Abstract
There currently are no tests available for early diagnosis or for the identification of patients at risk for development of pancreatic cancer. We report the discovery of single nucleotide polymorphism (SNP) in the cholecystokinin B receptor (CCKBR) gene predicts survival and risk of pancreatic cancer. Growth of human pancreatic cancer is stimulated by gastrin through the CCKBR and an alternatively spliced isoform of the CCKBR gene called CCKCR. One hundred and ten surgically resected benign and malignant pancreatic tissues as well as normal pancreas were prospectively evaluated for CCKBR genotype and protein expression. Analysis demonstrated the expression of the spliced isoform, CCKCR, was associated with a (SNP) (C > A) at position 32 of the intron 4 (IVS 4) of the CCKBR gene. Since the SNP is within an intron, it has not previously been identified in the GWAS studies. Only patients with the A/A or A/C genotypes, exhibited immunoreactivity to a selective CCKCR antibody. Survival among pancreatic cancer patients with the A-SNP was significantly shorter (p = 0.0001, hazard ratio = 3.63) compared with individuals with C/C genotype. Other variables such as surgical margins, lymph node status, histologic grade or adjuvant chemotherapy were not associated with survival. Furthermore, having one or two of the A-alleles was found to increase the risk of pancreatic adenocarcinoma by 174% (p = 0.0192) compared with the C/C wild type. Cancer cells transfected to overexpress the CCKCR demonstrated increased proliferation over controls. Genetic screening for this SNP may aid in early detection of pancreatic cancer in high risk subjects.
Introduction
Pancreatic cancer is a fatal malignancy estimated to have occurred in approximately 42,470 Americans last year and ranks as the fourth most common cause of cancer-related mortality in the United States.Citation1 With a 5 y survival rate of less than 1%, pancreatic cancer survival remains the lowest of all malignancies, and median survival is 3–6 mo with only 10% of patients surviving 2 y.Citation1,Citation2 Reasons for the aggressive behavior of pancreatic cancer are related to the inability to diagnose the disease in early stages and poor understanding of the factors involved in regulating growth of pancreatic cancer. Pancreatic cancer is most commonly diagnosed in advanced stages, and therefore, chemotherapy remains the major treatment modality for this disease.Citation3 Because of the overall poor response to chemotherapeutic agents, researchers have been evaluating novel approaches to treat pancreatic cancer by targeting specific markers.Citation4
Unlike many other malignancies that have serological markers for earlier cancer detection (prostate) or radiographic tests for screening large populations for cancer (breast cancer), there are no proven early screening tests that are useful for pancreatic cancer. The current standard serum marker for pancreatic cancer, CA19-9, lacks sufficient sensitivity (50–75%) and specificity (83%).Citation5 In fact, other serum markers including HCG β and CA72-4 are stronger prognostic factors in pancreatic cancer than either CA19-9 or CEACitation6 Other secreted proteins including macrophage inhibitory cytokine-1 (MIC-1) 7 and osteopontinCitation7,Citation8 are being evaluated; however, MIC-1 was not found to be significantly better than CA19-9 in distinguishing those with chronic pancreatitis from those with pancreatic cancer. Over the past several years, investigators have turned to molecular markers as potential tools for early screening of pancreatic cancer and recent reviews have been published.Citation9,Citation10 Genetic markers have been more thoroughly investigated in pancreatic juices than in serum. Some of these markers include K-ras, p53, and mitochondrial mutations, as well as DNA methylation alterations.Citation11 Although K-ras mutations are found in up to 90% of pancreatic adenocarcinomas, they are not specific for invasive pancreatic cancer.Citation12 In a recent review,Citation13 Korc describes the four major “driver mutations” in pancreatic cancer including the activation of K-ras and loss of function in P16/CDKN2A, TP53 and SMAD4/DPC4 genes.
Two classic G-protein coupled CCK receptors have been identified:Citation14–Citation17 CCKAR (or CCK1 receptor) and the CCKBR (or CCK2 receptor). Although both the CCKAR and CCKBR have been associated with pancreatic cancer,Citation18–Citation21 receptor antagonist studiesCitation18,Citation19 demonstrated that the receptor associated with pancreatic cancer growth is the CCKBR rather than the CCKAR. A third receptor isoform has been identified in pancreatic cancer cells, the CCKCR, that is a splice variant of the CCKBR.Citation22,Citation23 The CCKCR alternatively spliced variant receptor retains intron 4 (IVS 4) and adds 69 amino acids to the 3rd intracellular loop which is predicted to markedly alter the protein structure and presumably function. Interestingly, transcripts encoding CCKCR have also been described in a few human colon cancer surgical specimens, but were not detected in normal adjacent colon tissues from the same specimen.Citation24 One group suggestedCitation25 that alternative splicing of CCKBR to produce CCKCR transcripts in pancreatic cancer cells may be related to reduced activity of U2AF35 nuclear ribonucleoprotein particle splicing factor. In addition to the alternatively spliced CCKCR, investigators have identified other splice variants of the CCKBRCitation26,Citation27 and have termed these “the short and long CCK-B isoforms” depending on the presence or absence of five amino acids in the corresponding protein products.
