Abstract
Purpose: The objective of this study was to determine the antitumor effects of alternate dosing schedules of topotecan in prostate cancer.
Results: A concentration-dependent increase in cytotoxicity was observed in PC-3 and LNCaP cells after topotecan treatment using conventional and metronomic protocols. A significant increase in potency (2.4-18 fold, after 72 hr) was observed following metronomic dosing compared to conventional dosing administration in both cell lines. Metronomic dosing also increased the percentage of PC-3 cells in the G2/M, compared to control, but did not alter LNCaP cell cycle distribution. Metronomic dosing increased p21 protein expression in LNCaP and PC-3 cells compared to conventional dosing. The observed in vitro activity was confirmed using an in vivo model of human prostate cancer. Metronomic dosing and continuous infusion decreased tumor volume significantly (p < 0.05) compared to control and conventional topotecan treatment, but had no effect on tumor vascular staining.
Methods: The cytotoxicity of topotecan after conventional or metronomic dosing was determined by examining cellular morphology, mitochondrial enzymatic activity (MTT), total cellular protein (SRB), annexin V and propidium iodine (PI) staining, cell cycle and western blot analysis in human prostate cancer cell lines (PC-3 and LNCaP) and the effects metronomic or continuous infusion on tumor growth in an in vivo tumor xenograft model.
Conclusions: These data support the hypothesis that low-dose continuous administration of topotecan increases potency compared to conventional dosing in prostate cancer. These data also suggest the novel finding that the enhanced antitumor activity of topotecan following low-dose exposure correlates to alterations in cell cycle and increased p21 expression.
Introduction
Prostate cancer is the second leading cause of non-cutaneous cancer related deaths in men in the United States (www.cancer.org). Organ-confined prostate cancers are generally treated with surgery and/or radiation, and residual disease is managed with systemic therapies.Citation1–Citation3 In cases of inoperable tumors, evidence of metastases or unresponsive to radiation, chemotherapy may be the only treatment option. The location, grade and type of tumor limit the effectiveness of therapy. Androgen ablation is the standard therapy for primary tumors and metastatic spread.Citation4 Unfortunately, most of the later patients will eventually develop castration-refractory prostate tumor, for which there are no effective treatments.Citation5
Advanced prostate cancers also do not respond well to current treatment protocols, which include anti-cancer drug therapy, docetaxel and prednisolone,Citation6 in combination with hormone ablation and/or surgery. Conventional administration schedules of traditional chemotherapeutic (e.g., DNA-damaging or microtubule inhibitors) agents at or near their maximum tolerated dose (MTD) is based on their selectivity for rapidly dividing cells.Citation7,Citation8 The effectiveness of most chemotherapeutic agents is limited by the slow rate of tumor growth, non-target tissue toxicity, poor or heterogeneous intra-tumor distribution of drug and development of drug resistance.Citation6,Citation9,Citation10 Thus, effective chemotherapeutic strategies for treating prostate cancer and other slow growing solid malignancies are needed.
Continuous or frequent low-dose administration (i.e., metronomic or fractionated dosing) of some chemotherapeutic agents (e.g., trofosfamide, cyclophosphamide, methotrexate, capecitabine, docetaxel and paclitaxel) decreases tumor growth.Citation7,Citation11–Citation14 In vitro studies using human endothelial cells (ECs), human umbilical vein endothelial cells (HUVEC) and the human dermal microvascular endothelial cells (HMVEC-d)Citation15,Citation16 and in vivo studies show that metronomic dosing schedules inhibit tumor angiogenesis and decrease tumor vascular density and tumor growth.Citation17–Citation19 However, not all of the benefits of metronomic dosing directly correlate to antiangiogenic activity. For example, a recent report showed that concurrent administration of metronomic dosing of cyclophosphamide and tirapazamine reduced gliosarcoma tumor size without impacting tumor vasculature.Citation20 Although the mechanism(s) responsible for this activity are not fully known, developing dosing schedules that exploit both direct antitumor and antiangiogenic effects may improve treatment outcomes.
The objective of this study was to determine the antitumor effects of alternate dosing schedules of topotecan in prostate cancer. To accomplish this goal the effects of low doses of topotecan administered metronomically or infused continuously in the case of in vivo studies, were compared with the effects of topotecan administered using conventional protocols. A secondary objective of this study was to gain mechanistic insights into topotecan's cellular activity after both conventional and metronomic administration to support development of optimal dosing schedules for in vivo testing.
Topotecan and other camptothecin derivatives, e.g., gimatecan and irinotecan (CPT-11), exert antiangiogenic activity when administered frequently at low doses.Citation15,Citation21–Citation23 Camptothecins are potent cell cycle dependent inhibitors of DNA synthesis that induce DNA double strands break through topoisomerase I inhibition.Citation24 Topotecan is a semi-synthetic water-soluble derivative of camptothecin. The US FDA has approved topotecan for the treatment of relapsed small cell lung cancer, metastatic carcinoma of the ovary and carcinoma of the cervix. Topotecan use in prostate cancer has been limited, in part, due to its low efficacy and high non-target toxicity.Citation25,Citation26 Currently Phase I and II trials of oral topotecan administered metronomically for the treatment of gynecologic and other solid tumors are ongoing to evaluate its safety and efficacy (trials: NCT00382733 and NCT00800345 at http://clinicaltrial.gov/); however, no studies exist assessing the effect of metronomic dosing of topotecan on prostate cancer.
Results
Topotecan exposure.
The effect of conventional and metronomic administration of topotecan on prostate cancer cells were assessed by measurement of MTT and SRB absorbance after 24, 48 and 72 h treatments. Cell lines (PC-3 and LNCaP) were seeded in 96-well plates and allowed to attach for 24 h prior to media aspiration and replacement with fresh media containing topotecan (0.04–10,000 nM). Metronomic dosing was simulated by aspirating and replacing with fresh media and drug after 24 and 48 h. Metronomic exposure of PC-3 cells to topotecan resulted in significant (p ≤ 0.05) time-dependent decreases in SRB and MTT staining as well as IC50 after 24, 48 and 72 h, compared with conventional treatment (). A similar decrease in MTT and SRB staining, i.e., increased potency, was observed in LNCaP cells following metronomic dosing compared with conventional exposure (). Differences in IC50 values obtained from MTT and SRB staining are not unexpected as the time course of enzymatic (MTT) changes may not agree with total cellular protein (SRB).
demonstrates that metronomic dosing with topotecan increased the potency (lowered the IC50, by MTT and SRB) of topotecan significantly compared with conventional dosing in both LNCaP and PC-3 cells. In PC-3 cells, metronomic dosing decreased the IC50 8.6-fold (9.3 vs. 93.5 nM at 72 h by MTT) and 4.5-fold (17 vs. 76.3 nM at 72 h by SRB) in comparison to conventional dosing. Topotecan was more potent in LNCaP cells and metronomic dosing decreased the IC50 by 18-fold (1.4 vs. 26 nM by MTT) and 2.4-fold (3.6 vs. 8.5 nM by SRB) after 72 h.
Morphological and nuclear assessment.
PC-3 and LNCaP cells were exposed to metronomic and conventional dosing of topotecan at the calculated IC50 for each treatment regimen at 72 h and cellular morphology was assessed using phase-contrast microscopy. In agreement with SRB and MTT assays, micrographs of PC-3 and LNCaP cells exposed to both conventional and metronomic dosing regimens showed similar decreases in cell density compared with control cells (). In both cell lines, conventional treatment schedule resulted in an increased number of detached cells and cellular debris, whereas there were fewer detached cells in metronomically treated groups. Assessment of nuclear morphology, using DAPI staining, of attached cells did not suggest morphological changes indicative of apoptosis (nuclear fragmentation and chromatin condensation) ().
Assessment of cell death.
