Abstract
The antibody trastuzumab and the tyrosine kinase inhibitor lapatinib are approved by the FDA for the treatment of HER2-overexpressing breast cancer. These anti-HER2 drugs are changing the natural history of HER2-overexpressing breast cancer. However, therapeutic resistance to trastuzumab or lapatinib, as either single-agents or in combination with chemotherapy in the metastatic setting, typically occurs within months of starting therapy. Several mechanisms of trastuzumab-resistance have been reported that include signaling from other HER receptors, signaling from receptor tyrosine kinases (RTKs) outside of the HER (ErbB) family, increased phosphatidylinositol 3-kinase signaling, and the presence of truncated forms of HER2. Mechanisms of resistance to lapatinib also point to increased phosphatidylinositol 3-kinase signaling as well as derepression/activation of compensatory survival pathways. In this review, we discuss how these models and mechanisms enhance our understanding of the clinical resistance to HER2-directed therapies.
Introduction
HER2 (ErbB2) is a member of the ErbB family of transmembrane receptor tyrosine kinases, which also includes the epidermal growth factor receptor (EGFR, ErbB1), HER3 (ErbB3) and HER4 (ErbB4). Binding of ligands to the extracellular domain of EGFR, HER3 and HER4 induces the formation of kinase active homo- and heterodimers to which activated HER2 is recruited as a preferred partner.Citation1 HER2 does not bind any of the ErbB ligands directly; however, its catalytic activity can potently amplify signaling by ErbB-containing heterodimers via increasing ligand binding affinity and/or receptor recycling and stability.Citation2–Citation5 HER2/HER3 heterodimers are the most transforming of this receptor network.Citation6,Citation7 HER3, which lacks intrinsic kinase activity, is able to potently activate the phosphatidylinositol-3 kinase (PI3K)/Akt signaling pathwayCitation8,Citation9 via its six docking sites for the p85 adaptor subunit of PI3K, whereas HER2 is unable to directly bind to and thus activate PI3K-Akt. Loss of HER3 inhibits viability of HER2-overexpressing breast cancer cellsCitation10,Citation11 and HER2-overexpressing cells are particularly sensitive to apoptosis induced by PI3K inhibitors,Citation12 thus suggesting the HER3-PI3K axis is essential for survival of HER2-dependent cells.
Amplification of the HER2 gene occurs in approximately 25% of invasive breast cancers and is associated with poor patient outcome.Citation13 HER2 is an appealing therapeutic target in breast cancers because of the correlation between overexpression and poor prognosis and because normal cells have relatively low HER2 expression. Trastuzumab (Herceptin), a humanized monoclonal IgG1 that binds to the juxtamembrane region of HER2, induces clinical responses in HER2-overexpressing breast cancers and prolongs patient survival (see below). The clinical efficacy of trastuzumab appears limited to breast cancers that overexpress HER2 as measured by intense membrane staining in the majority of tumor cells with HER2 antibodies (3+ by immunohistochemistry [IHC]) or excess copies of the HER2 gene determined by fluorescent in situ hybridization (FISH). Therefore, HER2 overexpression by IHC and/or FISH is the biomarker predictive of good odds of response to treatment with the antibody.
Resistance to Trastuzumab
Trastuzumab binds to an epitope in the juxtamembrane region of the HER2 receptor tyrosine kinase. This binding induces uncoupling of ligand-independent HER2-HER3 heterodimers and inhibition of downstream signalingCitation14 as well as antibody-dependent, cell-mediated cytotoxicity (ADCC).Citation15 Several large randomized adjuvant trials (NCCTG N9831, NSABP B-31, BCIRG 006 and HERA) have shown that the addition of trastuzumab to standard chemotherapy reduces disease recurrence and the risk of death compared to chemotherapy alone in patients with surgically-resected tumors.Citation16–Citation19 In the N-9831 trial, a recent interim analysis showed that the benefit of concurrent trastuzumab and chemotherapy was more pronounced than that of chemotherapy followed by trastuzumab.Citation20 Based on these data, the addition of trastuzumab to adjuvant chemotherapy has become standard of care in women with HER2+ early breast cancer. Although it is anticipated that many patients treated with adjuvant trastuzumab will be cured of their disease, it is also expected that many will recur. Trastuzumab in combination with chemotherapy is also indicated for the treatment of HER2+ metastatic breast cancer.Citation21 Nevertheless, response rates to single-agent trastuzumab are short lived.Citation16 Thus, a large proportion of patients with HER2+ tumors either does not respond to trastuzumab or develops acquired tolerance to the antibody, suggesting both de novo and acquired mechanisms of drug resistance.
Most preclinical models have reported that HER2 gene amplification and RNA/protein overexpression are maintained in trastuzumab-resistant HER2+ clones,Citation22,Citation23 thus implying that HER2-overexpressing tumor cells that bypass trastuzumab action continue to depend on the HER2 oncogene. Several studies have reported potential mechanisms of resistance to trastuzumab, including signaling from RTKs outside of the HER (ErbB) family, increased PI3K signaling, amplification of signaling by other ErbB receptors and the presence of altered forms of HER2 that are not recognized or bound by trastuzumab.
Cross-talk with heterologous RTKs and amplification of ErbB signaling.
