Abstract
We review the rationale for seeking inhibitors of homologous recombination (HR) repair for use in cancer therapy. Cells use HR as one way to repair DNA double-strand breaks that arise directly from treatments such as radiotherapy, or indirectly during replication when forks encounter other damage. HR occurs during the S and G2 phases of the cell cycle and is therefore more significant in dividing cancer cells than in non-dividing cells of healthy tissue, giving a potential therapeutic advantage to inhibiting the process. Also, some tumors consist of cells that are defective in other DNA repair pathways, and such cells may be sensitive to HR repair inhibitors because of synthetic lethality, in which blocking two alternative pathways that a cell can use to reach a needed end-point has a much bigger impact than blocking either pathway alone. We review strategies for identifying HR inhibitors and discuss current progress.
Acknowledgments
This work was supported by the NIH grant P01 CA-067166 to J.M.B.
Figures and Tables
Figure 1 Homologous recombination participates in resolution of two main types of double-strand breaks (DSBs) from endogenous and exogenous sources: (A) directly produced two-ended DSBs (like the ones produced by ionizing radiation) and (B) one-ended DSBs that result from single-strand breaks (SSBs) encountering replication forks. SSBs are common intermediates in the repair of many DNA lesions.
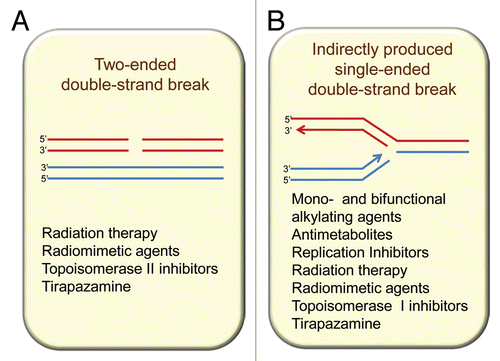
Figure 2 Double-strand break repair in mammalian cells. NHEJ, HR, MMEJ and SSA pathways compete for the repair of DSBs. Proteins involved in various stages of DSB repair are shown in teal. References for proteins involved at different stages can be found in references Citation7 and Citation21–Citation23. NHEJ is involved throughout the cell cycle, and HR is involved after replication, when the sister chromatid is available. A simple DSB occurring within euchromatin will be most likely repaired by NHEJ, while more complex DSBs or those in heterochromatin will initiate the full DNA damage response involving MRE11/RAD50/NBS1 (MRN) complex and ATM protein. MRN at the DSB recruits ATM, which phosphorylates BRE1A/B complex leading to relaxation of chromatin, facilitating repair by either NHEJ or HR. MRN recruits CtIP to initiate resection. Extensive resection is performed upon recruitment of EXO1 or DNA2 and the resulting single-stranded DNA (ssDNA) 3′-overhang is rapidly coated by RPA. The ssDNA-RPA recruits ATRIP leading to activation of ATR. The RPA then is displaced by Rad51 to form a nucleoprotein filament and initiate synapsis, a search for homologous template and DNA strand invasion, leading to formation of a D-loop intermediate. Synapsis is central to all HR reactions, and subsequent resolution of the D-loop is accomplished by at least three different pathways: double-strand break repair (DSBR), synthesis-dependent strand annealing (SDSA), and break-induced replication (BIR). An alternative pathway, microhomology-mediated end joining (MMEJ), which is active throughout the cell cycle, anneals short homologous sequences revealed by limited resection. Single-strand annealing (SSA) mediates repair when perfect repeats are revealed during end resection.
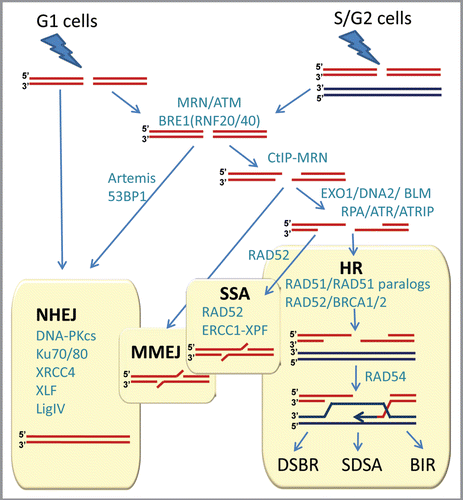
Figure 3 Key proteins involved in homologous recombination (HR). The main engine of HR is Rad51, which catalyzes the key reactions of synapsis-homology search and DNA strand invasion. Altogether, there are more than 200 proteins involved in different aspects of HR, providing a sufficient number of context-specific factors to explain the extraordinary versatility of HR, i.e., its functions in the repair of double-strand breaks (DSBs), DNA interstrand crosslinks (ICLs), and in resolution of stalled/collapsed replication forks. Any of the proteins are potentially targetable for cancer therapy.

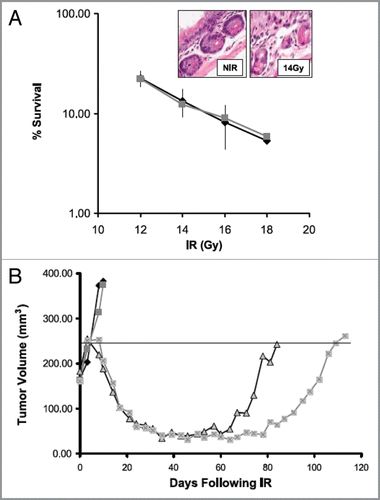
Figure 4 Imatinib increases radiotherapy-induced tumor growth delay without increasing intestinal toxicity in a PC-3 prostate cancer xenograft. (A) Intestinal crypt cell survival in BALB/c mice following 1–18 Gy in vivo in the presence of 50 mg/kg/d for 5 d. Surviving crypts were scored 3 d after irradiation based on three mice per treatment group. Points, mean survival; bars, SD. Inset, H&E-stained section of intestinal crypts before and after 14 Gy. (B) Plot of growth delay of PC3 xenografts treated with PBS alone, imatinib (50 mg/kg/d × 8 d) alone, 4 Gy × 5 plus PBS, or 4 Gy × 5 plus imatinib. Irradiated mice were either pretreated with 3 d PBS (sham) or 3 d imatinib (50 mg/kg/d i.p.) before irradiation and received daily PBS or imatinib i.p. dosing during 4 Gy daily radiation fractions for a total treatment period of 8 d similar to drug alone. For clarity, the median animal per group is shown. The calculated mean growth delay based on 9–10 mice per group was 100 ± 7 d for the combined treatment group, compared with 74 ± 14 d for the radiotherapy alone group (p = 0.003). There was no difference in growth delay between the imatinib alone and control groups. (Adapted and reprinted with permission from the American Association for Cancer Research: Choudhury et al., “Targeting homologous recombination using imatinib results in enhanced tumor cell chemosensitivity and radiosensitivity,” Mol Cancer Ther 2009; 8:203–13).