It has been proposed that the alternatively spliced CCKCR is related to cell proliferationCitation28,Citation29 and may render the receptor constitutively active in the absence of ligand.Citation24 Some have also tried to target the CCKCR with radiolabeled CCK-8.Citation30 Unfortunately, all these studies were performed with Balb3T3 and HEK-293 cells transfected to overexpress the spliced variant receptor rather than in colon or pancreatic cancer cells or tissues. Since use of RT-PCR to detect the splice variant receptor requires differentiation between the genomic DNA and the cDNA that retains the 4th intron, accurate detection of the CCKCR in some studies may be problematic. Thus, analysis at the protein level is very likely to be a more direct and accurate measure of CCKCR expression than RT-PCR or qRT-PCR. Previously, we found CCKCR immunoreactivity in pancreatic cancer surgical specimens but not normal pancreas using a polyclonal antibody that had been raised to the unique 69-amino acid fragment.Citation22,Citation23,Citation31 The primary aim of this investigation was to evaluate the genetic basis for CCKCR expression and determine if its presence correlates with disease behavior or patient survival. The second aim of this investigation was to develop a consistent and reliable monoclonal antibody specific to the CCKCR, and use that antibody to determine if CCKCR could be used as a diagnostic tool for detection of pancreatic cancer.
Results
Tumor and normal pancreas CCKCR IVS 4 SNP genotyping.
DNA analysis of tissues from patients with pancreatic cancer demonstrated the splice variant to be associated with an IVS 4 +32 C > A SNP. This SNP corresponds to base pair 11700 of the Vega Chromosome Sequence (Chromosome 11: 6,280,966-6,293,357), rs1800843, a known SNP located within IVS 4 of the CCKBR gene. Allelic frequency was studied in 110 specimens including 39 normal pancreas donors, 51 patients with pancreatic ductal adenocarcinoma, and 20 subjects with other pancreatic diseases (). The histologic categories of the other pancreatic diseases included neuroendocrine tumors (n = 5), IPMN (n = 5), benign serous cyst adenomas (n = 6), pancreatitis (n = 2) and other (n = 2; one GIST tumor and one pseudo-papillary tumor). Genotype frequency in normal tissues and benign pancreatic lesions was comparable to that reported in the SNP database (www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs=1800843) for A and C alleles of the rs1800843 SNP. However, in pancreatic cancer samples, the frequency of the SNP A-allele was increased (0.25) over that found in normal (0.12) and benign disorders (0.10). The risk of adenocarcinoma was 2.74 times or 174% increased with one or two copies of the A-allele (p = 0.0192). Matching SNP genotypes were ascertained from non-tumor tissue DNA, which confirms that the SNP was not a result of genetic changes in the cancerous tissue. SNP genotyping of the receptor in cultured pancreatic cancer cells revealed that MIA PaCa-2 and PANC-1 cells expressed the A/C genotype while BxPC-3, SW-1990 and AsPC-1 have the C/C genotype.
Specificity and characterization of CCKCR monoclonal antibodies.
Monoclonal antibodies were developed and characterized, specific to the human CCKCR. Isotyping of these monoclonal antibodies revealed both to be IgG1 isotype antibodies. Protein gel blot analysis and immunofluorescent staining of transiently transfected HEK-293 cells or COS-1 cells demonstrated that the antibody has both selectivity and specificity for the human CCKCR. The specificity of the CCKCR antibody was confirmed by protein gel blotting (), where the monoclonal antibody recognized the CCKCR protein but not the CCKBR protein. Further confirmation of the antibody's specificity was shown by immunocytochemistry (). COS-1 cells that transiently expressed the CCKBR reacted with the anti-CCKBR antibody but did not react with the CCKCR specific antibody. The anti-CCKBR antibody, which recognizes an epitope common to both forms of the receptor, stained COS-1 cells which expressed either form of the receptor.
CCKCR cellular location.
