Abstract
Cisplatin is an effective anticancer drug used to treat many types of cancer, including non-small cell lung carcinoma (NSCLCs), but development of resistance is the primary impediment in cancer treatment. Insulin-like growth factor-binding protein 7 (IGFBP7) is a secreted tumor suppressor that is inactivated in human lung cancer. IGFBP7 is known to alter sensitivity to interferon-based anticancer therapy, and here, we examined loss of IGFBP7 as a potential contributor to chemo-resistance to cisplatin. The transcriptional level of IGFBP7 was decreased in cisplatin-resistant human cancer cell lines and NSCLC xenografts. IGFBP7 knock-down increased cellular resistance to cisplatin and increased the level of mitogen-activated protein kinase phosphatases (MKP) 3 levels. The expression of MKP3 increased in a cisplatin-resistant NSCLC cell line and lung xenografts. MKP3 knock-down increased IGFBP7 level, indicating that MKP3 regulates IGFBP7. These findings suggest a novel molecular mechanism responsible for the tumor suppressive function of IGFBP7 in cisplatin-resistant human lung cancer and could lead to the development of IGFBP7 as a cisplatin-sensitizing agent.
Introduction
Lung cancer is a major cause of cancer mortality, and accounts for about 20% of all cancer deaths worldwide. The high mortality of lung cancer mostly results from being diagnosed at a highly disseminated stage with rare curative therapeutic options. Most therapies for lung cancer are currently focused on chemotherapy with drugs. Although chemotherapy modalities are widely used, many advanced cases are resistant to anticancer drugs.
Cis-diamminedichloroplatinum (II) (CDDP, cisplatin) is one of the most potent anticancer drugs, and widely used in human epithelial cancers (e.g., ovarian, head/neck and lung). It is included in most protocols for the treatment of advanced non-small-cell lung carcinomas (NSCLCs), the most frequent and therapy-refractive sub-class of lung cancer. However, the development of drug resistance is a major obstacle to the cisplatin-based therapy, and ultimately limits the life expectancy of the patient for a median survival time of approximately 1 y from the time of diagnosis. Like other anticancer agents, cisplatin induces a constitutive activation of the mitogen-activated protein (MAP) kinases, N-terminal-c-Jun kinase (JNK) and p38.Citation1
Insulin-like growth factor-binding protein 7 (IGFBP7) is one of the 16 IGFBP superfamily members, a large group of secreted proteins.Citation2 IGFBP7 regulates various cellular processes such as cell proliferation, cell adhesion, cellular senescence, differentiation, and angiogenesis. IGFBP7 is a downstream target of p53,Citation3 and loss of IGFBP7 is a critical step in the development of human tumors.Citation4–Citation7 IGFBP7 acts through autocrine/paracrine pathways to inhibit BRAF-MEK-Erk signaling to induce senescence or apoptosis.Citation4 In BRAF-positive human primary melanoma, IGFBP7 is epigenetically silenced, and restoration of IGFBP7 function by the addition of recombinant IGFBP7 (rIGFBP7) induces cell growth inhibition and apoptosis.Citation4,Citation8 In human metastatic melanomas, IGFBP7 is also epigenetically silenced at an even higher frequency than that found in primary melanomas.Citation5 Systemic administration of rIGFBP7 in mouse xenografts suppresses the growth of BRAF-positive primary and metastatic melanoma,Citation4 indicating that rIGFBP7 can function as an anticancer agent for human malignancy. IGFBP7 depletion renders cells more resistant to apoptosis,Citation9 and IGFBP7 alters sensitivity to interferon-based anticancer therapy;Citation10 however, few studies on IGFBP7 in cisplatin resistance have been reported to date.
Mitogen-activated protein kinase (MAPK) phosphatases (MKPs) are dual specificity phosphatases that negatively regulate MAPK activity by dephosphorylating the essential threonine and tyrosine residues in the activation loop.Citation11,Citation12 The different MKPs have distinct substrate specificities enabling the cell to control different MAPK pathways.Citation12 Repression of MKP1, a negative regulator of JNK/SAPK, increases sensitivity of NSCLC cells to cisplatin,Citation13,Citation14 and dephosphorylation and inactivation of JNK by MKP1 results in protection against cisplatin-induced apoptosis.Citation1 MKP3, a negative regulator of Erk, increases resistance to tamoxifen treatment in breast cancer,Citation15 but little is known about MKP3 in cisplatin resistance in lung cancer. Both MKP1 and MKP3 inhibit transcriptional activation of c-Jun, the principal physiological substrate of JNK.Citation16
In this study, we investigated the role of IGFBP7 in cisplatin-resistance. The expression of IGFBP7 decreased in cisplatin-resistant human cancer cell lines, and IGFBP7 gene knock-down increased cellular resistance to cisplatin. In contrast to IGFBP7, the expression of MKP3 increased in a cisplatin-resistant NSCLC cell line, and MKP3 gene knock-down increased IGFBP7 expression. These findings will contribute to our understanding of the molecular mechanism responsible for tumor suppressive function of IGFBP7 in cisplatin-resistant human lung cancer.