A potential mechanism of trastuzumab resistance involves RTKs outside of the HER family modulating levels of the Cdk inhibitor p27KIP1, such as the IGF-I receptor. For example, overexpression of IGF-IR or increased levels of IGF-IR/HER2 heterodimers,Citation24,Citation25 which potently activate PI3K and its downstream effector AKT, abrogate trastuzumab action when transfected into antibody-sensitive breast cancer cells. In a neoadjuvant trial of chemotherapy plus trastuzumab, high levels of IGF-IR as measured by IHC correlated with a poor clinical response.Citation26 MET (HGF receptor) has also been implicated in trastuzumab resistance. HER2 overexpressing cells upregulate MET following exposure to trastuzumab. Further, activation of MET protects cells against trastuzumab by abrogating the induction of p27.Citation27 In a cohort of patients with HER2+ breast cancers, overexpression of the EphA2 RTK was associated with reduced disease-free and overall survival. Treatment of resistant cells with trastuzumab induced phosphorylation of Src and EphA2 resulting in the activation of PI3K/AKT and MAPK. Administration of a neutralizing EphA2 antibody restored sensitivity to trastuzumab in vivo.Citation28 Finally, the receptor for erythropoietin (EpoR) is co-expressed in a proportion of cell lines and primary tumors that also harbor HER2 gene amplification. In those cells, treatment with recombinant human erythropoietin (rHuEPO) activates Jak and Src leading to inactivation of PTEN and attenuation of the response to trastuzumab. Interestingly, the concurrent administration of rHuEPO and trastuzumab correlated with a shorter progression-free and overall survival in patients with HER2+ metastatic breast cancer.Citation29
Other members of the ErbB receptor network are thought to play a role in trastuzumab resistance. Exogenous ligands of the EGFR and HER3/4 co-receptors have been shown to rescue from the anti-proliferative effect of the antibody.Citation30,Citation31 This is consistent with structural data using ErbB receptor ectodomains, which show that trastuzumab is unable to block ligandinduced EGFR/HER2 and HER2/HER3 heterodimers.Citation32,Citation33 Our laboratory reported trastuzumab-resistant HER2-overexpressing BT-474 human breast cancer cells generated in vivo. The resistant cells retained HER2 gene amplification and trastuzumab binding. They exhibited higher levels of phosphorylated EGFR and HER3 and EGFR/HER2 heterodimers as well as overexpression of EGFR, TGFα, HB-EGF and heregulin RNAs compared to the parental trastuzumab-sensitive cells,Citation23 thus suggesting enhanced EGFR- and HER3-mediated activation of HER2. The HER2 tyrosine kinase inhibitor (TKI) lapatinib and the HER2 antibody, pertuzumab, which blocks HER2 heterodimerization with ErbB co-receptors,Citation34,Citation35 inhibited growth of the antibody-resistant cells suggesting that, although resistant to trastuzumab, the cells were still dependent on HER2-dependent interactions with the ErbB receptor network.Citation23 In line with this report, the activation of TGFβ receptors, a pathway amplified in metastatic mammary tumors, has been shown to induce phosphorylation of the sheddase TACE/ADAM17 resulting in increased secretion of TGFα, amphiregulin and heregulin. These changes are followed by enhanced coupling of p85 and HER3, activation of PI3K/AKT and resistance to trastuzumab. Further, a gene signature induced by expression of a constitutively active, mutant type I TGFβ receptor correlated with resistance to trastuzumab in a panel of HER2+ breast cancer cells lines and with poor clinical outcome in patients with invasive breast cancer.Citation36
Amplification of the PI3K/AKT pathway.
Resistance to trastuzumab may occur as a result of aberrant activation of signaling pathways downstream of the receptor, such as PI3K/Akt. Molecular alterations involving this pathway are considered the most frequent in breast cancer, together encompassing over 30% of invasive tumors. Alterations in breast cancer resulting in hyperactivity of the PI3K pathway include gain-of-function mutations in PIK3CA (the gene encoding the PI3K catalytic subunit p110α),Citation37,Citation38 mutations in AKT1,Citation39 amplifications of AKT2,Citation40 loss of the PTEN lipid phosphatase,Citation41,Citation42 and loss of the tumor suppressor INPP4B (inositol polyphosphate 4-phosphatase type II).Citation43 PIK3CA mutations in primary breast tumors have been associated with lymph node metastases, the presence of ER and PgR and HER2 overexpression.Citation44,Citation45 It is generally accepted that antiHER2 therapies should inhibit PI3K/Akt signaling downstream the HER2 receptor in order to inhibit tumor growth.Citation14,Citation46
Using a large-scale RNA interference screen, Berns et al. identified PTEN as the only gene whose knockdown resulted in trastuzumab resistance,Citation47 consistent with the previous observation that in antibody-sensitive cells, trastuzumab increases the phosphatase activity of PTEN via inhibition of Src and Src-mediated (inhibitory) phosphorylation of PTEN.Citation48 This same report also showed that oncogenic mutants of PIK3CA, the gene encoding for the catalytic subunit of PI3K p110α, conferred resistance to trastuzumab to cells in culture. In patients with breast cancer, the presence of oncogenic PIK3CA mutations and low PTEN expression measured by IHC identified those patients with the worst outcome following chemotherapy plus trastuzumab.Citation47 Supporting aberrant PI3K signaling and causality to drug resistance, in more recent preclinical studies, the addition of PI3K inhibitors to trastuzumab has inhibited growth of HER2+/PIK3CA mutant tumors resistant to anti-HER2 therapy.Citation49–Citation51 Interestingly, inhibitors of mTOR, a serine-threonine kinase downstream PI3K, have shown activity after progression on trastuzumab. Dalenc et al. recently reported a multicenter phase II study of 55 women with HER2+ MBC whose tumors were resistant to trastuzumab and taxanes. Patients were treated with the TOR inhibitor everolimus, paclitaxel and trastuzumab, exhibiting an impressive partial response rate of 19% and an overall clinical benefit rate of 81%.Citation52
Alterations in binding of trastuzumab to HER2.
Another potential mechanism of resistance is the presence of truncated forms of HER2 that trastuzumab does not recognize. Anido et al. reported the presence of HER2 C-terminal fragments, which result from alternative translation initiation from methionines near the transmembrane domain of the full-length receptor molecule.Citation53 These fragments are kinase-active but lack the trastuzumab binding epitope and therefore, can potentially allow the cancer cell to escape antibody action.Citation54 Analysis of a cohort of patients with HER2+ metastatic breast cancer treated with trastuzumab and chemotherapy showed a very low response rate in tumors with cytosolic p95HER2 compared to those without.Citation54 Lapatinib has been shown to inhibit the catalytic activity of p95HER2. Therefore, patients with p95HER2-positive breast cancers treated with lapatinib alone or in combination with capecitabine exhibited a similar progression-free survival and overall response rate compared to p95HER2-negative tumors,Citation55 suggesting a clinical setting where a HER2 TKI might be advantageous over trastuzumab ().