A question was addressed regarding the CCKCR and whether the addition of 69 amino acids shifts the domain topology of this 7-membrane spanning receptor. To address this question, the CCKCR monoclonal antibody was used to determine sub-cellular localization of the additional amino acids in the CCKCR by confocal and electron microscopy. CCKCR immunoreactivity was only identified in cells that were permeabilized with Triton-X 100 () and not in nonpermeabilized cells (), indicating that the CCKCR-specific epitope is intracellular within the plasma membrane. The topology of the CCKCR was further verified using electron microscopy. Using the CCKCR monoclonal antibody and a gold-labeled secondary antibody, electron micrographic images showed that the CCKCR antibody bound to the intracellular region of the plasma membrane (). Secondary antibody alone showed no immunoreactivity ().
Detection of the CCKCR in human tissue specimens by immunohistochemistry.
Using the CCKCR monoclonal antibody, it was discovered that only tumors with the A allele (either the A/A or A/C genotype) possess immunoreactivity for the CCKCR (). This suggests that the presence of one or two A-SNP alleles is associated with the alternative splicing and retention of IVS 4 and the unique amino acids to which the antibody was generated. A representative tumor section from a patient with genotype A/A is shown in Figure 3A. Evidence of tumor invasion of nerve tissue is revealed by positive staining of the CCKCR monoclonal antibody. Immunoreactivity was also seen in patients with the A/C heterozygous genotype (). The surrounding stroma of samples from subjects with an A/A or A/C genotype was void of any immunoreactivity to this receptor suggesting that the alternatively spliced receptor is only present in the cancer epithelial cells from subjects with a A/A or A/C genotype. In contrast, surgical histologic sections from C/C genotype subjects () lacked immunoreactivity to the CCKCR. Normal human pancreatic tissue from A/A genotype subjects from tissue procured for transplantation did not react with the CCKCR antibody indicating the specificity of this receptor phenotype expression only in individuals with malignancy (). PANC-1 cells which were A/C genotype demonstrated CCKCR immunoreactivity (); however, the AsPC-1 cells which are C/C genotype did not stain with the CCKCR antibody (). Additionally, subjects with an A-allele and benign lesions, chronic pancreatitis, or neuroendocrine tumors of the pancreas lacked CCKCR immunoreactivity (data not shown). This demonstrates the specificity of the CCK-C receptor expression only in the presence of ductal adenocarcinoma.
The C/C genotype predicts prolonged survival.
Surgical reports were reviewed from 71 patients for tissue histology (benign pancreatic lesions vs. malignant ductal adenocarcinoma) and patient survival. All patients with benign or other pancreatic histology besides ductal adenocarcinoma (n = 20) were alive with the exception of one patient with a neuroendocrine tumor, C/C SNP genotype, who died at the age of 70 after surviving 61 mo. Of the 51 patients with pancreatic ductal adenocarcinoma who underwent surgical resection, 13 patients were still alive at the time of this report and 85% of these (n = 11) possessed the C/C genotype. Survival was much shorter in patients with A/C or A/A genotype compared with those with genotype C/C (p = 0.0001, ). Overall survival is 55% greater with the C/C genotype (n = 30) compared with those with the A/C genotype (n = 16), and 67% greater than those with the homozygous A/A (n = 5). These differences are significant from the C/C group but not statistically different from each other (i.e., A/C vs. A/A) due to the low number of A/A patients. Age at the time of diagnosis occurred slightly earlier in those with the A/A or A/C genotypes (66 vs. 70 y) suggesting perhaps a more aggressive tumor behavior; however, this was not statistically significant (p = 0.17).
Patient case records were reviewed for tumor characteristics (positive surgical margins, lymph node involvement with tumor and histologic grade of cancer) and patient demographics (gender, age, diabetes, smoking history, family history of pancreatic cancer and adjuvant chemotherapy) and these factors are shown in . The hazard rate in the presence of the A-SNP is estimated to be 3.63 times of the hazard rate of the C/C genotype (p = 0.0004) in Cox proportional hazards model. Positive lymph nodes at the time of resection elevated the hazard rate to be 2.03-fold of those with negative lymph nodes. After adjusting for lymph node status, the hazard ratio for the A/A or A/C genotypes remains basically the same at 3.6 (), which is substantially larger than 1. The positive lymph nodes are not as important as the presence of the A-SNP (p = 0.05 vs. p = 0.0005, hazard ratio 2.0 vs. 3.6) in their joint Cox model. Similar results for the effect of genotype were obtained after adjusting for other predictors (data not shown). Therefore, the difference between the two genotypes in survival is not due to the confounding effect with other predictors. These factors support the finding that the SNP genotype of the CCK receptor is the most important predictive factor for survival in patients undergoing resection for pancreatic cancer.
