase II and histone deacetylase inhibitors delay the G2/M transition by triggering the p38 MAPK checkpoint pathway. J Cell Biol 2004; 166:517-26; PMID:15302851; http://dx.doi.org/10.1083/jcb.200405167
- Martin GR. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc Natl Acad Sci U S A 1981; 78:7634-8; PMID:6950406; http://dx.doi.org/10.1073/pnas.78.12.7634
- Xu C, Rosler E, Jiang J, Lebkowski JS, Gold JD, O’Sullivan C, Delavan-Boorsma K, Mok M, Bronstein A, Carpenter MK. Basic fibroblast growth factor supports undifferentiated human embryonic stem cell growth without conditioned medium. Stem Cells 2005; 23:315-23; PMID:15749926; http://dx.doi.org/10.1634/stemcells.2004-0211
- Bogdanov AM, Bogdanova EA, Chudakov DM, Gorodnicheva TV, Lukyanov S, Lukyanov KA. Cell culture medium affects GFP photostability: a solution. Nat Methods 2009; 6:859-60; PMID:19935837; http://dx.doi.org/10.1038/nmeth1209-859
- Bogdanov AM, Kudryavtseva EI, Lukyanov KA. Anti-fading media for live cell GFP imaging. [Electronic Resource]. PLoS One 2012; 7:e53004; PMID:23285248; http://dx.doi.org/10.1371/journal.pone.0053004