Results
IGFBP7 is downregulated in primary and metastatic lung cancers.
To compare IGFBP7 expression between patients with lung cancer (LT) and non-cancer patients (NN), we assessed transcriptional level of IGFBP7 by qRT-PCR. At a cut-off set at relative expression value of 1, the level of IGFBP7 displaying over the cut-off was observed in 8 of 9 NN (88%) and in 5 of 14 LT (35%) (p = 0.029, Fisher's exact test) (). We then examined IGFBP7 level in cDNA prepared from tumor (PT) and corresponding normal tissues (PN) of individual lung cancer patients. We observed downregulation of IGFBP7 in 4 of 5 cases of lung cancer patients compared with their matched PN (), indicating a specific decrease of IGFBP7 transcription in lung cancer. To further investigate IGFBP7 level in advanced lung cancer, we examined IGFBP7 level in primary and metastatic tissue cDNA prepared from a lung cancer patient. We found a 4-fold decrease of IGFBP7 level in metastatic lung cancer compared with primary cancer (p = 0.047) (). These results indicate that alteration of IGFBP7 expression might be involved in advanced lung cancer progression.
Downregulation of IGFBP7 is associated with cisplatin resistance.
To investigate the expression level of IGFBP7 in cisplatin resistance, we performed qRT-PCR in nine pairs of parental cisplatin-sensitive (CP-S) and isogenic, cisplatin-resistant (CP-R) cell lines derived from human head and neck, cervix, ovary, and lung cancers. Although no correlation was observed in 2−ΔCt values (ΔCt = Ct,IGFBP7 − Ct,β-actin) between two groups of cell lines (p = 0.798, Pearson correlation coefficients), a significant decrease of IGFBP7 level was observed between seven out of nine pairs of CP-R and CP-S cells when a relative IGFBP7 expression (fold-change) calculated by ΔΔCt methods (2−ΔΔCt) was compared between each pair of CP-S and CP-R cells (). To investigate whether IGFBP7 level was modulated by cisplatin treatment, we treated cells derived from NSCLC (PC-9, PC-9/CP, PC-14 and PC-14/CP) and small cell lung cancer (SCLC) (SBC-3 and SBC-3/CP) with cisplatin at approximate IC50 of each parental cells (2 µg/ml for PC-9, 5 µg/ml for PC-14 and 4 µg/ml for SBC-3) (). Cisplatin significantly increased IGFBP7 level in all sensitive lung cancer cell lines tested, but not in the cisplatin-resistance NSCLC cells (), indicating depletion of IGFBP7 in the NSCLC cells with acquired resistance to cisplatin. However, no significant difference of IGFBP7 level between SBC-3 and SBC-3/CP cells was observed. In addition, increase of IGFBP7 by cisplatin was observed in SBC-3/CP cells, indicating that IGFBP7 is not depleted in the SCLC cell line by cisplatin.
IGFBP7 knock-down renders cells resistance to cisplatin.
To examine the role of IGFBP7 in cisplatin resistance, we transfected the PC-9 and PC-14 cells with control or IGFBP7 siRNA (10 nM) for 24 h, and then exposed cells to increasing concentrations of cisplatin (0∼10 µg/ml) for 48 h. IGFBP7 knockdown itself had little effect on the cell growth (data not shown), but shifted the cell survival curve from the left to the right (), indicating that downregulation of IGFBP7 confers cellular resistance to cisplatin. In addition, we examined the cellular growth after cisplatin treatment for 3 d. We observed a significant increase of cell growth in the PC-9 and PC-14 cells with IGFBP7 knock-down ( and D). To further examine the effect of IGFBP7 on cisplatin resistance, we treated the PC-9/CP and PC-14/CP cells with rIGFBP7 with cisplatin (0∼50 µg/ml). rIGFBP7 alone (100 ng/ml) had a negligible effect on cell growth in both cell lines (Fig S2), but clearly enhanced drug sensitivity to cisplatin at the concentration of 50 µg/ml ( and S2).
IGFBP7 and MKP3 are inversely regulated in cisplatin resistant cell lines.