Expression of the CCKCR increases the growth of pancreatic cancer cells.
When the CCKCR was overexpressed in AsPC-1 pancreatic cancer cells (which are the C/C genotype) cell growth significantly increased using the MTS assay in vitro by up to 60% compared with wild-type and vector-transfected cells (). Further support of the growth advantage of cells with the CCKCR was demonstrated by increased BrdU uptake in cells overexpressing this receptor compared with controls without the receptor (). No significant change in either growth by the MTS assay or BrdU incorporation was seen in AsPC-1 cancer cells overexpressing the CCKBR receptor ().
Overexpression of mRNA for CCKBR and CCKCR was confirmed by qRT-PCR. CCKCR protein expression in cultured transfected pancreatic cancer cells was confirmed by flow cytometry () using the CCKCR monoclonal antibody.
Discussion
The current investigation involves the discovery of a genetic alteration in patients with pancreatic ductal adenocarcinoma that predicts risk and survival. The presence of the SNP was an independent factor that predicted outcome regardless of tumor histologic grade, surgical margins, adjuvant chemotherapy, and lymph node status in patients undergoing radical tumor excision. In fact, the presence of one or two A-alleles may increase the risk for development of pancreatic cancer significantly compared with the C/C genotype perhaps making this variation an important screening test for pancreatic cancer. The rs1800843 C > A SNP results in retention of IVS 4 in CCKBR transcripts that leads to the variant receptor. Since the rs1800843 SNP occurs within an intron, this genetic alteration has not been previously described as part of large Genome-Wide Association Studies (GWAS).Citation33,Citation34
Survival is correlated with the SNP-genotype in patients with pancreatic ductal adenocarcinoma. Few patients with the A/A SNP were identified in the tumors examined from surgical resection. This finding may suggest that those patients with A/A genotype have more aggressive disease and are not deemed to be candidates for surgical resection. However, since our genetic analysis of normal tissues also revealed a low frequency of the A/A genotype, we believe this finding supports that the A/A genotype is relatively infrequent in the general population and the C/C is the predominant form. Survival among subjects with benign lesions of the pancreas was not dependent on the presence of the SNP and immunohistochemical staining of the tissues obtained from subjects with the A-SNP who had benign pancreatic lesions were negative for the CCKCR. Our data suggests that the A-allele is associated with the protein expression of the variant receptor only in malignant tissue. Therefore, CCKCR staining may be a helpful tool to diagnose early cancers in high risk patients.
When the retained IVS 4 is translated, it lengthens the third internal loop of the CCKBR; an important site for intracellular regulation and GTP coupled processes. It is possible that intracellular signaling pathways may be altered by the additional amino acids. Carter and colleaguesCitation35 describe a PIK3 kinase tumor suppressor gene SNP, PIK3CG / R839C which has the second highest scoring predicted driver mutation among newly described set of genes identified as drivers in pancreatic cancer. Korc suggestedCitation13 that there may be a connection between mutations in PIK3CG and activation of the CCKBR and CCKCR since PI3Kγ transmits signals downstream from these G-protein coupled receptors.
In fact when HEK cells were transiently transfected with the CCKCR, it was suggested that the receptor became constitutively activeCitation24 even in the absence of the receptor's ligand, gastrin. Furthermore, the in vitro growth studies in this current investigation support the contention that cancer cells with this variant receptor have a more aggressive phenotype and growth advantage.
Because RT-PCR detection of the unspliced intron in CCKCR mRNA may be prone to false-positive results due to the presence of contaminating genomic DNA, we suggest that use of a monoclonal antibody directed toward the receptor protein is a more reliable and specific test. Imaging studies with the confocal and electron microscopes verify that the antibody recognized an epitope on the intracellular plasma membrane confirming topographic predictions for this variant receptor. Since the antibody binding site is intracellular, it would not be useful for imaging in live cells or targeting cancer cells with a humanized monoclonal antibody, which typically bind to extracellular proteins. However, intracellular receptor domains have been shown to be ideal targets for development of vaccinations rendering this unique receptor a potential target for a cancer vaccine.Citation36,Citation37 Perhaps vaccination of pancreatic cancer patients with the A/A or A/C genotype could theoretically improve the outcome.