PC-3 and LNCAP cells were exposed to metronomic and conventional dosing of topotecan at the IC50 for each treatment regimen over 72 h followed by assessment of annexin V (marker of apoptosis) and PI (marker of necrosis) using flow cytometry (). Because conventional treatments induced significant cell detachment we assessed both attached and detached cells concurrently. Treatment of PC-3 cells with a conventional dose of topotecan (80 nM) did not significantly increase the percent cells staining positive for annexin V alone after 72 h (), however, significant increases in cells staining positive for annexin V and PI were observed (). Interestingly, metronomic dosing of PC-3 cells with topotecan (10 nM) significantly increased the percent cells staining positive for both annexin V and PI, suggesting an increase in late apoptosis.
Treatment of LNCaP cells with a conventional dose of topotecan, 10 nM () significantly increased the percent of cells staining positive for annexin V alone (apoptotic cells) after 72 h, as well as increasing those staining positive for annexin V and PI (late apoptosis) (). Metronomic (2 nM) dosing slightly increased the percent of LNCaP cells staining positive for annexin V alone, compared with control (), as well as annexin V and PI (), however neither differed significantly from control cells. The percent of PC-3 and LNCaP cells staining positive for PI alone in metronomic was not significantly different than those exposed to conventional dosing ().
Cell cycle analysis.
PC-3 cells were exposed to topotecan at the calculated IC50 for both treatment protocols. Treatment of PC-3 cells with topotecan at the conventional calculated IC50 resulted in apparent G2/M arrest and cell death, (described above) in both cell lines; this is in agreement with topotecan effects on cell cycle in other cell lines.Citation27 Treatment of PC-3 cells metronomically at IC50 (10 nM) increased the percentage of cells in G2/M phase significantly (), with a concomitant decrease in G1 population. An increase in the S phase was also observed, but this difference was not statistically significant. The only differences in cell cycle observed in LNCaP cells after conventional or metronomic dosing at any time point was a slight increase in sub G1 cells compared with control ().
Assessment of p21 and p53 expression.
Our previous studies in LNCaP and PC-3 cells using other lactone derivatives demonstrated that G2/M arrest correlated with activation of p21, and that p21 could be activated in the absence of p53.Citation28,Citation29 Thus, we assessed the hypothesis that changes in cell cycle induced by metronomic dosing regimens were mediated by p21 using immunoblot analysis. We also assessed p53 expression in LNCaP cells as these underwent a slight, but significant sub G1 arrest after 72 h, which can be mediated by p53. The expression of GAPDH was used as a loading control. An increase in p21 expression was detected following topotecan exposure in both cell lines ( and B) compared with untreated controls. Overall p21 expression was lower in PC-3 cells compared with LNCaP cells, which is consistent with our previous studies and the lack of p53 in PC-3 cells. Both conventional and metronomic treatment protocols increased p21 expression in each cell lines. Increases in p21 expression in PC-3 cells were lower than that in LNCaP cells, and were highest after 72 h exposure to metronomic schedules at both the metronomic and conventional IC50 ( and D). In contrast, increases in p21 expression were detected after 48 h in LNCaP cells. A marked increase in p21 expression in cells exposed to metronomic treatments vs. conventional treatment was also observed. A significant (p < 0.05) increase in p21 expression in cells exposed to metronomic treatments vs. untreated controls was observed (). No increases in p53 expression were detected in LNCaP cells under any condition tested (data not shown). The expression of p53 in PC-3 cells was not studied, as these cells are known to be, and were experimentally determined to be, p53 null.
Topotecan in vivo tumor xenograft activity.
A pilot study was completed to determine the effect of daily, intra-tumor metronomic dosing of topotecan on tumor growth in vivo using a xenograft tumor model of human prostate cancer implanted in male (NCr) nude mice. Conventional dosing of topotecan (160 µg/kg) was performed weekly by direct intra-tumor injection, while metronomic dosing of topotecan (20 µg/kg) was performed by daily direct intra-tumor injection. As described previously, the metronomic dose was based on calculated IC50 for each dosing schedule and assumption of a tumor volume of 1 mm3 and the conventional dose based on a previous study examining the effect of intra-cerebral administration of topotecan for the treatment of brain tumors.Citation30 After 17 d of treatment, animals subjected to metronomic dosing with topotecan had a significantly (p ≤ 0.05) smaller tumor volume (65.4% ± 11.2) compared with control animals (136% ± 14) and those treated conventionally (138% ± 10) (). At the conclusion of the study animals subjected to the metronomic dosing regimens of topotecan had significantly (p ≤ 0.05) smaller tumor volumes (54.8% ± 16.5) than animals receiving conventional dosing (144% ± 11) or the control group (207% ± 26). No significant differences in animal weights were observed during treatment (data not shown). Histopathological examination revealed treatment mediated differences between control and treatment groups. Control ( and B) and animals treated conventionally ( and D) had larger tumors and evidence of necrosis, regions of hypereosinophilia and loss of tissue architecture, compared with metronomically ( and F) dosed groups. Animals treated metronomically had tumors that were smaller in size with no evidence of necrosis.
The effect of low-dose systemic exposure of topotecan compared with conventional I.V. administration on tumor volume, animal weight and tumor vascular staining was determined flowing implantation of ALZET® micro-osmotic pumps in nude mice after tumors xenografts reached 200–300 mm3. ALZET® pumps were primed to delivery 2.45 mg/kg/day to achieve plasma concentrations at the experimentally determined IC50, i.e., 4–5 ng/mL and 0.10 mg/kg/day (4% the IC50 concentration), as described in methods. After 21 d of treatment, animals implanted with 2.45 and 0.1 mg/kg/day topotecan had significantly (p ≤ 0.05) smaller tumor volumes compared with ALZET® control animals (). No significant differences were detected between animals treated I.V. conventionally with topotecan and the control groups, and between conventional and ALZET® control groups. No significant differences in animal weights were observed between each treatment group (data not shown). Tumors were harvested at the completion of the study and the vascular was stained for factor VIII (von Williebrand factor). There was strong, but no appreciable differences in tumor vascular staining in all treatment groups, as determined by a pathologist blinded to tissue identify (). In all treatment groups there was also no evidence of apoptosis in tumor sections as assessed by TUNEL staining (Fig. S1). This is not unexpected as tissues were harvested 3–7 d after the last dose and evidence of cells undergoing apoptosis can be cleared rapidly in vivo.
Discussion
Treatment options for prostate cancers are limited and no effective chemotherapy for advanced prostatic cancer (castration resistant) is available. The use of conventional high dose topotecan for the treatment of prostate cancer clinically is limited due to its low efficacy and high incidence of side effects.Citation25 Thus, we hypothesized that low-dose continuous administration of topotecan may be used to achieve comparable or increased potency compared with conventional high-dose drug schedules. This hypothesis is supported by metronomic dosing studies in normal human endothelial cells in vitro and in vivo studies using mouse corneal revascularization models.Citation21,Citation22 Further, topotecan significantly inhibited tumor growth following low-dose exposure in experimental Wilms tumor.Citation31 Data reported in this study advances previous work by suggesting the novel finding that metronomic dosing increases the efficacy of topotecan in models of human prostate cancer. Furthermore this study suggests that the mechanisms of these events correlate to changes in cell cycle, expression of p21 and annexin V and PI staining, but did not appear to be mediated by alterations tumor vascularization.
Understanding the effect of metronomic low-dose exposure on the activity of topotecan against cancer cells can be used as a guide to optimize dosing regimens of topotecan and to enhance the activity of topotecan on angiogenesis and directly on tumor parenchyma. To understand differences in drug activity against different prostate cancer stages, we used two types of prostate cancer cell lines, LNCaP and PC-3. LNCaP cells demonstrate the characteristics of prostate cancer in early stages of malignancy and were used to evaluate the potential role of topotecan metronomic schedule in the treatment of early stage of prostate cancer.Citation32 In contrast, PC-3 cells were used to evaluate changes associated with different topotecan dosing regimens on advanced prostatic cancer.Citation33 Data reported above suggest that metronomic dosing schedules increased the efficacy of topotecan in both local and advanced prostate cancer and may be effective at treating metastatic spread. The fact that metronomic dosing increased the efficacy of topotecan in both LNCaP and PC-3 cells suggests that metronomic dosing increases efficacy using mechanisms that are independent of androgen-signaling and p53 status.