Basal expression of IGFBP7 protein was clearly observed in the PC-14 () and PC-9 cells (data not shown), but downregulated in the PC-14/CP () and PC-9/CP cells (data not shown). Increased IGFBP7 protein level after cisplatin treatment in the PC-14 cells but not in the PC-14/CP cells was also observed (). The basal expression of MKP3 protein in the PC-14 cells was clearly observed, but that of MKP1 was not. Both MKP1 and MKP3 levels increased in the PC-14/CP cells compared with those in the PC-14 cells, but under cisplatin treatment, MKP1 and MKP3 were regulated in a different way. MKP1 was increased in the PC-14 cells and decreased in the PC-14/CP cells by cisplatin, whereas MKP3 was decreased in the PC-14 cells, and not changed in the PC-14/CP cells by cisplatin. Decreased MKP3 level by cisplatin was also observed in the PC-9 and SBC-3 cells (data not shown), indicating that IGFBP7 and MKP3 protein levels are inversely regulated in cisplatin resistance. In spite of the increased MKP3 expression, Erk phosphorylation in the PC-14/CP cells was higher than in the PC-14 cells. In addition, cisplatin treatment in the PC-14 cells increased phosphorylation of Erk, but not in the PC-14/CP cells, indicating that Erk may not be regulated by MKP3 in the PC-14/CP cells that acquired resistance to cisplatin.
Activation of Stat is found in a variety of human cancers, and high expression of Stat3 is associated with cisplatin resistance in certain types of human cancer.Citation17,Citation18 Transcriptional level of Stat3 increases in cisplatin-resistant human NSCLC cell lines,Citation19 suggesting that Stat3 mediates cisplatin resistance in NSCLC. However, in our results, the level of both Stat3Y705 phosphorylation that induces Stat3 activation (dimerization, nuclear translocation, and DNA binding)Citation20,Citation21 and total Stat3 level in the PC-14/CP cells were downregulated compared with those in the PC-14 cells. Moreover, cisplatin decreased Stat3Y705 phosphorylation in both cell lines, implicating that cisplatin resistance at least in the PC-14/CP cells may involve inactivation of Stat3.
MKP3 knock-down increases IGFBP7 expression.
The inverse regulation of MKP3 and IGFBP7 protein in cisplatin resistance prompted us to investigate whether MKP3 regulates IGFBP7 expression. We found that MKP3 knock-down increased both IGFBP7 mRNA and protein levels in the PC-14/CP cells ( and C), indicating that IGFBP7 may be directly or indirectly regulated by MKP3. The IGFBP7 promoter contains a consensus binding site for the dimeric AP-1 (c-Jun/c-Fos) which stimulates IGFBP7 transcription.Citation4 Transcriptional activity of c-Jun is regulated by phosphorylation at Ser63 and Ser73 of c-Jun.Citation16 Significantly, c-Jun is activated through RAF-MEK-Erk signalingCitation22 as IGFBP7,Citation4 and physically interacts with Stat3 and binds to the AP-1 site together.Citation23 In our results, MKP3 depletion increased Stat3Y705 and c-JunS73 phosphorylation (), suggesting that the interplay of Stat3 and c-Jun on the AP-1 site may be responsible for the increase of IGFBP7 transcription. In contrast, Erk phosphorylation was not affected by MKP3 depletion, confirming that Erk may not be under the control of MKP3 in the PC-14/CP cells.
IGFBP7 and MKP3 are inversely regulated in primary lung xenografts.
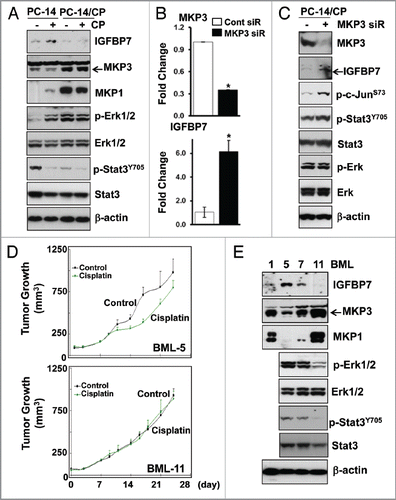
Directly implanted human low passage xenografts have been shown to retain key features of the original tumor including drug sensitivity, and accurately represent the heterogeneity of the disease.Citation24 To investigate whether IGFBP7 downregulation in the cisplatin-resistant cells could be validated in tumor samples from patients, we initially tested the response to cisplatin of the four patient-derived lung xenografts (BML-1, BML-5, BML-7 and BML-11) that were directly xenografted into nude mice. While tumor size of the BML-1, -7 and -11 under cisplatin treatment (1.5 mg/kg, i.p., for 3∼4 weeks) were bigger than or similar to that of the control under no treatment (), tumors of the BML-5 was relatively smaller than the control (tumor growth inhibition, 22%), indicating that the BML-5 responds to cisplatin with moderate sensitivity. Then, we determined the expression of IGFBP7, MKP1 and MKP3 in protein lysates extracted from those BMLs. Among four BMLs, IGFBP7 level was highest but MKP1 and MKP3 were lowest in the BML-5 (). In addition, an inverse expression of IGFBP7 and MKP3, but not MKP1, was clearly observed in the lung xenografts. Among BML-5, -7 and -11, the highest Erk and Stat3 phosphorylation were also observed in the BML-5, implicating that Erk and Stat3 are inactivated in intrinsic resistance to cisplatin.