In addition to truncated forms of HER2, overexpression of the membrane-associated glycoprotein mucin-4 (MUC4),Citation56,Citation57 has been shown to mask trastuzumab binding epitopes in the HER2 receptor, resulting in acquired resistance. Finally, an oncogenic splice isoform with an in-frame deletion of exon 16 (HER2Δ16) is found in some HER2-overexpressing breast cancer cell lines and primary breast cancers.Citation58,Citation59 Loss of exon 16 results in a constitutively dimerized and active HER2 receptor, enhanced Src activity and accelerated transformation. Cells expressing HER2Δ16 are resistant to trastuzumab; this resistance is abrogated by co-treatment with Src inhibitors.Citation60 It has not been shown yet whether HER2Δ16 is a mechanism of resistance to trastuzumab in patients with HER2+ tumors.
Resistance to Tyrosine Kinase Inhibitors
Another approach to block HER2 is the use of ATP-competitive, small molecule TKIs. The dual EGFR/HER2 TKI lapatinib is active as first line monotherapy in patients with HER2+ metastatic breast cancer and in combination with chemotherapy improves progression free survival compared to chemotherapy alone.Citation61,Citation62 In the latter registration trial, fewer brain metastases occurred in women in the combination than in the monotherapy arm, suggesting a potential difference between lapatinib and trastuzumab as it applies to recurrences in the CNS.Citation62 In the registration study and in a second randomized trial of paclitaxel ± lapatinib in patients with metastatic breast cancer, the clinical benefit of lapatinib was limited to patients with HER2 overexpression as scored by IHC and/or FISH.Citation63 Like lapatinib, the HER2/EGFR dual TKI neratinibCitation64 has shown clinical activity in patients with HER2+ metastatic breast cancer who have progressed on trastuzumab. As with trastuzumab, it is generally accepted that in order to exert an antitumor effect in HER2+ cancers, treatment with lapatinib should inhibit the PI3K/Akt pathway.Citation23,Citation65
Proposed mechanisms of resistance to lapatinib involve recovery through derepression and/or activation of compensatory survival pathways. For example, in HER2-overexpressing BT474 cells selected for acquired resistance to lapatinib, the resistant cells continued to show inhibition of HER2, HER3, MAPK and AKT phosphorylation upon treatment with lapatinib. In these cells, inhibition of AKT with lapatinib resulted in derepression of FoxO3a thus leading to increased ERα transcription and ER signaling.Citation66,Citation67 Co-treatment with lapatinib and the ER down-regulator fulvestrant prevented the outgrowth of drug resistant cells. Further, lapatinib was shown to induce ER signaling in tumor biopsies from patients with HER2+/ER+ but not HER2+/ER−negative breast cancers. The same group also found calcium-dependent increased levels of phosphorylated RelA, the pro-survival subunit of NFκB, upon lapatinib treatment of HER2+ breast cancer cell lines.Citation68 Using either small interfering RNA constructs targeting RelA or an intracellular calcium chela-tor enhanced the apoptotic effects of lapatinib, suggesting a possible role for RelA in adaptation to the HER2 TKI.
Using HER2+ cells selected in culture, another study identified overexpression of AXL as a mechanism of resistance to lapatinib.Citation69 AXL is an RTK with a kinase domain closely resembling MET and an extracellular domain resembling neural cell adhesion molecules.Citation70 BT474 cells rendered drug-resistant by chronic exposure to lapatinib exhibited increased expression and activation of AXL. GSK1363089 (foretinib), a multikinase inhibitor of AXL, MET and VEGFR, restored lapatinib and trastuzumab sensitivity in the AXL-overexpressing, drug-resistant cells.Citation69
Other studies have shown upregulation of HER3 transcription and protein levels and recovery of HER3 phosphorylation after short-term inhibition of HER2 with the TKIs gefitinib and lapatinib.Citation71,Citation72 As with ERα, this recovery of HER3 is also explained by derepression of FoxO upon inhibition of PI3K/AKT downstream of HER2 and upregulation of FoxO-dependent HER3 transcription (Garret et al. submitted). In one study, HER3-PI3K-AKT activity was completely inhibited by higher, pulsatile doses of lapatinib both in vitro and in vivo.Citation72 While the complete inactivation of the HER2 kinase may require doses of lapatinib that are not tolerated in vivo, it is still possible that high intermittent doses might be more effective than the currently approved (lower dose) daily regimens. These alternative treatment schedules are currently being explored in clinical trials.
As with trastuzumab, activating PIK3CA mutations, loss of the tumor suppressor PTEN and alternative signaling pathways that activate PI3K-AKT are reported mechanisms for escape from lapatinib. Using a large-scale loss-of-function short hairpin RNA screen to identify novel modulators of resistance to lapatinib, Eichhorn et al. identified the tumor suppressor PTEN as a gene whose loss reduced the sensitivity to the TKI both in vitro and in vivo.Citation50 In addition, two dominant activating mutations in PIK3CA (E545K and H1047R), which are prevalent in breast cancer, also conferred resistance to lapatinib. These authors also showed that the resistance to lapatinib induced by the PI3K mutants can be abrogated through the use of BEZ235, a dual inhibitor of PI3K/mTOR.Citation50
HER2 Gene Mutations: Possible Role in Acquired Resistance
A common mechanism of clinical resistance to TKIs that target RTKs other than HER2 is the development of mutations in the targeted receptor. For example, some lung cancers that acquire resistance to EGFR inhibitors harbor T790M alleles that comprise <5% of the total EGFR alleles;Citation73 these mutations are generally detected in cancers that progress after an initial response to an EGFR TKI.Citation74–Citation76 Additional examples include mutations in BCR/ABL and c-kit in chronic myeloid leukemia and gastrointestinal stromal tumors, respectively, which result in resistance to imatinib, a specific BCR/ABL and c-kit kinase inhibitor.Citation77,Citation78 It is possible that as a result of selective pressure from anti-HER2 therapies, breast cancers will acquire or will be ‘enriched’ for mutations in HER2, which may be present in only a fraction of the HER2 alleles. Intragenic somatic mutations in the HER2 gene were reported in about 4% of non-small-cell lung cancers (NSCLC). These involve in-frame duplications/insertions in a small stretch within exon 20 of HER2.Citation79,Citation80 Two studies did not find HER2 kinase domain mutationsCitation79,Citation81 though it is possible that direct gene sequencing methods used in these studies may have missed mutations present in a few HER2 alleles in tumors with HER2 gene amplification. Only one report has identified a low frequency of HER2 mutations in breast cancer.Citation82 Interestingly, one of these mutations, a YVMA insertion at G776 in exon 20, was found to confer de novo resistance to trastuzumab and lapatinib. Cells expressing this mutant still responded to CI-1033, an irreversible covalent inhibitor of the HER2 kinase.Citation83 This inhibitor is very similar to neratinib, the irreversible HER2 kinase inhibitor in late clinical development.Citation64 These acquired alterations have yet to be detected in metastatic lesions recurring after an initial response to primary anti-HER2 therapy or following adjuvant therapy.