The primary mechanism of cell death induced by conventional dosing of topotecan in LNCaP cells appears to be apoptotic as evidence by the increase in attached and detached cells staining positive for annexin V only. An increase in cells staining positive for annexin V and PI after 72 h is likely the result of cells progressing to late apoptosis. Metronomic dosing resulted in a slight increase in cells staining positive for annexin V and annexin V and PI compared with control cells, but this was not significantly different. These data suggest that following conventional dosing cells die through apoptosis, in agreement with known mechanism of topotecan.Citation27 The decrease in apoptosis in LNCaP cells exposed to metronomic doses may be mediated by a decrease in the rate of cell death or induction of cytostasis. In contrast to LNCaP cells, there was a decrease in annexin V staining in PC-3 cells following conventional and metronomic (p ≤ 0.05) treatment. The increase in late apoptosis, in the absence of necrosis, supports the hypothesis that topotecan is inducing apoptosis in PC-3 cells after both conventional and metronomic treatments. In contrast to LNCaP cells, metronomic dosing of topotecan did not decrease cell death. Reasons for these differences are not known, but these data suggests that there are schedule dependent differences in the antitumor effects of topotecan in LNCaP and PC-3 cells. Further, the differences may be related to differences in gene expression of p53 in LNCaP, but not PC-3 cells.
The tumor suppressor protein p53 and the cell cycle inhibitor protein p21 can mediate cell death.Citation34,Citation35 Activation of p53 typically induces cell death and cell cycle arrest.Citation36 p53 can induce p21 during renal cell injury, but p21 can also be activated independently of p53.Citation37 The mechanisms of p53-meidated activation of p21 are under study, but may involve increases in transcription. We previously demonstrated that both p53 and p21 are activated in LNCaP during cell death and cytostasis.Citation29 Data reported herein also show that both metronomic and conventional dosing of topotecan activated p21 in both LNCaP and PC-3 cells. The activation of p21 correlates to the cell cycle arrest. Further, these data suggest that the mechanism of p21 induction in both cells types is p53 independent. These data also suggest that the increased efficacy of metronomic dosing against prostate cancer cell death can be mediated by p53-independent mechanisms.
Using attached cells we also showed that metronomic dosing of topotecan increased significantly the percentage of PC-3 cells in G2/M of PC-3 cells, while only slight differences in cell cycle were observed in LNCaP cells. Whereas there is strong G2/M arrest following conventional dosing leading to apoptosis and cell death. It should be noted that metronomic dosing increased potency in LNCaP cell greater than PC-3 cells and that this may be related to differences in cell cycle alterations.
Even though the cell death studies suggested the presence of apoptosis, no evidence of apoptosis, as indicated by increases in a sub-G0/G1 peak, were observed during the cell cycle analysis. It should be noted that the cell death studies were conducted using both attached and detached cells, whereas cell cycle analysis was conducted on attached cells only. Examination of nuclear morphology of attached cells showed no evidence of apoptosis, which agrees with the lack of a sub-G0/G1 peak in attached cell. Thus, these data support the literature that conventional dosing of topotecan leads to apoptosisCitation27 and suggests that these apoptotic cells detach.
We hypothesized that metronomic dosing may mediate direct antitumor effects in addition to its documented antiangiogenic activity. Expression of p21 plays important roles in regulating cell cycle and its overexpression results in G2/M arrest.Citation38 Increases in p21 expression are associated with earlier clinical stages of pancreatic adenocarcinoma and enhanced survival rate.Citation39 Another study suggests p21 expression improved survival in prostate cancer patients and is a better prognostic factor for advanced gastric carcinoma.Citation40,Citation41 Thus, it's possible that, in addition to inhibition of angiogenesis, increases in efficacy induced by metronomic dosing can be mediated by increased p21 expression. It is also possible that p21 alterations are induced by the same mechanism that mediates antiangiogenic activity. Further studies are needed to test these hypotheses.
The mechanisms by which metronomic dosing regimens increase p21 expression are still under study. Increases in p21 protein were observed after both conventional and metronomic treatments, but were greater in metronomic treated cells. This suggests that metronomic dosing of topotecan enhanced p21 activation pathways. The identity of these pathways is the subject future studies.
We demonstrated that low-dose daily administration of topotecan enhanced its antitumor activity in a model of human prostate cancer after metronomic—direct tumor injection and via continuous systematic exposure using implantable ALZET® micro-osmotic pumps. Our data also supports previous studies that have demonstrated conventional high-dose administration of topotecan is only marginally effective compared with control tumor growth. However, the antitumor activity of topotecan administered at lower doses daily injection or continuous low-dose exposure was highly efficacious and appeared to have no statistically significant effect on body weight, a gross measure of systemic toxicity or tumor vascular staining. We believe that these data are the first to demonstrate such a finding in prostate cancer for topotecan.
It is also interesting to note that histopathological analysis of tumors from animals treated with intra-tumor conventional, high-dose treatment, had evidence of necrosis compared with the groups treated following a metronomic schedule. According to the Gleason score, histopathological evidence of necrosis is correlated with increased tumor malignancy and poor prognosis resulting from the rapid growth of tumor and poor diffusions of nutrients and removal of waste products.Citation42 Cells treated metronomically showed no evidence of necrosis compared with control and conventional treatment groups. This further suggests that metronomic dosing alters tumor growth phenotype and decreases its malignancy compared with control and conventional treatment.
Data reported herein show that metronomic dosing or continuous low-dose infusions increase the efficacy of topotecan both in vitro and in vivo. These results support existing preclinical and clinical studies that have shown metronomic dosing of topotecan orally in non-prostate cancer models increases efficacy and is well tolerated with lower side effects compared with conventional administration.Citation43 However, to our knowledge, this is first time metronomic dosing of topotecan has been shown to increase efficacy against prostate cancer cells in vitro and an in vivo model. These data suggest that frequent low-dose administration of topotecan may be useful for treatment of prostate cancer and supports further investigation.
In conclusion, we showed that low-dose frequent administration of topotecan increased its anticancer efficacy in both in vitro and in vivo models of prostate cancer. Data in this study also demonstrate that the observed increase in efficacy was independent of the androgen receptivity of the prostate cancer as well as its p53 status and appears to be independent of its known vascularization effects. Immunoblot analysis showed that metronomic dosing increased p21 expression, compared with conventional dosing regimens, suggesting that differential effects of metronomic dosing in cancer cell growth compared with conventional dosing may be mediated by this protein.
Materials and Methods
Chemicals and reagents.
FBS (fetal bovine serum) and trypsin (0.25% w/v) were purchased from (Hyclone, Thermo Fisher Scientific Inc., Rockford, IL). F-12K Nutrient Mixture (Kaighn's Mod.) was obtained from (Mediatech, Manassas, VA). Topotecan was purchased from 21st Century Global E-Commerce Network (East Sussex, UK). Dimethyl sulfoxide (DMSO), sulforhodamine B (SRB), TRIS buffer, acetic acid, permount and Pierce ECL protein gel blotting substrate for chemiluminescence were obtained from Thermo Fisher Scientific Inc. (Rockford, IL). DAPI was purchased from Calbiochem (San Diego, CA). 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), propidium iodide (PI), phosphate buffered saline (PBS), glucose and RNase A were purchased from Sigma-Aldrich Inc. (St. Louis, MO). Absolute ethanol was purchased from Pharmco-AAPER. Mouse anti-human p53, p21 and GAPDH antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The anti-mouse secondary antibody was purchased from Promega (Madison, WI).
Cell lines.