Discussion
In this study, we showed that loss of IGFBP7 was associated with chemo-resistance to cisplatin in human cancer cell lines and lung xenografts. To the best of our knowledge, this is the first study to explore the alterations of IGFBP7 in cisplatin resistance. A previous study reported decreased expression of IGFBP7 in most cancer cell lines and over 46% primary lung tumor tested,Citation25 which examined the IGFBP7 level in lung cancer cell lines and tissue. In our study, we included normal tissue derived from patients without cancer (NN) and matched tumor (PT) and normal (PN) lung tissues in addition to lung tumor (LT). We determined IGFBP7 transcription by qRT-PCR, and found that IGFBP7 expression decreased in human primary and metastatic lung cancer compared with normal lung tissues. We also observed downregulation of IGFBP7 in the isogenic, cisplatin-resistant NSCLC cells compared with the parental sensitive cells. Although information about whether lung cancer patients received chemotherapy is not available, we could compare average values of 2−ΔCt (ΔCt = Ct,IGFBP7 − Ct,β-actin) in each group of tissues with those in lung CP-S and CP-R cell lines. The average level of IGFBP7 in NN was highest among groups, and the average IGFBP7 levels of primary tumors (LT and PT) and CP-S cell lines were higher than that of CP-R cell lines (Fig. S1). Therefore, downregulation of IGFBP7 may be involved in cisplatin-resistance and can serve as a biomarker for aggressive lung behavior, which needs our further study with a large numbers of tissue cohorts.
IGFBP7 is inactivated by DNA hypermethylation in human lung cancer.Citation25 We recently reported that methylation and transcriptional silencing of certain genes are associated with the development of cisplatin resistance.Citation26 We thus analyzed the methylation status of IGFBP7 in the parental and cisplatin-resistant cells derived from ovarian and lung cancer by bisulfite-sequencing analysis. We examined a CpG island region near the transcription start site (TSS) in the IGFBP7 promoter. The IGFBP7 promoter region was densely methylated in all cell lines tested (data not shown), indicating that DNA methylation in the IGFBP7 promoter, at least in the region near the TSS, is not involved in the decrease of IGFBP7 expression during development of cisplatin resistance.
In our search in literature for proteins that could be differentially expressed by IGFBP7, MKP3 attracted our attention; overexpression of MKP3 increases tumor growth and resistance to cisplatin-mediated cell death in human glioblastomas.Citation27 MKP3 is a cytoplasmic dual-specificity phosphatase 6 (DUSP6) specific for the MAP kinases Erk1/2, and the MEK-Erk axis exerts a negative feedback control on its own signaling through regulation of MKP3.Citation28 The MEK-Erk pathway plays a pivotal role in various cellular responses, including cellular growth, differentiation, survival and motility. Constitutive activation of the Erk pathway has been linked to the development and progression of human cancers. IGFBP7 is also regulated through MEK-Erk signaling and inhibits the signaling to induce senescence or apoptosis.Citation4 In our results, MKP3 and IGFBP7 proteins were inversely regulated in the NSCLC cell lines and lung xenografts, and MKP3 depletion increased the level of IGFBP7 protein. An inverse regulation of IGFBP7 and MKP3 in the mRNA level was observed between PC-14 and PC-14/CP cell lines, which was statistically significant (data not shown). Although no significance was observed, similar trends were also observed in other CP-S and CP-R cell lines (data not shown). In addition, knock-down of IGFBP7 gene in the PC-14 cells also increased transcriptional level of MKP3 (data not shown). These results suggest that MKP3 and IGFBP7 reciprocally regulate each other during the development of cisplatin resistance in NSCLC. The involvement of Erk pathway in the reciprocal regulation between MKP3 and IGFBP7 will be investigated in our future study. Moreover, decreased Stat3 activation in a cisplatin-resistant NSCLC cell line and xenografts in our results challenges previous reports that high expression of Stat3 is associated with cisplatin resistance in human cancer,Citation17,Citation18 which will be also clarified.
In our study, loss of IGFBP7 expression had a functional role in cisplatin resistance in human lung cancer, which may represent a possible basis for therapeutic strategies. Further studies are warranted to demonstrate an association of IGFBP7 expression in cisplatin resistance in a large cohort, which will lead to the development of IGFBP7 as a novel biomarker for cisplatin-resistant, human NSCLC. Elaboration of the biological role and the molecular mechanism of IGFBP7 in cisplatin resistance will also result in identification of IGFBP7 as a chemo-sensitizing agent in cisplatin resistant human cancer, which will benefit research on cisplatin resistance.