Dual HER2 Blockade and Abrogation of Drug Resistance
As indicated above, HER2 TKIs have shown clinical activity in patients with HER2+ breast cancer who progress on trastuzumab. These data suggest that trastuzumab-resistant tumors continue to be dependent on the HER2 tyrosine kinase after escaping trastuzumab action. However, the clinical responses to single agent TKIs such as lapatinib or neratinib tend to be short-lived.Citation61,Citation62 Further, these patients may still need trastuzumab beyond progression as suggested by a recent study where the combination of lapatinib and trastuzumab was superior to lapatinib alone at improving progression-free survival, clinical response and overall survival in patients with HER2+ metastatic breast cancer who had progressed on trastuzumab.Citation84 The activity of trastuzumab beyond progression is not limited to combinations with TKIs as it has also been shown in a study where the combination of trastuzumab plus capecitabine was clearly superior to capecitabine alone.Citation85
A second piece of evidence supporting continued dependence on HER2 after progression on anti-HER2 therapy is provided by clinical data with the antibody-toxin fusion trastuzumab-DM1 T-DM1, T-DM1 is an antibody-drug conjugate in which one molecule of trastuzumab is covalently coupled via a non-cleavable linker to three molecules of the microtubule polymerization inhibitor Derivative of Maytansine 1 (DM1).Citation86 T-DM1 binds to HER2 with similar affinity as trastuzumab. It is proposed that after binding to the receptor, the T-DM1/HER2 complex is internalized followed by degradation in the lysosome, release of DM1 and subsequent cell lysis. Although used at lower doses and frequency than trastuzumab, T-DM1 retains the ability to inhibit signaling and engaging immune effectors that mediate ADCC and is active against lapatinib-resistant xenografts.Citation87 Phase I–II studies of T-DM1 demonstrated mild, reversible toxicity and a remarkable clinical response rate in excess of 25% in patients with heavily pretreated HER2-overexpressing metastatic breast cancer who had progressed after trastuzumab and lapatinib.Citation88,Citation89 T-DM1 is being further evaluated in two large phase III randomized studies in the first- and second-line metastatic disease settings.
Taken together, these data imply that even in advanced stages, HER2+ breast cancers remain dependent on HER2 and that single-agent trastuzumab and lapatinib are not adequate to inhibit the HER2 signaling network completely. They also imply that the use of combinations of HER2-targeted agents delivered early against HER2+ breast cancer should be widely considered. Several preclinical and early clinical data have already suggested combinations of HER2 inhibitors, which because of their distinct properties and (thus) mechanisms by which they interact with HER2 cells, provide an opportunity for synergy. These characteristics and mechanisms are summarized for four anti-HER2 agents in .
Along these lines, Adjuvant Lapatinib and/or Trastuzumab Treatment Optimization (ALTTO) and Neo-ALTTO are two ongoing large international adjuvant and neoadjuvant studies, respectively, comparing trastuzumab vs. lapatinib vs. dual HER2 blockade using both drugs. Results on the neoadjuvant study were reported recently. Neo-ALTTO is a 450 patient study in which HER2+ tumors measuring >2 cm were randomized to trastuzumab, lapatinib or the combination for 6 weeks, at which time paclitaxel is added to each of the arms for an additional 12 weeks. After surgery, patients in all three arms receive adjuvant chemotherapy with FEC followed by the respective HER2 inhibitor either alone or in combination for 34 weeks. About half of the patients enrolled had ER+ tumors. There was increased but manageable toxicity in the lapatinib arms, mostly diarrhea and transaminitis. Pathological complete response (path CR, defined as no invasive cancer in the breast or only DCIS in the breast specimen) was significantly higher in the combination arm (51.3%) vs. 29.5 and 24.7% in the trastuzumab and lapatinib arms, respectively. In all three arms, the pathological CR rate was lower in the ER+ vs. the ER− tumors.Citation90 This study is the best demonstration to date of superiority of dual HER2 blockade over single agent trastuzumab or lapatinib. Whether the combination is superior at abrogating resistance and, hence, prolonging disease-free and overall survival compared to single agent anti-HER2 therapy, as suggested by the higher pathological CR rate in Neo-ALTTO, would require longer follow-up.
A second approach that improves inactivation of the HER2 network is the combination of trastuzumab and pertuzumab. For example, in trastuzumab-resistant xenografts and in patients with HER2+ breast cancer that have progressed on trastuzumab, only the combination of both but not each antibody alone exhibited clinical activity.Citation91,Citation92 These data suggest that both HER2 antibodies, each binding to a different epitope in HER2 (), might be required to completely inhibit HER2-HER3 dimerization in situ, potentially explaining their clinical activity in combination. To test this hypothesis, the phase III Cleopatra study is currently randomizing patients with HER2+ metastatic breast cancer to trastuzumab and docetaxel ± pertuzumab as first line therapy in the metastatic setting using progression-free survival as a primary endpoint. Notably, in the recently reported NeoSphere trial in patients with HER2+ primary breast cancer, the pathological CR rate was 45.8 vs. 29% (p = 0.01) in patients treated with neoadjuvant docetaxel/trastuzumab/pertuzumab vs. docetaxel/trastuzumab, respectively.Citation93 Currently, the HER3 monoclonal antibodies AMG-888,Citation94 and MM-121,Citation95 are completing phase I testing. We anticipate that like pertuzumab, they may also exert a synergistic effect in combination with trastuzumab or lapatinib in patients with HER2+ breast cancer.