Androgen-independent human prostate cancer epithelial cells (PC-3) and androgen-dependent human prostate cancer epithelial cells (LNCaP) were obtained from American Type Culture Collection (ATCC) (Rockville, MD). PC-3 cells were maintained in 10% (v/v) fetal bovine serum supplemented F-12K and LNCaP cells were maintained in 10% (v/v) fetal bovine serum supplemented RPMI-1640 media at 37°C, 21% O2 and 5% CO2 in a humidified cell culture chamber (NuAire Inc., Plymouth, MN). Cells were sub-cultured when they reached approximately 80–90% confluence.
Treatment protocols.
The effect of topotecan on prostate cell growth inhibition studies was conducted at 24, 48 and 72 h. Three studies (n = 3) were performed with five replicates for each concentration of topotecan. Cell growth was assessed at each time point using SRB and MTT cellular staining and examination of cellular and nuclear morphology.
Conventional dosing of topotecan.
Prostate cancer cell lines (PC-3 and LNCaP) were seeded at 2 x 103 and 4 x 103 cells in 96 well plates with a 10% (v/v) fetal bovine serum supplemented F-12K and RPMI-1640 media, respectively. Plates were incubated for 24 h prior to media change and replacement with serum supplemented media containing topotecan (0.04–10,000 nM). Plates were then incubated at the same conditions for an additional 24, 48 or 72 h.
Metronomic dosing of topotecan.
Prostate cancer cell lines (PC-3 and LNCaP) were seeded as described in conventional dosing. To simulate metronomic dosing, cells were dosed at t = 0, i.e., 24 h after seeding as described for conventional exposures above; however, at 24 and 48 h after initial dosing, media was removed and cells were exposed to freshly prepared serum supplemented media containing topotecan (0.04–10,000 nM). Media for controls cells was changed daily and did not contain drug.
Assessment of cell growth and viability.
Cell growth and viability were assessed by measuring MTT and SRB staining at 24, 48 and 72 h post initial topotecan exposure. SRB and MTT staining were performed as described previously in reference Citation44 and Citation45. Briefly, MTT reagent was added to each well and plates were returned to the same conditions for 2 h. The resultant formazan crystals were dissolved in 200 µL DMSO and absorbance measured at 550 nm using a Synergy HT Multi-Mode Microplate Reader (BioTek Instruments, Inc., Winooski, VT). SRB staining was performed by fixing live cells in cold 10% (w/v) TCA for one hr at 4°C. Fixed cells were washed with water and stained with SRB dye for 5 min. Excess SRB was removed by washing plates with 1% (v/v) acetic acid and SRB dye bound to cellular protein was dissolved in 200 µL TRIS (10 mM). Absorbance was measured at 490 nm using a Synergy HT Multi-Mode Microplate Reader. The potency (IC50) of each dosing schedule was calculated by fitting all data (naïve pooled data approach) from the growth inhibition studies against topotecan concentration (C) to an inhibitory maximum effect (Emax) model with a baseline (Eo) effect parameter, i.e., E = Emax − (Emax − Eo) × C/(C + IC50), using WinNonlin professional software, version 5.2 (Pharsight corporation, Mountain View, CA), pharmacodynamic models were chosen to fit the data from three independent studies (n = 5/study).
Assessment of cellular and nuclear morphology.
Cell morphology was assessed using phase-contrast microscopy. PC-3 and LNCaP cells were seeded in 6-well plates (8 x 104 for PC-3 cells and 1.6 x 105 for LNCaP cells) and exposed to topotecan at the calculated IC50 for 72 h (MTT) using each treatment protocol (conventional and metronomic), which was calculated from growth inhibition experiments. PC-3 cells were exposed to 80 and 10 nM, while LNCaP were exposed to 30 and 2 nM. At least three areas with approximately equal cell densities were identified in each well and images of these areas were captured with a Nikon AZ100 stereo-fluorescent microscope mounted with a Nikon Digital Sight DS-QiMc camera utilizing NIS-Elements image analysis software (Nikon, Melville, NY).
Cells used for nuclear staining were grown on glass coverslips in 12-well plates, treated and stained with DAPI as described previously in reference Citation36. In brief, after 72 h cells were washed with PBS, fixed for 10 min, and then washed three times with fresh PBS, 10 min/each. Cells were incubated with DAPI, 34 µM diluted in PBS. After 20 min on a shaker cells were washed three times with PBS and applied to glass slides using mounting media and sealed with permount. DAPI staining was visualized by fluorescence microscopy at 350/486 nm (ex/em) using a Nikon AZ100 stereo-fluorescent microscope.
Measurement of cell death.
Annexin V and PI staining plus flow cytometry was used to assess apoptosis and necrosis.Citation36 Briefly, cells seeded in 6 well plates at concentrations described above and exposed to topotecan using the IC50 for both conventional and metronomic treatments. After 72 h annexin and PI staining were assessed in both attached and detached cells. Binding buffer contained annexin V-FITC (25 µg/ml) and PI was (25 µg/ml) as well as 10 mM HEPES, 140 mM NaCl, 5 mM KCl, 1 mM MgCl2 and 1.8 mM CaCl2 (pH = 7.4). Cells were incubated for 10 min., followed by three washes in binding buffer. Both detached and attached cells were combined and staining was quantified using a Dako Cyan flow cytometer. For each measurement 10,000 events were counted.
Assessment of cell cycle.
Cell cycle was assessed at the calculated IC50 of topotecan after each treatment protocol (conventional and metronomic) of PC-3 and LNCaP. Cells were seeded in 6-well plates (8 x 104 for PC-3 cells and 16 x 104 for LNCaP cells) and exposed to topotecan (conventional and metronomic dosing) at the corresponding IC50 of each treatment protocol, which was calculated from growth inhibition experiments. After 24, 48 and 72 h of exposure, cells were harvested and fixed in 70% (v/v) ethanol and stored at 4°C until staining. Cells were prepared for cell cycle analysis by staining with PI (50 µg/ml) in sample buffer [PBS + 1% (w/v) glucose], containing RNase A (100 units/ml) for 30 min at room temperature. Cells (1 x 104) were sorted and analyzed by flow cytometry using a Becton Dickinson FACSCalibur flow cytometer (BD Biosciences, San Jose, CA). Cell cycle data were analyzed using FlowJo software (Tree Star, Inc., Ashland, OR).
Assessment of p21 and p53 expression.
Treatment mediated effects on p21 and p53 expression were assessed at the calculated IC50 of topotecan for each treatment protocol in PC-3 and LNCaP cell lines 72 h after initial topotecan exposure. Cells were seeded in 6-well plates (8 x 104 for PC-3 cells and 16 x 104 for LNCaP cells) and exposed to topotecan (conventional and metronomic) at the corresponding IC50 of each treatment protocol. At each time point, media was aspirated from the wells and cells were lysed in immunoblot buffer (0.25 M TRIS-HCl, pH 6.8, 10% glycerol, 1 mg/ml bromophenol blue and 0.5% (v/v) 2-mercaptoenthanol). Samples were then sonicated and heated for 10 min at 80°C before being loaded onto a 12% SDS-polyacrylamide gel. Proteins were separated under reducing conditions and then transferred to a nitrocellulose membrane. Transfer efficiency was confirmed with Ponceau S staining. Nonspecific binding was limited by incubating the membrane in blocking buffer (2.5% (w/v) casein, pH 7.6, 150 mM NaCl, 10 mM TRIS-HCl and 0.02% sodium azide) for 3 h. Following blocking, the membrane was incubated with the primary antibody overnight (p53 1:500, p21 1:250, Bax 1:10,000 and GAPDH 1:1,000) and then with the appropriate secondary antibody (1:10,000) for 1.5 h at room temperature. Bands were detected using Pierce ECL Protein gel blotting substrate (Thermo Scientific, Rockford, IL) for chemiluminescence.
Activity of topotecan in vivo.