Materials and Methods
Cell lines and tissues.
cDNAs of primary and metastatic cancer tissue derived from a patient with lung cancer were purchased from BioChain Institute, Inc. (C8235152-p.m.). cDNA derived from patients with lung cancer (n = 14) and patients without cancer (n = 9) were described previously.Citation29 The human squamous carcinoma cell lines SCC-25 and its derivative SCC-25/CP were described previously.Citation26,Citation30 The human epidermoid carcinoma cell line KB-3-1, a subclone of the human HeLa cervical adenocarcinoma cell line, and two clones of cisplatin-resistant KB-CP cells were described previously.Citation26,Citation31 Three human ovarian cancer cell lines resistant to cisplatin (IGROV1/CP, A2780/CP and 2008/CP) and their parental cell lines were kindly provided by Dr. Stephen B. Howell (Moores UCSD Cancer Center). Three human lung cancer cell lines resistant to cisplatin (PC-9/CDDP, PC-14/CDDP and SBC-3/CDDP) and their parental cell lines were generously provided by Dr. Fumiaki Koizumi (National Cancer Center Research Institute, Japan). Human lung xenografts were provided by Champions Biotechnologies. This study was approved by the Institutional Review Board of the Johns Hopkins University.
Quantitative real-time PCR (qRT-PCR) analysis.
One microliter of each cDNA was used for real-time RT-PCR using QuantiFast SYBR Green PCR Kit from Qiagen (204052), and PCR condition was followed as described previously.Citation32 Results were normalized to the β-actin level using the comparative Ct method, and fold changes of mRNA levels were calculated by the equation 2−ΔΔCt (ΔΔCt methods).Citation32 IGFBP7 primers were 5′-CACTGGTGCCCAGGTGTACT-3′ (forward) and 5′-TTGGATGCATGGCACTCATA-3′ (reverse) and β-actin primers were 5′-TGGCACCACACCTTCTACAATGAGC-3′ (forward) and 5′-GCACAGCTTCTCCTTAATGTCACGC-3′ (reverse).
Knockdown of IGFBP7 and MKP3, and in vitro drug sensitivity assay.
ON-TARGETplus SMARTpool control small interfering RNA (siRNA), and siRNAs targeting IGFBP7 (L-008675-00-0005) or MKP3 (L-003964-00-0005) were purchased from Dharmacon. Cells were plated at a density of 3,000 per well in 96-well plates. The following day, cells were transfected with siRNAs as manufacturer's instructions and incubated for 24 h. Cells were then treated with increasing concentrations of cisplatin for 48 h after the removal of transfection media, and cellular sensitivity to cisplatin was examined by the MTT assay.Citation32 In order to plot cell growth curve, cisplatin was treated every day for 3 d. The results were expressed as a percentage of MTT reduction in samples compared with control.Citation32
Protein gel blot analyses.
Whole cell lysates extracted in RIPA buffer were separated on 4–12% gradient SDS-PAGE and transferred to nitrocellulose membrane. The blots were incubated with specific antibodies for each protein for 2 h at room temperature or 4°C overnight. After antibody washing, the blots were reacted with their respective secondary antibody and detected with enhanced chemiluminescence reagents (Amersham) according to the supplier's protocol. All antibodies were purchased from Cell Signaling except for the anti-IGFBP7 (Abcam, ab51392), anti-Erk (sc-135900) and anti-p-Erk (sc-81492) antibodies (Santa Cruz Biotechnologies)
Statistical analysis.
All data were statistically analyzed using the two-tailed t-test from at least three independent experiments for comparison with the control group. Data were expressed as the mean ± SD p values less than 0.05 were considered significant. All statistical analyses were conducted using STATA Version 9 (STATA Inc.).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Abbreviations
| IGFBP7 | = | insulin-like growth factor-binding protein 7 |
| MKP | = | mitogen-activated protein kinase phosphatases |
| NSCLC | = | non-small cell lung cancer |
Figures and Tables
Figure 1 IGFBP7 expression in normal and tumor lung. IGFBP7 expression was examined in cDNAs prepared from patients with lung cancer (LT) and without cancer (NN) (A), in pairs of normal and tumor cDNAs from five lung cancer patients (B), and in primary and metastatic lung tumor cDNAs from a patient with metastatic lung cancer (C) by qRT-PCR. 2^-(), expression of IGFBP7 relative to β-actin calculated based on the threshold cycle (Ct) as 2−ΔCt (ΔCt = Ct,IGFBP7 − Ct,β-actin). Experiments were done in duplicate, and values indicate means ± SD *p < 0.05 in t-test.