Conclusions
At this time, only trastuzumab and lapatinib are approved by the FDA for the treatment of patients with HER2+ overexpressing breast cancer. Although not approved in combination, data summarized above clearly suggest dual HER2 blockade is feasible and superior to each HER2 inhibitor alone. In addition, there are a plethora of agents that either target HER2 by different mechanisms or inhibit those mechanisms of resistance summarized above. All these drugs () are currently in different phases of clinical development. It is anticipated that an increasing number of these agents will eventually be combined with the approved anti-HER2 therapies. We propose that the increasing use of dual HER2 blockade with trastuzumab and lapatinib as well as the development of novel anti-HER2 combinations will markedly limit or eventually abrogate acquired resistance to primary anti-HER2 therapy. On the other hand, molecular profiling of HER2+ metastatic recurrences following anti-HER2 therapy should provide important leads as to which of the molecular mechanisms of resistance summarized above are the more relevant and targetable in the clinic.
Figures and Tables
Figure 1 Diagram of mechanisms of resistance to trastuzumab. Trastuzumab can block HER2 homodimers. However, it is unable to interfere with ligand-activated EGFR-HER2 and HER2–HER3 heterodimers (dotted lines). The HER2/EGFR dual inhibitor lapatinib should be able to block the signaling output of HER2-containing heterodimers. Trastuzumab cannot bind kinase-active cytosolic fragments of HER2 (p95HER2), whereas lapatinib and other irreversible TKIs, such as neratinib and BIBW2992, can inhibit the catalytic activity of p95HER2.
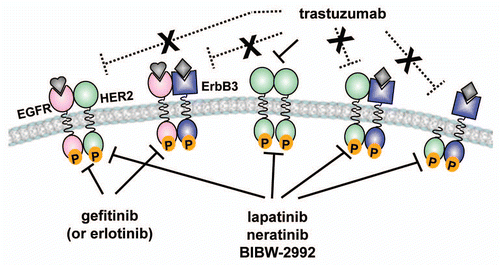
Table 1 Molecular and clinical characteristics and mechanisms of antitumor action of HER2-targeted drugs
Table 2 Drugs in development that target HER2 or pathways proposed to contribute or to mediate resistance to trastuzumab and/or lapatinib
Acknowledgements
This work was supported by R01 grant CA80195, ACS Clinical Research Professorship Grant CRP-07-234, The Lee Jeans Translational Breast Cancer Research Program, Breast Cancer Specialized Program of Research Excellence (SPORE) P50 CA98131 and Vanderbilt-Ingram Cancer Center Support Grant P30 CA68485. J.T.G. is partially supported by grant T32DK007563 and ACS 118813-PF-10-070-01-TBG and DOD BC093376 post-doctoral fellowship awards.
References
- Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2001; 2:127 - 137
- Graus-Porta D, Beerli RR, Daly JM, Hynes NE. ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J 1997; 16:1647 - 1655
- Pinkas-Kramarski R, Soussan L, Waterman H, Levkowitz G, Alroy I, Klapper L, et al. Diversification of Neu differentiation factor and epidermal growth factor signaling by combinatorial receptor interactions. EMBO J 1996; 15:2452 - 2467
- Wang LM, Kuo A, Alimandi M, Veri MC, Lee CC, Kapoor V, et al. ErbB2 expression increases the spectrum and potency of ligand-mediated signal transduction through ErbB4. Proc Natl Acad Sci USA 1998; 95:6809 - 6814
- Worthylake R, Opresko LK, Wiley HS. ErbB-2 amplification inhibits downregulation and induces constitutive activation of both ErbB-2 and epidermal growth factor receptors. J Biol Chem 1999; 274:8865 - 8874
- Alimandi M, Romano A, Curia MC, Muraro R, Fedi P, Aaronson SA, et al. Cooperative signaling of ErbB3 and ErbB2 in neoplastic transformation and human mammary carcinomas. Oncogene 1995; 10:1813 - 1821
- Wallasch C, Weiss FU, Niederfellner G, Jallal B, Issing W, Ullrich A. Heregulin-dependent regulation of HER2/neu oncogenic signaling by heterodimerization with HER3. EMBO J 1995; 14:4267 - 4275
- Moasser MM. The oncogene HER2: its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene 2007; 26:6469 - 6487
- Hsieh AC, Moasser MM. Targeting HER proteins in cancer therapy and the role of the non-target HER3. Br J Cancer 2007; 97:453 - 457
- Lee-Hoeflich ST, Crocker L, Yao E, Pham T, Munroe X, Hoeflich KP, et al. A central role for HER3 in HER2-amplified breast cancer: implications for targeted therapy. Cancer Res 2008; 68:5878 - 5887
- Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF 3rd, Hynes NE. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc Natl Acad Sci USA 2003; 100:8933 - 8938
- Brachmann SM, Hofmann I, Schnell C, Fritsch C, Wee S, Lane H, et al. Specific apoptosis induction by the dual PI3K/mTor inhibitor NVP-BEZ235 in HER2 amplified and PIK3CA mutant breast cancer cells. Proc Natl Acad Sci USA 2009; 106:22299 - 22304
- Ross JS, Fletcher JA. The HER-2/neu oncogene in breast cancer: prognostic factor, predictive factor and target for therapy. Stem Cells 1998; 16:413 - 428
- Junttila TT, Akita RW, Parsons K, Fields C, Lewis Phillips GD, Friedman LS, et al. Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell 2009; 15:429 - 440
- Park S, Jiang Z, Mortenson ED, Deng L, Radkevich-Brown O, Yang X, et al. The therapeutic effect of anti-HER2/neu antibody depends on both innate and adaptive immunity. Cancer Cell 2010; 18:160 - 170
- Vogel CL, Cobleigh MA, Tripathy D, Gutheil JC, Harris LN, Fehrenbacher L, et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol 2002; 20:719 - 726
- Romond EH, Perez EA, Bryant J, Suman VJ, Geyer CE Jr, Davidson NE, et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med 2005; 353:1673 - 1684