The effect of conventional and metronomic dosing of topotecan on prostate tumor growth was determined by implanting PC-3 cells subcutaneously in 7–8 week old male nude (NCr) mice that were acclimated 2 weeks after receipt from Taconic Farms, Inc. (Germantown, NY). Animals were housed and maintained in accordance with an approved Institutional Animal Care and Use Committee (IACUC) protocol at the University of Georgia and in accordance with the US. Public Health Service (PHS) Policy on Humane Care and Use of Laboratory Animals, updated 1996. Animals were housed in pathogen-free cages within a light and temperature-controlled isolated room and provided with autoclaved rodent chow and autoclaved water ad libitum. For tumor implantation, sub confluent PC-3 cells grown in 10% fetal bovine serum supplemented F-12K were harvested using 0.25% trypsin (v/v). Cells were counted and re-suspended in serum free media to a final concentration of 1 x 107 cells/mL. Media was mixed with ice-cold Matrigel (1:1, v/v) and 200 µL of the mixture was injected subcutaneously into the mouse flank. Tumors were allowed to grow and mice were monitored every other day. Tumor diameters were measured using digital calipers, recorded and tumor volumes were calculated according to the following formula: (larger dimension) x (smaller dimension)Citation2 x 0.5 as described previously in reference Citation46 and Citation47. When tumors reached ∼400 mm3 mice were randomly treated with intra-tumor injections of topotecan, 20 µg/kg/day x 30 d (metronomic dosing) or 160 µg/kg/week x 5 weeks (conventional dosing). The metronomic dose was based on calculated IC50 for each dosing schedule and assumption of a tumor volume of 1 mm3 and the conventional dose based on a previous study examining the effect of intra-cerebral administration of topotecan for the treatment of brain tumors.Citation30 Individual tumor volumes were normalized to their tumor volume on the day treatment was initiated. Treatment continued for 4 weeks, tumor dimension and animals weights were measured every other day. Animals were euthanized 3 and 7 d after the last treatment. Tumors were collected, fixed for 24 h in 10% (v/v) formalin and embedded in paraffin. Tumors were sectioned and stained with hematoxylin and eosin (H&E) for histopathological examination and for factor VIII to visualize endothelial cells. Apoptosis was assessed based on the staining of terminal deoxynucleotidyle transferase dUTP end labels (TUNEL) as determined using the GenScript (Piscataway, NJ) TUNEL apoptosis detections kit, following the manufactures instructions. Two positive controls were prepared, (i) untreated PC-3 tumor slices were incubated with DNAase for 60 min and (ii) rat kidney tissue sections after treatment with bromate, which we have shown to induce apoptosis in rat kidneys (Cummings, BS, unpublished data). Micrographs were captured using a Nikon AZ100 stereo-fluorescent microscope mounted with a Nikon DS-Qi1Mc color camera and processed using NIS-Elements image analysis software.
The effect of continuous systemic exposure to topotecan was also determined in this xenograft model. ALZET® micro-osmotic pumps were implanted subcutaneously according to the manufacturer instructions. The pumps were designed to deliver specific volume over a period of 28 d. The dose required to achieve the calculated IC50 was determined in nude mice (four to five mice/dose level) implanted subcutaneously with ALZET® pumps after priming. Blood samples were collected using repetitive sampling from the mouse cheek (submandibular region) and analyzed according to the method described below. The subcutaneous implantation of these ALZET® pumps, with a constant-infusion of 2.45 mg/kg/day, achieved plasma concentrations of 3–4 ng/mL (data not shown). These plasma concentrations are similar to the experimentally determined IC50 of topotecan in PC-3 cells in vitro (4.57 ng/mL) after 72 h exposure.
The antitumor activity was determined in another group of mice (four to five animals in each cohort) were implanted with PC-3 cells in the mouse flank as described previously. When the tumor reached a volume of 200–300 mm3, mice were assigned to receive 2.45, 0.10 or 0.0 (vehicle control) mg/kg/day continuous exposure of topotecan and compared with conventional I.V. dosing, of 4 or 0.0 (vehicle control) mg/kg, every 4 d (q4d). The conventional dose was selected based on other studies that evaluated topotecan at or below its maximum tolerated dose and in a variety of models.Citation48–Citation50
Measurement of topotecan plasma concentration.
Total topotecan was extracted from plasma samples according to a modified published procedure.Citation51 Plasma samples (20 µL) were extracted using protein precipitation with 60 µL ice-cold methanol and by acidification with 20 µL 100 mM phosphoric acid (H3PO4) to allow the detection of total topotecan. SN-38 was used as an internal standard. Samples were vortexed, kept on ice for 5 min then centrifuged at 10,000x g for 10 min at 4°C. The resultant supernatants were transferred to low volume glass vial inserts. The supernatant (20 µL) was injected onto the HPLC column. Calibration standards of total topotecan were prepared in drug free mouse plasma and extracted as described above. A concentration range of 625 to 2.44 ng/mL of topotecan was used to construct standard curve and the ratios of topotecan area to the internal standard area were used to prepare calibration curves.
Measurement of topotecan was performed on an Agilent 1100 HPLC system coupled to fluorescence detector (Santa Clara, CA). Topotecan was separated on an Agilent Eclipse plus C18 column (3.5 µm, 100 x 4.6 mm) from Agilent technologies (Santa Clara, CA) equipped with a Phenomenex Security C18 guard column (4.0 x 3.0 mm). Analytes were eluted from the column using a gradient elution. Mobile phase A was composed of 75 mM ammonium acetate and 7.5 mM tetrabutylammonium bromide (TBAB) with pH adjusted to 6.4 using glacial acetic acid. Acetonitrile was used as mobile phase B. Gradient elution started from 12% B and reached 30% B at 13 min at a flow rate of 1 ml/min. Analytes signals were measured using a fluorescence detector (at excitation/emission wavelengths of 370/520 nm).
Statistical analysis.
A one-way ANOVA followed by Bonferroni t-tests was used to assess differences between the calculated IC50's and cell cycle distributions. Differences were considered statistically significant if a p-value was ≤0.05. SigmaStat for Windows version 3.11 (Systat Software, Inc.) was used for statistical analysis.
Abbreviations
| DAPI | = | 4′,6-diamidino-2-phenylindole |
| DMSO | = | dimethyl sulfoxide |
| FBS | = | fetal bovine serum |
| IC50 | = | the half maximal inhibitory concentration |
| LNCaP | = | androgen-dependent human prostate cancer epithelial cells |
| MTT | = | 3-(4,5-dimethylthiazol-2-yl)2,5-diphenyltetrazolium bromide |
| PBS | = | phosphate buffered saline |
| PC-3 | = | androgen-independent human prostate cancer epithelial cells |
| PI | = | propidium iodide |
| q4d | = | every 4 days |
| Rnase A | = | ribonuclease A |
| SRB | = | sulforhodamine B |
| TCA | = | trichloro acetic acid |
| TRIS | = | tris(hydroxymethyl)aminomethane buffer |
Figures and Tables
Figure 1 Effect of treatment on cellular and nuclear morphology. Cellular morphology was assessed at the calculated 72 h IC50 of each treatment protocol, i.e., 80 nM conventional and 10 nM metronomic in PC-3 and 30 nM conventional and 2 nM metronomic in LNCaP cells (A). Nuclear morphology of attached cells grown on coverslips and stained with DAP I was assessed using the treatment schedule above (B). Scale bars represent 50 µm for PC-3 and LNCaP cells.