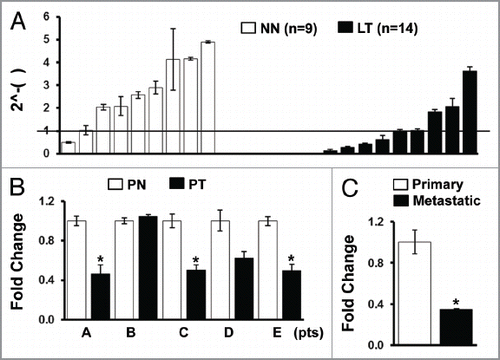
Figure 2 Downregulation of IGFBP7 in cisplatin resistant cell lines. (A) Expression of IGFBP7 was compared between the parental, cisplatin-sensitive (CP-S) and isogenic, cisplatin-resistant (CP-R) cell lines by qRT-PCR. H and N, cells derived from head and neck cancer. (B) Cells were treated with cisplatin for 48 h and qRT-PCR was performed. NSCLC, non-small cell lung cancer; SCLC, small cell lung cancer. Experiments were done in duplicate, and values indicate means ± SD *p < 0.05 in t-test.
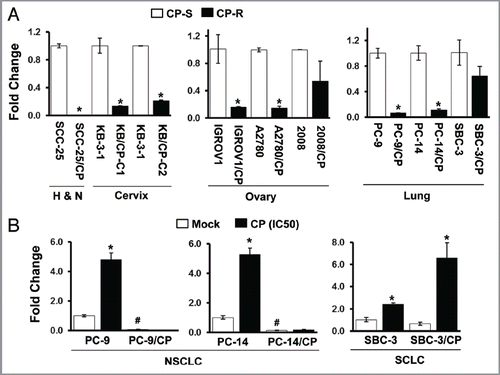
Figure 3 Increased resistance to cisplatin by IGFBP7 knock-down in the parental cell lines. (A) PC-9 and PC-14 cells were transfected with non-targeting control (Cont siR) or IGFBP7 siRNA (IGFBP7 siR) for 24 h, and then treated with cisplatin for 48 h. The MTT assay was performed to assess cellular sensitivity to cisplatin. (B) Cells were treated with cisplatin at IC50 for 3 d after transfection with non-targeting control or IGFBP7 siRNA. Cell growth was assessed by the MTT assay. (C) IGFBP7 knock-down was confirmed by qRT-PCR. Experiments were done in duplicate, and values indicate means ± SD *p < 0.05 in t-test. Pictures of PC-14 (D), PC-9/CP and PC-14/CP cells (E) on tissue culture plates were taken after DMSO was added to dissolve MTT crystals. Growth curves of PC-9/CP and PC-14/CP cells after rIGFBP7 treatment is shown in Figure S2.
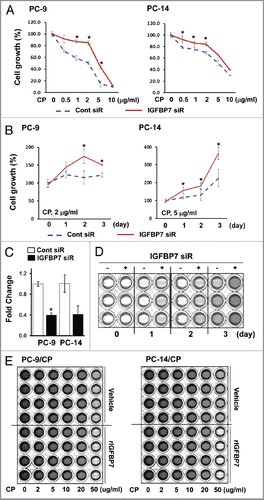
Figure 4 Decreased IGFBP7 expression in cisplatin-resistant NSCLC. (A) Whole protein lysates were extracted from PC-14 and PC-14/CP cells with or without treatment with cisplatin (5 µg/ml), and immunoblot analysis was performed with the indicated antibodies. The PC-14/CP cells were transfected with non-targeting control or MKP siRNA for 48 h, and qRT-PCR (B) and immunoblot (C) analyses were performed. Experiments were done in duplicate, and values indicate means ± SD *p < 0.05 in t-test. (D) Tumor growth was monitored for 25 d after cisplatin injection in nude mice bearing the human NSCLC xenografts (1.5 mg/Kg, i.p.), and tumor volume was calculated. (E) Whole protein lysates were extracted from human lung xenografts (BML-1, -5, -7 and -11), and immunoblot analysis was performed. Erk and Stat3 levels were not examined in BML-1 due to insufficient quantity of protein.
Additional material
Download Zip (165.8 KB)Acknowledgments
This study was supported by the Flight Attendant Medical Research Institute Young Clinical Scientist Award, and in part by National Cancer Institute grant (U01-CA84986).