- Smith I, Procter M, Gelber RD, Guillaume S, Feyereislova A, Dowsett M, et al. 2-year follow-up of trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer: a randomised controlled trial. Lancet 2007; 369:29 - 36
- Robert N, Leyland-Jones B, Asmar L, Belt R, Ilegbodu D, Loesch D, et al. Randomized phase III study of trastuzumab, paclitaxel and carboplatin compared with trastuzumab and paclitaxel in women with HER-2-overexpressing metastatic breast cancer. J Clin Oncol 2006; 24:2786 - 2792
- Perez EA, Suman VJ, Davidson NE. Results of chemotherapy alone, with sequential or concurrent addition of 52 weeks of trastuzumab in the ncctg n9831 her2-positive adjuvant breast cancer trial. Cancer Res 2009; 69:80
- Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001; 344:783 - 792
- Nahta R, Takahashi T, Ueno NT, Hung MC, Esteva FJ. p27(kip1) downregulation is associated with trastuzumab resistance in breast cancer cells. Cancer Res 2004; 64:3981 - 3986
- Ritter CA, Perez-Torres M, Rinehart C, Guix M, Dugger T, Engelman JA, et al. Human breast cancer cells selected for resistance to trastuzumab in vivo overexpress epidermal growth factor receptor and ErbB ligands and remain dependent on the ErbB receptor network. Clin Cancer Res 2007; 13:4909 - 4919
- Lu Y, Zi X, Zhao Y, Mascarenhas D, Pollak M. Insulin-like growth factor-i receptor signaling and resistance to trastuzumab (herceptin). J Natl Cancer Inst 2001; 93:1852 - 1857
- Nahta R, Yuan LX, Zhang B, Kobayashi R, Esteva FJ. Insulin-like growth factor-I receptor/human epidermal growth factor receptor 2 heterodimerization contributes to trastuzumab resistance of breast cancer cells. Cancer Res 2005; 65:11118 - 11128
- Harrris LN, You F, Schnitt SJ, Witkiewicz A, Lu X, Sgroi D, et al. Preoperative therapy for HER2-overexpressing early-stage breast cancer: Multigene profiling may identify predictors of resistance to trastuzumab and vinorelbine therapy. Clin Cancer Res 2006; 13:1198 - 1207
- Shattuck DL, Miller JK, Carraway KL 3rd, Sweeney C. Met receptor contributes to trastuzumab resistance of Her2-overexpressing breast cancer cells. Cancer Res 2008; 68:1471 - 1477
- Zhuang G, Brantley-Sieders DM, Vaught D, Yu J, Xie L, Wells S, et al. Elevation of receptor tyrosine kinase EphA2 mediates resistance to trastuzumab therapy. Cancer Res 2010; 70:299 - 308
- Liang K, Esteva FJ, Albarracin C, Stemke-Hale K, Lu Y, Bianchini G, et al. Recombinant human erythropoietin antagonizes trastuzumab treatment of breast cancer cells via Jak2-mediated Src activation and PTEN inactivation. Cancer Cell 2010; 18:423 - 435
- Motoyama AB, Hynes NE, Lane HA. The efficacy of ErbB receptor-targeted anticancer therapeutics is influenced by the availability of epidermal growth factor-related peptides. Cancer Res 2002; 62:3151 - 3158
- Moulder SL, Yakes FM, Muthuswamy SK, Bianco R, Simpson JF, Arteaga CL. Epidermal growth factor receptor (HER1) tyrosine kinase inhibitor ZD1839 (Iressa) inhibits HER2/neu (erbB2)-overexpressing breast cancer cells in vitro and in vivo. Cancer Res 2001; 61:8887 - 8895
- Cho HS, Mason K, Ramyar KX, Stanley AM, Gabelli SB, Denney DW Jr, et al. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 2003; 421:756 - 760
- Agus DB, Akita RW, Fox WD, Lewis GD, Higgins B, Pisacane PI, et al. Targeting ligand-activated ErbB2 signaling inhibits breast and prostate tumor growth. Cancer Cell 2002; 2:127 - 137
- Adams CW, Allison DE, Flagella K, Presta L, Clarke J, Dybdal N, et al. Humanization of a recombinant monoclonal antibody to produce a therapeutic HER dimerization inhibitor, pertuzumab. Cancer Immunol Immunother 2006; 55:717 - 727
- Franklin MC, Carey KD, Vajdos FF, Leahy DJ, de Vos AM, Sliwkowski MX. Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell 2004; 5:317 - 328
- Wang SE, Xiang B, Guix M, Olivares MG, Parker J, Chung CH, et al. Transforming growth factor beta engages TACE and ErbB3 to activate phosphatidylinositol-3-kinase/Akt in ErbB2-overexpressing breast cancer and desensitizes cells to trastuzumab. Mol Cell Biol 2008; 28:5605 - 5620
- Campbell IG, Russell SE, Choong DY, Montgomery KG, Ciavarella ML, Hooi CS, et al. Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer Res 2004; 64:7678 - 7681
- Bachman KE, Argani P, Samuels Y, Silliman N, Ptak J, Szabo S, et al. The PIK3CA gene is mutated with high frequency in human breast cancers. Cancer Biol Ther 2004; 3:772 - 775
- Carpten JD, Faber AL, Horn C, Donoho GP, Briggs SL, Robbins CM, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature 2007; 448:439 - 444
- Bellacosa A, de Feo D, Godwin AK, Bell DW, Cheng JQ, Altomare DA, et al. Molecular alterations of the AKT2 oncogene in ovarian and breast carcinomas. Int J Cancer 1995; 64:280 - 285
- Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast and prostate cancer. Science 1997; 275:1943 - 1947
- Saal LH, Johansson P, Holm K, Gruvberger-Saal SK, She QB, Maurer M, et al. Poor prognosis in carcinoma is associated with a gene expression signature of aberrant PTEN tumor suppressor pathway activity. Proc Natl Acad Sci USA 2007; 104:7564 - 7569
- Gewinner C, Wang ZC, Richardson A, Teruya-Feldstein J, Etemadmoghadam D, Bowtell D, et al. Evidence that inositol polyphosphate-4-phosphatase type II is a tumor suppressor that inhibits PI3K signaling. Cancer Cell 2009; 16:115 - 125