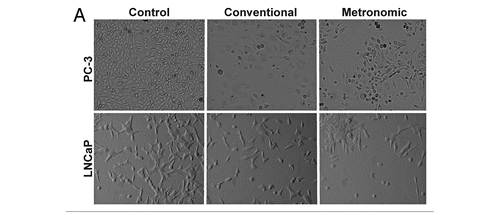
Figure 2 Effect of topotecan on PC-3 and LNCaP cell death. PC-3 cells were exposed to 10 nM topotecan using different protocols (conventional vs. metronomic) for 72 h and LNCaP cells were exposed in a similar fashion using 2 nM topotecan. Media containing fresh topotecan was changed daily to simulate metronomic dosing. After 72 h cells were harvested and stained with annexin-FITC and PI using flow cytometry. Representative plots of annexin and PI staining of PC-3 (A–C) and LNCaP (D–F) are shown. (G) shows alterations in cells staining positive only for annexin V (apoptotic cells), while (H) shows cells staining positive for both annexin V and PI (late apoptosis). Data are presented as the mean ± SEM of three separate experiments (n = 5/study). Means noted with (*) are significantly (p ≤ 0.05) different in comparison to control and (#) from metronomic dosing.
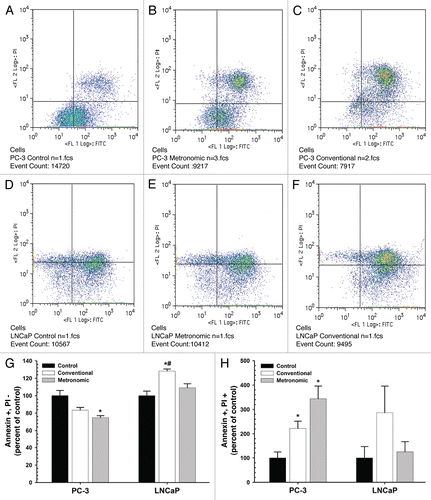
Figure 3 Effect of topotecan on pC-3 and LNCaP cell cycle. PC-3 cells (A–C) were exposed to 10 nM topotecan using different protocols (conventional vs. metronomic) for 24, 48 and 72 h and LNCaP cells (D–F) were exposed to 2 nM topotecan, respectively. Media containing fresh topotecan was changed daily to simulate metronomic dosing. At each time point, cells were harvested and stained with PI and cell cycle phases were assessed by flow cytometry. Data are presented as the mean ± SEM of three separate experiments (n = 5/study). Means noted with asterisk (*) are significantly (p ≤ 0.05) different in comparison to control.
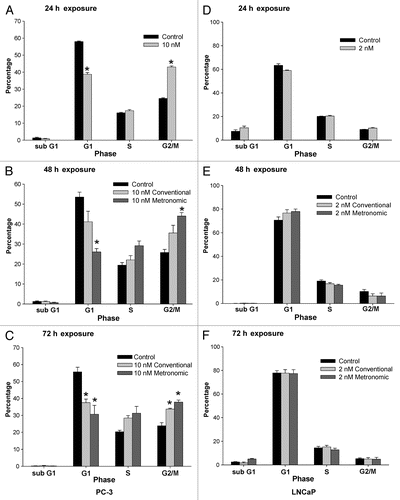
Figure 4 Effect of topotecan on p21 expression. (A) LNCaP and (B) PC-3 cells were exposed to topotecan at the calculated IC50 of each treatment protocol (conventional vs. metronomic) for 72 h and p21 expression was determined by immunoblot analysis. The expression of glyceraldehydes-3-phosphate dehydrogenase (GAPDH) is shown as a loading control. (C and D) show the densitometry for p21 expression normalized to GAPDH for (C) LNCaP and (D) PC-3 cells. Data in (C and D) are presented as the mean ± SEM of four separate experiments. Means noted with asterisk (*) are significantly (p ≤ 0.05) different in comparison to control.
Figure 5 Effect of topotecan dosing schedules on tumor volume in a PC-3 xenograft model. Six to seven week old NCr nude mice (four to five per group) were implanted subcutaneously with PC-3 cells and treated using direct intra-tumor injections, continuous infusion or conventional I.V. of topotecan or control. (A) Tumor volume were determine following direct intra-tumor injections of topotecan following a conventional schedule (160 mg/kg/week x 5, represented by diamonds on x-axis) or a metronomic schedule (20 mg/kg/day x 30, represented by squares along x-axis). A significant (p ≤ 0.05) reduction in tumor volume was observed following metronomic exposure of topotecan vs. untreated control and conventional treatments. In a separate study, (B) tumor volume were determine following topotecan administered continuously using subcutaneously implanted ALZET® micro-osmotic pumps (at doses of 2.45, 0.10 and 0.0 (vehicle control) mg/kg/day, (continuous exposure of topotecan is represented by a solid line along x-axis) compared with I.V. injection at 4 mg/kg q4d x 4, represented by diamonds on x-axis) or control (normal saline). A significant (p ≤ 0.05) reduction in tumor volume was observed following continuous exposure of topotecan at 2.45 and 0.10 mg/kg/day vs. ALZET® control and conventional treatments. Data for both studies are presented as the mean ± SEM (n = 4 to 5). Means noted with (*) are significantly (p ≤ 0.05) different in comparison to controls and (#) are significantly (p ≤ 0.05) different in comparison to conventional treatment. (C) The effect of topotecan dosing on tumor vascular (arrowhead) using Factor VIII staining was also determined from samples obtained from the Alzet study; topotecan conventional I.V. administration at 4.0 (C1) and administered continuously using ALZET® micro-osmotic pumps at 2.45 (C2), 0.10 (C3) and 0.0 (C4) vehicle control, mg/kg/day (scale bars = 100 µm). No appreciable differences in vascular density, as indicated by Factor VIII staining were observed. q4d-every 4 d.
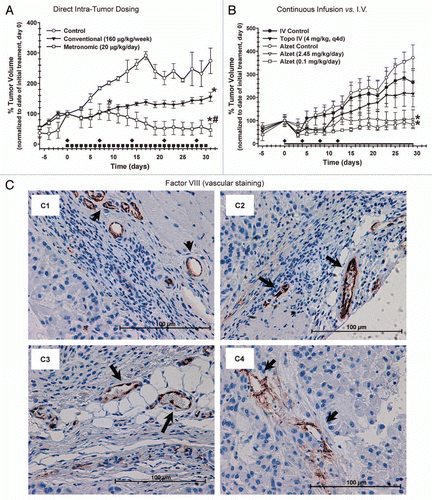
Figure 6 Representative histological images of mouse PC-3 tumor xenografts treated with different dosing regimens of topotecan and stained with hematoxylin and eosin (H&E). When tumors reached 400 mm3 mice were treated with intra-tumor injections of topotecan 20 µg/kg/day x 30 (metronomic) or 160 µg/kg/week x 5 (conventional dosing); control groups received no treatment. Micrographs of representative tumor sections at low [20x, (A, C and E) scale bars = 500 µm] and high [100x, (B, D and F) scale bars = 100 µm] magnification were examined 3 d after the last treatment. (A) Control tumors had large areas of necrosis within the tumor. (B) The necrotic area (NECR) is clearly distinct from the viable tissue and characterized by hypereosinophilia and loss of tissue architecture. (C and D) Conventionally treated tumors had evidence of necrosis. Note the necrotic area in the center of the image characterized by hypereosinophilia and loss of tissue architecture. (E and F) Metronomically treated tumors had no appreciable necrosis visible within the tumor. Metronomically treated tumor cells were arranged in haphazard fashion (including looser arrangement on the right side of the image), but with no evidence of necrosis.