References
- Sánchez-Pérez I, Martínez-Gomariz M, Williams D, Keyse SM, Perona R. CL100/MKP-1 modulates JNK activation and apoptosis in response to cisplatin. Oncogene 2000; 19:5142 - 5152; PMID: 11064451; http://dx.doi.org/10.1038/sj.onc.1203887
- Burger AM, Leyland-Jones B, Banerjee K, Spyropoulos DD, Seth AK. Essential roles of IGFBP-3 and IGFBP-rP1 in breast cancer. Eur J Cancer 2005; 41:1515 - 1527; PMID: 15979304; http://dx.doi.org/10.1016/j.ejca.2005.04.023
- Suzuki H, Igarashi S, Nojima M, Maruyama R, Yamamoto E, Kai M, et al. IGFBP7 is a p53 responsive gene specifically silenced in colorectal cancer with CpG island methylator phenotype. Carcinogenesis 2010; 31:342 - 349; PMID: 19638426; http://dx.doi.org/10.1093/carcin/bgp179
- Wajapeyee N, Serra RW, Zhu X, Mahalingam M, Green MR. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell 2008; 132:363 - 374; PMID: 18267069; http://dx.doi.org/10.1016/j.cell.2007.12.032
- Wajapeyee N, Kapoor V, Mahalingam M, Green MR. Efficacy of IGFBP7 for treatment of metastatic melanoma and other cancers in mouse models and human cell lines. Mol Cancer Ther 2009; 8:3009 - 3014; PMID: 19861408; http://dx.doi.org/10.1158/1535-7163.MCT-09-0470
- Hinoue T, Weisenberger DJ, Pan F, Campan M, Kim M, Young J, et al. Analysis of the association between CIMP and BRAF in colorectal cancer by DNA methylation profiling. PLoS ONE 2009; 4:e8357; PMID: 20027224; http://dx.doi.org/10.1371/journal.pone.0008357
- Tomimaru Y, Eguchi H, Wada H, Kobayashi S, Marubashi S, Tanemura M, et al. IGFBP7 down-regulation is associated with tumor progression and clinical outcome in hepatocellular carcinoma. Int J Cancer 2011; In press PMID: 21328580; http://dx.doi.org/10.1002/ijc.25994
- Nousbeck J, Sarig O, Avidan N, Indelman M, Bergman R, Ramon M, et al. Insulin-like growth factor-binding protein 7 regulates keratinocyte proliferation, differentiation and apoptosis. J Invest Dermatol 2010; 130:378 - 387; PMID: 19710688; http://dx.doi.org/10.1038/jid.2009.265
- Smith P, Nicholson LJ, Syed N, Payne A, Hiller L, Garrone O, et al. Epigenetic inactivation implies independent functions for insulin-like growth factor binding protein (IGFBP)-related protein 1 and the related IGFBPL1 in inhibiting breast cancer phenotypes. Clin Cancer Res 2007; 13:4061 - 4068; PMID: 17634530; http://dx.doi.org/10.1158/1078-0432.CCR-06-3052
- Tomimaru Y, Eguchi H, Wada H, Noda T, Murakami M, Kobayashi S, et al. Insulin-like growth factor-binding protein 7 alters the sensitivity to interferon-based anticancer therapy in hepatocellular carcinoma cells. Br J Cancer 2010; 102:1483 - 1490; PMID: 20407444; http://dx.doi.org/10.1038/sj.bjc.6605669
- Camps M, Nichols A, Arkinstall S. Dual specificity phosphatases: a gene family for control of MAP kinase function. FASEB J 2000; 14:6 - 16; PMID: 10627275
- Farooq A, Zhou MM. Structure and regulation of MAPK phosphatases. Cell Signal 2004; 16:769 - 779; PMID: 15115656; http://dx.doi.org/10.1016/j.cellsig.2003.12.008
- Chattopadhyay S, Machado-Pinilla R, Manguan-García C, Belda-Iniesta C, Moratilla C, Cejas P, et al. MKP1/CL100 controls tumor growth and sensitivity to cisplatin in non-small-cell lung cancer. Oncogene 2006; 25:3335 - 3345; PMID: 16462770; http://dx.doi.org/10.1038/sj.onc.1209364
- Cortes-Sempere M, Chattopadhyay S, Rovira A, Rodriguez-Fanjul V, Belda-Iniesta C, Tapia M, et al. MKP1 repression is required for the chemosensitizing effects of NF-kappaB and PI3K inhibitors to cisplatin in non-small cell lung cancer. Cancer Lett 2009; 286:206 - 216; PMID: 19553005; http://dx.doi.org/10.1016/j.canlet.2009.05.029
- Cui Y, Parra I, Zhang M, Hilsenbeck SG, Tsimelzon A, Furukawa T, et al. Elevated expression of mitogen-activated protein kinase phosphatase 3 in breast tumors: a mechanism of tamoxifen resistance. Cancer Res 2006; 66:5950 - 5959; PMID: 16740736; http://dx.doi.org/10.1158/0008-5472.CAN-05-3243