- Saal LH, Holm K, Maurer M, Memeo L, Su T, Wang X, et al. PIK3CA mutations correlate with hormone receptors, node metastasis and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer Res 2005; 65:2554 - 2559
- Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, Neve RM, Kuo WL, Davies M, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN and AKT mutations in breast cancer. Cancer Res 2008; 68:6084 - 6091
- Yakes FM, Chinratanalab W, Ritter CA, King W, Seelig S, Arteaga CL. Herceptin-induced inhibition of phosphatidylinositol-3-kinase and Akt is required for antibody-mediated effects on p27, cyclin D1 and antitumor action. Cancer Res 2002; 62:4132 - 4141
- Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, Beelen K, et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell 2007; 12:395 - 402
- Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, Sahin AA, et al. PTEN activation contributes to tumor inhibition by trastuzumab and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 2004; 6:117 - 127
- Serra V, Markman B, Scaltriti M, Eichhorn PJ, Valero V, Guzman M, et al. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res 2008; 68:8022 - 8030
- Eichhorn PJ, Gili M, Scaltriti M, Serra V, Guzman M, Nijkamp W, et al. Phosphatidylinositol-3-kinase hyper-activation results in lapatinib resistance that is reversed by the mTOR/phosphatidylinositol 3-kinase inhibitor NVP-BEZ235. Cancer Res 2008; 68:9221 - 9230
- Chakrabarty A, Rexer BN, Wang SE, Cook RS, Engelman JA, Arteaga CL. H1047R phosphatidylinositol-3-kinase mutant enhances HER2-mediated transformation by heregulin production and activation of HER3. Oncogene 2010; 29:5193 - 5203
- Dalenc F, Campone M, Hupperets P. Everolimus in combination with weekly paclitaxel and trastuzumab in patients (pts) with HER2-overexpressing metastatic breast cancer (MBC) with prior resistance to trastuzumab and taxanes: A multicenter phase II clinical trial. J Clin Oncol 2010; 28:15
- Anido J, Scaltriti M, Bech Serra JJ, Santiago Josefat B, Todo FR, Baselga J, et al. Biosynthesis of tumorigenic HER2 C-terminal fragments by alternative initiation of translation. EMBO J 2006; 25:3234 - 3244
- Scaltriti M, Rojo F, Ocana A, Anido J, Guzman M, Cortes J, et al. Expression of p95HER2, a truncated form of the HER2 receptor and response to anti-HER2 therapies in breast cancer. J Natl Cancer Inst 2007; 99:628 - 638
- Scaltriti M, Chandarlapaty S, Prudkin L, Aura C, Jimenez J, Angelini PD, et al. Clinical benefit of lapatinib-based therapy in patients with human epidermal growth factor receptor 2-positive breast tumors coexpressing the truncated p95HER2 receptor. Clin Cancer Res 2010; 16:2688 - 2695
- Price-Schiavi SA, Jepson S, Li P, Arango M, Rudland PS, Yee L, et al. Rat Muc4 (sialomucin complex) reduces binding of anti-ErbB2 antibodies to tumor cell surfaces, a potential mechanism for herceptin resistance. Int J Cancer 2002; 99:783 - 791
- Nagy P, Friedlander E, Tanner M, Kapanen AI, Carraway KL, Isola J, et al. Decreased accessibility and lack of activation of ErbB2 in JIMT-1, a herceptinresistant, MUC4-expressing breast cancer cell line. Cancer Res 2005; 65:473 - 482
- Kwong KY, Hung MC. A novel splice variant of HER2 with increased transformation activity. Mol Carcinog 1998; 23:62 - 68
- Castiglioni F, Tagliabue E, Campiglio M, Pupa SM, Balsari A, Menard S. Role of exon-16-deleted HER2 in breast carcinomas. Endocr Relat Cancer 2006; 13:221 - 232
- Mitra D, Brumlik MJ, Okamgba SU, Zhu Y, Duplessis TT, Parvani JG, et al. An oncogenic isoform of HER2 associated with locally disseminated breast cancer and trastuzumab resistance. Mol Cancer Ther 2009; 8:2152 - 2162
- Gomez HL, Doval DC, Chavez MA, Ang PC, Aziz Z, Nag S, et al. Efficacy and safety of lapatinib as first-line therapy for ErbB2-amplified locally advanced or metastatic breast cancer. J Clin Oncol 2008; 26:2999 - 3005
- Geyer CE, Forster J, Lindquist D, Chan S, Romieu CG, Pienkowski T, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med 2006; 355:2733 - 2743
- Press MF, Finn RS, Cameron D, Di Leo A, Geyer CE, Villalobos IE, et al. HER-2 gene amplification, HER-2 and epidermal growth factor receptor mRNA and protein expression and lapatinib efficacy in women with metastatic breast cancer. Clin Cancer Res 2008; 14:7861 - 7870
- Burstein HJ, Sun Y, Dirix LY, Jiang Z, Paridaens R, Tan AR, et al. Neratinib, an irreversible ErbB receptor tyrosine kinase inhibitor, in patients with advanced ErbB2-positive breast cancer. J Clin Onco 2010; l 28:1301 - 1307
- Faber AC, Wong KK, Engelman JA. Differences underlying EGFR and HER2 oncogene addiction. Cell Cycle 9:851 - 852
- Xia W, Bacus S, Hegde P, Husain I, Strum J, Liu L, et al. A model of acquired autoresistance to a potent ErbB2 tyrosine kinase inhibitor and a therapeutic strategy to prevent its onset in breast cancer. Proc Natl Acad Sci USA 2006; 103:7795 - 7800
- Guo S, Sonenshein GE. Forkhead box transcription factor FOXO3a regulates estrogen receptor alpha expression and is repressed by the Her-2/neu/phosphatidylinositol-3-kinase/Akt signaling pathway. Mol Cell Biol 2004; 24:8681 - 8690
- Xia W, Bacus S, Husain I, Liu L, Zhao S, Liu Z, et al. Resistance to ErbB2 tyrosine kinase inhibitors in breast cancer is mediated by calcium-dependent activation of RelA. Mol Cancer Ther 2010; 9:292 - 299