![Figure 6 Representative histological images of mouse PC-3 tumor xenografts treated with different dosing regimens of topotecan and stained with hematoxylin and eosin (H&E). When tumors reached 400 mm3 mice were treated with intra-tumor injections of topotecan 20 µg/kg/day x 30 (metronomic) or 160 µg/kg/week x 5 (conventional dosing); control groups received no treatment. Micrographs of representative tumor sections at low [20x, (A, C and E) scale bars = 500 µm] and high [100x, (B, D and F) scale bars = 100 µm] magnification were examined 3 d after the last treatment. (A) Control tumors had large areas of necrosis within the tumor. (B) The necrotic area (NECR) is clearly distinct from the viable tissue and characterized by hypereosinophilia and loss of tissue architecture. (C and D) Conventionally treated tumors had evidence of necrosis. Note the necrotic area in the center of the image characterized by hypereosinophilia and loss of tissue architecture. (E and F) Metronomically treated tumors had no appreciable necrosis visible within the tumor. Metronomically treated tumor cells were arranged in haphazard fashion (including looser arrangement on the right side of the image), but with no evidence of necrosis.](/cms/asset/a3285abe-75b1-4987-9c28-0976463eb339/kcbt_a_10915950_f0006.gif)
Table 1 Effect of treatment and time of exposure on topotecan potency (IC50)
Additional material
Download Zip (179.5 KB)Acknowledgments
We would like to thank the Histological Laboratory at the College of Veterinary Medicine at the University of Georgia for embedding, processing and staining of slides for histological examination. This research was funded in part by an UGA Faculty Research Grant, an Interdisciplinary Toxicology Program equipment grant and Georgia Cancer Coalition Distinguished Scholar Grants to B.S.C. and R.D.A., a King Saud University Fellowship to I.A.A. and graduate stipend support to J.N.M. funded by a grant from the NIH, NIBIB-EB008153 to R.D.A./B.S.C.
References
- Gulley J, Dahut W. Novel clinical trials in androgen-independent prostate cancer. Clin Prostate Cancer 2002; 1:51 - 57; PMID: 15046713
- Thompson I, Thrasher JB, Aus G, Burnett AL, Canby-Hagino ED, Cookson MS, et al. Guideline for the management of clinically localized prostate cancer: 2007 update. J Urol 2007; 177:2106 - 2131; PMID: 17509297; http://dx.doi.org/10.1016/j.juro.2007.03.003
- Jani AB. Management strategies for locally advanced prostate cancer. Drugs Aging 2006; 23:119 - 129; PMID: 16536635; http://dx.doi.org/10.2165/00002512-200623020-00003
- Rumohr JA, Chang SS. Current chemotherapeutic approaches for androgen-independent prostate cancer. Curr Opin Investig Drugs 2006; 7:529 - 533; PMID: 16784023
- Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer 2001; 1:34 - 45; PMID: 11900250; http://dx.doi.org/10.1038/35094009
- Seruga B, Ocana A, Tannock IF. Drug resistance in metastatic castration-resistant prostate cancer. Nat Rev Clin Oncol 8:12 - 23; PMID: 20859283; http://dx.doi.org/10.1038/nrclinonc.2010.136
- Kerbel RS, Kamen BA. The anti-angiogenic basis of metronomic chemotherapy. Nat Rev Cancer 2004; 4:423 - 436; PMID: 15170445; http://dx.doi.org/10.1038/nrc1369
- Kerbel RS, Klement G, Pritchard KI, Kamen B. Continuous low-dose anti-angiogenic/metronomic chemotherapy: from the research laboratory into the oncology clinic. Ann Oncol 2002; 13:12 - 15; PMID: 11863092; http://dx.doi.org/10.1093/annonc/mdf093
- Jain RK. Barriers to drug delivery in solid tumors. Sci Am 1994; 271:58 - 65; PMID: 8066425; http://dx.doi.org/10.1038/scientificamerican0794-58
- Berry W, Eisenberger M. Achieving treatment goals for hormone-refractory prostate cancer with chemotherapy. Oncologist 2005; 10:30 - 39; PMID: 16368869; http://dx.doi.org/10.1634/theoncologist.10-90003-30
- Lennernäs B, Albertsson P, Damber JE, Norrby K. Albertsson P, Damber J, Norrby K. Antiangiogenic effect of metronomic paclitaxel treatment in prostate cancer and non-tumor tissue in the same animals: a quantitative study. APMIS 2004; 112:201 - 219; PMID: 15153162; http://dx.doi.org/10.1111/j.1600-0463.2004.apm1120306.x
- Vogt T, Hafner C, Bross K, Bataille F, Jauch K, Berand A, et al. Antiangiogenetic therapy with pioglitazone, rofecoxib and metronomic trofosfamide in patients with advanced malignant vascular tumors. Cancer 2003; 98:2251 - 2256; PMID: 14601096; http://dx.doi.org/10.1002/cncr.11775
- Colleoni M, Orlando L, Sanna G, Rocca A, Maisonneuve P, Peruzzotti G, et al. Metronomic low-dose oral cyclophosphamide and methotrexate plus or minus thalidomide in metastatic breast cancer: antitumor activity and biological effects. Ann Oncol 2006; 17:232 - 238; PMID: 16322118; http://dx.doi.org/10.1093/annonc/mdj066
- Kamat AA, Kim T, Landen C Jr, Lu C, Han L, Lin Y, et al. Metronomic chemotherapy enhances the efficacy of antivascular therapy in ovarian cancer. Cancer Res 2007; 67:281 - 288; PMID: 17210709; http://dx.doi.org/10.1158/0008-5472.CAN-06-3282
- Bocci G, Falcone A, Fioravanti A, Orlandi P, Di Paolo A, Fanelli G, et al. Antiangiogenic and anticolorectal cancer effects of metronomic irinotecan chemotherapy alone and in combination with semaxinib. Br J Cancer 2008; 98:1619 - 1629; PMID: 18443598; http://dx.doi.org/10.1038/sj.bjc.6604352
- Wang J, Lou P, Lesniewski R, Henkin J. Paclitaxel at ultra low concentrations inhibits angiogenesis without affecting cellular microtubule assembly. Anticancer Drugs 2003; 14:13 - 19; PMID: 12544254; http://dx.doi.org/10.1097/00001813-200301000-00003
- Fontana A, Galli L, Fioravanti A, Orlandi P, Galli C, Landi L, et al. Clinical and pharmacodynamic evaluation of metronomic cyclophosphamide, celecoxib and dexamethasone in advanced hormone-refractory prostate cancer. Clin Cancer Res 2009; 15:4954 - 4962; PMID: 19622584; http://dx.doi.org/10.1158/1078-0432.CCR-08-3317
- Bocci G, Nicolaou K, Kerbel R. Protracted low-dose effects on human endothelial cell proliferation and survival in vitro reveal a selective antiangiogenic window for various chemotherapeutic drugs. Cancer Res 2002; 62:6938 - 6943; PMID: 12460910
- Miller KD, Sweeney C, Sledge G Jr. Redefining the target: chemotherapeutics as antiangiogenics. J Clin Oncol 2001; 19:1195 - 1206; PMID: 11181686
- Doloff JC, Khan N, Ma J, Demidenko E, Swartz H, Jounaidi Y. Increased tumor oxygenation and drug uptake during anti-angiogenic weekly low dose cyclophosphamide enhances the anti-tumor effect of weekly tirapazamine (Sup. Material). Curr Cancer Drug Targets 2009; 9:777 - 788; PMID: 19754361; http://dx.doi.org/10.2174/156800909789271503
- O'Leary JJ, Shapiro RL, Ren CJ, Chuang N, Cohen HW, Potmesil M. Antiangiogenic effects of camptothecin analogues 9-amino-20(S)-camptothecin, topotecan and CPT-11 studied in the mouse cornea model. Clin Cancer Res 1999; 5:181 - 187; PMID: 9918217