- Kyriakis JM, Banerjee P, Nikolakaki E, Dai T, Rubie EA, Ahmad MF, et al. The stress-activated protein kinase subfamily of c-Jun kinases. Nature 1994; 369:156 - 160; PMID: 8177321; http://dx.doi.org/10.1038/369156a0
- Gu F, Ma Y, Zhang Z, Zhao J, Kobayashi H, Zhang L, et al. Expression of Stat3 and Notch1 is associated with cisplatin resistance in head and neck squamous cell carcinoma. Oncol Rep 2010; 23:671 - 676; PMID: 20127005
- Selvendiran K, Bratasz A, Kuppusamy ML, Tazi MF, Rivera BK, Kuppusamy P. Hypoxia induces chemoresistance in ovarian cancer cells by activation of signal transducer and activator of transcription 3. Int J Cancer 2009; 125:2198 - 2204; PMID: 19623660; http://dx.doi.org/10.1002/ijc.24601
- Ikuta K, Takemura K, Kihara M, Nishimura M, Ueda N, Naito S, et al. Overexpression of constitutive signal transducer and activator of transcription 3 mRNA in cisplatin-resistant human non-small cell lung cancer cells. Oncol Rep 2005; 13:217 - 222; PMID: 15643501
- Darnell JE Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994; 264:1415 - 1421; PMID: 8197455; http://dx.doi.org/10.1126/science.8197455
- Ihle JN. Cytokine receptor signalling. Nature 1995; 377:591 - 594; PMID: 7566171; http://dx.doi.org/10.1038/377591a0
- Leppä S, Saffrich R, Ansorge W, Bohmann D. Differential regulation of c-Jun by ERK and JNK during PC12 cell differentiation. EMBO J 1998; 17:4404 - 4413; PMID: 9687508; http://dx.doi.org/10.1093/emboj/17.15.4404
- Shi M, Liu D, Duan H, Han C, Wei B, Qian L, et al. Catecholamine up-regulates MMP-7 expression by activating AP-1 and STAT3 in gastric cancer. Mol Cancer 2010; 9:269; PMID: 20939893
- Gorelik B, Ziv I, Shohat R, Wick M, Hankins WD, Sidransky D, et al. Efficacy of weekly docetaxel and bevacizumab in mesenchymal chondrosarcoma: a new theranostic method combining xenografted biopsies with a mathematical model. Cancer Res 2008; 68:9033 - 9040; PMID: 18974149; http://dx.doi.org/10.1158/0008-5472.CAN-08-1723
- Chen Y, Cui T, Knösel T, Yang L, Zöller K, Petersen I. IGFBP7 is a p53 target gene inactivated in human lung cancer by DNA hypermethylation. Lung Cancer 2011; 73:38 - 44; PMID: 21095038; http://dx.doi.org/10.1016/j.lungcan.2010.10.015
- Chang X, Monitto CL, Demokan S, Kim MS, Chang SS, Zhong X, et al. Identification of hypermethylated genes associated with cisplatin resistance in human cancers. Cancer Res 2010; 70:2870 - 2879; PMID: 20215521; http://dx.doi.org/10.1158/0008-5472.CAN-09-3427
- Messina S, Frati L, Leonetti C, Zuchegna C, Di Zazzo E, Calogero A, et al. Dual-specificity phosphatase DUSP6 has tumor-promoting properties in human glioblastomas. Oncogene 2011; 30:3813 - 3820; PMID: 21499306; http://dx.doi.org/10.1038/onc.2011.99
- Bermudez O, Jouandin P, Rottier J, Bourcier C, Pagès G, Gimond C. Post-transcriptional regulation of the DUSP6/MKP-3 phosphatase by MEK/ERK signaling and hypoxia. J Cell Physiol 2011; 226:276 - 284; PMID: 20665674; http://dx.doi.org/10.1002/jcp.22339
- Park JC, Chae YK, Son CH, Kim MS, Lee J, Ostrow K, et al. Epigenetic silencing of human T (brachyury homologue) gene in non-small-cell lung cancer. Biochem Biophys Res Commun 2008; 365:221 - 226; PMID: 17980147; http://dx.doi.org/10.1016/j.bbrc.2007.10.144
- Caney C, Bulmer JT, Singh G, Lukka H, Rainbow AJ. Pre-exposure of human squamous carcinoma cells to low-doses of gamma-rays leads to an increased resistance to subsequent low-dose cisplatin treatment. Int J Radiat Biol 1999; 75:963 - 972; PMID: 10465362; http://dx.doi.org/10.1080/095530099139728
- Shen DW, Akiyama S, Schoenlein P, Pastan I, Gottesman MM. Characterisation of high-level cisplatin-resistant cell lines established from a human hepatoma cell line and human KB adenocarcinoma cells: cross-resistance and protein changes. Br J Cancer 1995; 71:676 - 683; PMID: 7710928; http://dx.doi.org/10.1038/bjc.1995.134
- Kim MS, Chang X, LeBron C, Nagpal JK, Lee J, Huang Y, et al. Neurofilament heavy polypeptide regulates the Akt-beta-catenin pathway in human esophageal squamous cell carcinoma. PLoS ONE 2010; 5:e9003; PMID: 20140245; http://dx.doi.org/10.1371/journal.pone.0009003