- Liu L, Greger J, Shi H, Liu Y, Greshock J, Annan R, et al. Novel mechanism of lapatinib resistance in HER2-positive breast tumor cells: activation of AXL. Cancer Res 2009; 69:6871 - 6878
- Hafizi S, Dahlback B. Signalling and functional diversity within the Axl subfamily of receptor tyrosine kinases. Cytokine Growth Factor Rev 2006; 17:295 - 304
- Sergina NV, Rausch M, Wang D, Blair J, Hann B, Shokat KM, et al. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature 2007; 445:437 - 441
- Amin DN, Sergina N, Ahuja D, McMahon M, Blair JA, Wang D, et al. Resiliency and vulnerability in the HER2-HER3 tumorigenic driver. Sci Transl Med 2010; 2:16 - 17
- Engelman JA, Mukohara T, Zejnullahu K, Lifshits E, Borras AM, Gale CM, et al. Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer. J Clin Invest 2006; 116:2695 - 2706
- Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2005; 2:73
- Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med 2005; 352:786 - 792
- Kosaka T, Yatabe Y, Endoh H, Yoshida K, Hida T, Tsuboi M, et al. Analysis of epidermal growth factor receptor gene mutation in patients with non-small cell lung cancer and acquired resistance to gefitinib. Clin Cancer Res 2006; 12:5764 - 5769
- Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 2001; 293:876 - 880
- Tamborini E, Bonadiman L, Greco A, Albertini V, Negri T, Gronchi A, et al. A new mutation in the KIT ATP pocket causes acquired resistance to imatinib in a gastrointestinal stromal tumor patient. Gastroenterology 2004; 127:294 - 299
- Stephens P, Hunter C, Bignell G, Edkins S, Davies H, Teague J, et al. Lung cancer: intragenic ERBB2 kinase mutations in tumours. Nature 2004; 431:525 - 526
- Shigematsu H, Takahashi T, Nomura M, Majmudar K, Suzuki M, Lee H, et al. Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer Res 2005; 65:1642 - 1646
- Stephens P, Edkins S, Davies H, Greenman C, Cox C, Hunter C, et al. A screen of the complete protein kinase gene family identifies diverse patterns of somatic mutations in human breast cancer. Nat Genet 2005; 37:590 - 592
- Lee JW, Soung YH, Seo SH, Kim SY, Park CH, Wang YP, et al. Somatic mutations of ERBB2 kinase domain in gastric, colorectal and breast carcinomas. Clin Cancer Res 2006; 12:57 - 61
- Wang SE, Narasanna A, Perez-Torres M, Xiang B, Wu FY, Yang S, et al. HER2 kinase domain mutation results in constitutive phosphorylation and activation of HER2 and EGFR and resistance to EGFR tyrosine kinase inhibitors. Cancer Cell 2006; 10:25 - 38
- Blackwell KL, Burstein HJ, Storniolo AM, Rugo H, Sledge G, Koehler M, et al. Randomized study of Lapatinib alone or in combination with trastuzumab in women with ErbB2-positive, trastuzumab-refractory metastatic breast cancer. J Clin Oncol 2010; 28:1124 - 1130
- von Minckwitz G, du Bois A, Schmidt M, Maass N, Cufer T, de Jongh FE, et al. Trastuzumab beyond progression in human epidermal growth factor receptor 2-positive advanced breast cancer: a german breast group 26/breast international group 03–05 study. J Clin Oncol 2009; 27:1999 - 2006
- Lewis Phillips GD, Li G, Dugger DL, Crocker LM, Parsons KL, Mai E, et al. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res 2008; 68:9280 - 9290
- Junttila TT, Li G, Parsons K, Phillips GL, Sliwkowski MX. Trastuzumab-DM1 (T-DM1) retains all the mechanisms of action of trastuzumab and efficiently inhibits growth of lapatinib insensitive breast cancer. Breast Cancer Res Treat 2010; In press
- Krop IE, Beeram M, Modi S, Jones SF, Holden SN, Yu W, et al. Phase I study of trastuzumab-DM1, an HER2 antibody-drug conjugate, given every 3 weeks to patients with HER2-positive metastatic breast cancer. J Clin Oncol 2010; 28:2698 - 2704
- Burris HA 3rd, Rugo HS, Vukelja SJ, Vogel CL, Borson RA, Limentani S, et al. Phase II study of the antibody drug conjugate trastuzumab-dm1 for the treatment of human epidermal growth factor receptor 2 (her2)-positive breast cancer after prior her2-directed therapy. J Clin Oncol 2011; 29:398 - 405
- Baselga J, Bradbury I, Eidtmann H. First results of the NeoALTTO trial (BIG 01–06/EGF 106903): A phase III, randomized, open label, neoadjuvant study of lapatinib, trastuzumab and their combination plust paclitaxel in women with HER2-positive primary breast cancer. Cancer Res 2010; 70:82
- Scheuer W, Friess T, Burtscher H, Bossenmaier B, Endl J, Hasmann M. Strongly enhanced antitumor activity of trastuzumab and pertuzumab combination treatment on HER2-positive human xenograft tumor models. Cancer Res 2009; 69:9330 - 9336
- Baselga J, Gelmon KA, Verma S, Wardley A, Conte P, Miles D, et al. Phase II trial of pertuzumab and trastuzumab in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer that progressed during prior trastuzumab therapy. J Clin Oncol 2010; 28:1138 - 1144
- Gianni L, Pienkowski T, Im YH. Neoadjuvant pertuzumab (P) and trastuzumab (T): Antitumor and safety analysis of randomized phase II study (NeoSphere). Cancer Res 2010; 70:82
- Freeman D, Ogbagabriel S, Rothe M. Fully human anti-HER3 monoclonal antibodies (mAbs) have unique in vitro and in vivo functional and anti-tumor activities versus othe HER family inhibitors. Proc Am Assoc Cancer Res 2008; 49
- Schoeberl B, Faber AC, Li D, Liang MC, Crosby K, Onsum M, et al. An ErbB3 antibody, MM-121, is active in cancers with ligand-dependent activation. Cancer Res 2010; 70:2485 - 2494