- Clements MK, Jones CB, Cumming M, Daoud SS. Antiangiogenic potential of camptothecin and topotecan. Cancer Chemother Pharmacol 1999; 44:411 - 416; PMID: 10501915; http://dx.doi.org/10.1007/s002800050997
- Petrangolini G, Pratesi G, De Cesare M, Supino R, Pisano C, Marcellini M, et al. Antiangiogenic effects of the novel camptothecin ST1481 (gimatecan) in human tumor xenografts. Mol Cancer Res 2003; 1:863 - 870; PMID: 14573787
- Hsiang YH, Liu L. Identification of mammalian DNA topoisomerase I as an intracellular target of the anticancer drug camptothecin. Cancer Res 1988; 48:1722 - 1726; PMID: 2832051
- Hudes GR, Kosierowski R, Greenberg R, Ramsey H, Fox S, Ozols R, et al. Phase II study of topotecan in metastatic hormone-refractory prostate cancer. Invest New Drugs 1995; 13:235 - 240; PMID: 8729952; http://dx.doi.org/10.1007/BF00873806
- Klein CE, Tangen CM, Braun TJ, Hussain MH, Peereboom DM, Nichols CR, et al. SWOG-9510: evaluation of topotecan in hormone refractory prostate cancer: a Southwest Oncology Group study. Prostate 2002; 52:264 - 268; PMID: 12210486; http://dx.doi.org/10.1002/pros.10118
- Tolis C, Peters GJ, Ferreira CG, Pinedo HM, Giaccone G. Cell cycle disturbances and apoptosis induced by topotecan and gemcitabine on human lung cancer cell lines. Eur J Cancer 1999; 35:796 - 807; PMID: 10505042; http://dx.doi.org/10.1016/S0959-8049(98)00425-0
- Sun B, Zhang X, Talathi S, Cummings BS. Inhibition of Ca2+-independent phospholipase A2 decreases prostate cancer cell growth by p53-dependent and independent mechanisms. J Pharmacol Exp Ther 2008; 326:59 - 68; PMID: 18441250; http://dx.doi.org/10.1124/jpet.108.138958
- Sun B, Zhang X, Yonz C, Cummings BS. Inhibition of calcium-independent phospholipase A2 activates p38 MAPK signaling pathways during cytostasis in prostate cancer cells. Biochem Pharmacol 2010; 79:1727 - 1735; PMID: 20171194; http://dx.doi.org/10.1016/j.bcp.2010.02.005
- Kaiser MG, Parsa A, Fine R, Hall J, Chakrabarti I, Bruce J. Tissue distribution and antitumor activity of topotecan delivered by intracerebral clysis in a rat glioma model. Neurosurgery 2000; 47:1391 - 1398; PMID: 11126910; http://dx.doi.org/10.1097/00006123-200012000-00026
- Soffer SZ, Kim E, Moore J, Huang J, Yokoi A, Manley C, et al. Novel use of an established agent: Topotecan is anti-angiogenic in experimental Wilms tumor* 1. J Pediatr Surg 2001; 36:1781 - 1784; PMID: 11733906; http://dx.doi.org/10.1053/jpsu.2001.28823
- Horoszewicz JS, Leong S, Kawinski E, Karr J, Rosenthal H, Chu T, et al. LNCaP model of human prostatic carcinoma. Cancer Res 1983; 43:1809 - 1818; PMID: 6831420
- Kaighn ME, Narayan K, Ohnuki Y, Lechner J, Jones L. Establishment and characterization of a human prostatic carcinoma cell line (PC-3). Invest Urol 1979; 17:16 - 23; PMID: 447482
- Price PM, Safirstein RL, Megyesi J. The cell cycle and acute kidney injury. Kidney Int 2009; 76:604 - 613; PMID: 19536080; http://dx.doi.org/10.1038/ki.2009.224
- Jiang M, Dong Z. Regulation and pathological role of p53 in cisplatin nephrotoxicity. J Pharmacol Exp Ther 2008; 327:300 - 307; PMID: 18682572; http://dx.doi.org/10.1124/jpet.108.139162
- Cummings BS, Schnellmann RG. Cisplatin-induced renal cell apoptosis: caspase 3-dependent and -independent pathways. J Pharmacol Exp Ther 2002; 302:8 - 17; PMID: 12065694; http://dx.doi.org/10.1124/jpet.302.1.8
- Megyesi J, Udvarhelyi N, Safirstein RL, Price PM. The p53-independent activation of transcription of p21WAF1/CIP1/SDI1 after acute renal failure. Am J Physiol 1996; 271:1211 - 1216; PMID:8997395; DOI:
- Niculescu AB III, Chen X, Smeets M, Hengst L, Prives C, Reed S. Effects of p21Cip1/Waf1 at both the G1/S and the G2/M cell cycle transitions: pRb is a critical determinant in blocking DNA replication and in preventing endoreduplication. Mol Cell Biol 1998; 18:629 - 643; PMID: 9418909
- Dergham S, Dugan M, Joshi U, Chen Y, Du W, Smith D, et al. The clinical significance of p21WAF1/CIP-1 and p53 expression in pancreatic adenocarcinoma. Cancer 1997; 80:372 - 381; http://dx.doi.org/10.1002/(SICI)1097-0142(19970801)80:3<372::AIDCNCR4>3.0.CO;2-U
- Aaltomaa S, Lipponen P, Eskelinen M, Ala-Opas M, Kosma V. Prognostic value and expression of p21(waf1/cip1) protein in prostate cancer. Prostate 1999; 39:8 - 15; PMID: 10221260; http://dx.doi.org/10.1002/(SICI)1097-0045(19990401)39:1<8::AID-PROS2>3.0.CO;2-N
- Gomyo Y, Ikeda M, Osaki M, Tatebe S, Tsujitani S, Ikeguchi M, et al. Expression of p21(waf1/cip1/sdi1), but not p53 protein, is a factor in the survival of patients with advanced gastric carcinoma. Cancer 1997; 79:2067 - 2072; PMID: 9179052; http://dx.doi.org/10.1002/(SICI)1097-0142(19970601)79:11<2067::AIDCNCR3>3.0.CO;2-M
- Humphrey PA. Gleason grading and prognostic factors in carcinoma of the prostate. Mod Pathol 2004; 17:292 - 306; PMID: 14976540; http://dx.doi.org/10.1038/modpathol.3800054
- Tillmanns T, Buller R, Stewart C, Mac Eachern J, Schaiquevich P, Walker M, et al. Daily oral topotecan: Utilization of a metronomic dosing schedule to treat recurrent or persistent solid tumors. Chemotherapy 2008; 3:19 [Presented at American Society of Clinical Oncology Annual Meeting]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983; 65:55 - 63; PMID: 6606682; http://dx.doi.org/10.1016/0022-1759(83)90303-4
- Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst 1990; 82:1107 - 1112; PMID: 2359136; http://dx.doi.org/10.1093/jnci/82.13.1107
- Davol PA, Frackelton AR Jr. Targeting human prostatic carcinoma through basic fibroblast growth factor receptors in an animal model: characterizing and circumventing mechanisms of tumor resistance. Prostate 1999; 40:178 - 191; PMID: 10398280; http://dx.doi.org/10.1002/(SICI)1097-0045(19990801)40:3<178::AIDPROS6>3.0.CO;2-I
- Geran R, Greenberg N, MacDonald M, Schumacher A, Abbott B. Protocols for screening chemical agents and natural products against animal tumors and other biological systems. Cancer Chemother Rep 1972; 3:1 - 103
- Tardi P, Choice E, Masin D, Redelmeier T, Bally M, Madden TD. Liposomal encapsulation of topotecan enhances anticancer efficacy in murine and human xenograft models. Cancer Res 2000; 60:3389 - 3393; PMID: 10910044
- Venditto VJ, Simanek EE. Cancer therapies utilizing the camptothecins: a review of the in vivo literature. Mol Pharm 2010; 7:307 - 349; PMID: 20108971; http://dx.doi.org/10.1021/mp900243b
- Shah DK, Shin BS, Veith J, Toth K, Bernacki RJ, Balthasar JP. Use of an anti-vascular endothelial growth factor antibody in a pharmacokinetic strategy to increase the efficacy of intraperitoneal chemotherapy. J Pharmacol Exp Ther 2009; 329:580 - 591; PMID: 19233938; http://dx.doi.org/10.1124/jpet.108.149443
- Zastre J, Anantha M, Ramsay E, Bally M. Irinotecancisplatin interactions assessed in cell-based screening assays: cytotoxicity, drug accumulation and DNA adduct formation in an NSCLC cell line. Cancer Chemother Pharmacol 2007; 60:91 - 102; PMID: 17009029; http://dx.doi.org/10.1007/s00280-006-0353-z