Pre-mRNA splicing is a highly regulated process that occurs following gene transcription and before mRNA translation. This reaction is performed by spliceosomes: multicomponent ribonucleoprotein complexes containing five small nuclear ribonucleoproteins (snRNPs) and an abundant number of other proteins.Citation38 Specific alterations in splicing patterns have been found in association with cancers, many of which may play a functional role in growth, drug sensitivity, and metastasis of tumors. Alternative splicing also appears to affect other important processes in tumors such as altered intracellular signaling, apoptosis, and cell-cell or cell-matrix interactions.Citation39 Alternative mRNA splicing is often modulated by SR (serine-arginine) proteins, a family of proteins that regulate multiple aspects of mRNA splicing, export and translation.Citation40 Binding of SR proteins to mRNA recruits U1 and U2 snRNPs and promotes formation of splicing complexes.Citation41 Although SR proteins function in constitutive mRNA splicing, they can also activate alternative mRNA splicing. Among the best studied SR proteins is SRp55, which in humans is encoded by the SFRS6 gene. This SR protein, SRp55, binds to the mRNA consensus sequence [U G/C C G A/C] as determined by functional SELEX screening.Citation40 Interestingly, the rs1800843 SNP A-allele falls within a predicted SRp55 binding sequence. The CCKCR SNP C-allele, “GCGA,” is predicted to bind to SRp55, while the A-allele, “GAGA,” is not predicted to not bind to SRp55 by ESE Finder 3.0 analysis (www.rulai.cshl.edu/cgibin/tools/ESE3/esefinder.cgi?process=home). This prediction suggests that the rs1800843 SNP may cause alternative splicing of IVS 4 by affecting a SRp55 binding site in CCKBR transcripts. Thus, the presence of the SNP could result in different alternative splicing patterns of A and C bearing allele mRNAs.
Personalized cancer therapy is a term used when an individual's cancer tissue is analyzed for specific tumor markers or genetic anomalies rendering it sensitive to selective therapy. Today we have the ability to genetically test patients and/or their tumors to select the optimal therapeutic regimens available. Knowing that a specific SNP genotype predicts tumor phenotype and behavior, patients could undergo DNA testing to determine their genotype for the CCK receptor SNP. Since new strategies are needed to help improve the prognosis of this malignancy, better understanding of the contribution of alternative splicing to the progression of pancreatic cancer may lead to insights that can aid in the earlier diagnosis, treatment, and prevention of pancreatic cancer.
Materials and Methods
Genotyping of human pancreatic ductal adenocarcinoma tumors, pancreatic cancer cells, other pancreatic neoplasms, and normal human pancreatic tissues for the CCKBR IVS 4.
Surgical specimens were obtained in a prospective fashion from patients deemed to be potentially “resectable and curable” for pancreatic ductal adenocarcinoma and from subjects undergoing surgical resection for other pancreatic conditions including benign cystic lesions, chronic pancreatitis, or neuroendocrine tumors of the pancreas. Corresponding normal tissue from the same patient was obtained either from adjacent normal pancreas or a buccal mucosa swab. Normal pancreas specimens were obtained from the Gift of Life Program and were procured pancreatic specimens without disease that were not used for transplantation. The protocol for tissue analysis was approved by the Institutional Research Committee at the Pennsylvania State University, and the first specimens were collected starting in 2004. Pancreatic cancer cell lines (MIA PaCa-2, BxPC-3, PANC-1, AsPC-1 and SW1990) were obtained from the ATCC and were maintained in the appropriate growth media in humidified air with 5% CO2.
DNA was isolated from approximately 20 mg of frozen tissue from surgical specimens or from an 80% confluent 100 mm plate of in vitro cancer cells using the DNeasy kit according to manufacturer's instructions (Qiagen). Two hundred nanograms of each resultant DNA sample was then amplified by CCKCR primers upstream (5′-GCC CTA CCC CGT GTA CAC TGT CGT G-3′) and downstream (5′-CGG TGT GCA CCC GGG CCA TCA AAG GCG C-3′) using the Phusion High-Fidelity PCR Master Mix with GC buffer (New England BioLabs) to produce a 1049 bp product. The PCR products were then purified using the QIAquick PCR Purification Kit (Qiagen) and sequenced using a 5′-GCA CTA TAA ACT GGC AAC CAA CAC AG-3′primer with an ABI 3130XL Capillary sequencer in the Penn State Hershey DNA sequencing core facility. DNA genotyping was also confirmed using a TaqMan SNP Genotyping Assay (C__22273290_10).
Development and characterization of the CCKCR selective monoclonal antibodies.
Since the SNP leads to mis-splicing of the fourth intron and addition of 69 amino acids to the receptor, selective murine monoclonal antibodies were raised to this CCKCR receptor epitope. BALB/c mice were immunized with a 20-amino acid synthetic peptide which corresponds to the beginning of the retained intron sequence of the CCKCR. Lymphocyte suspensions from inoculated mice collected and fused with P3X63-Ag8.653, a non-secreting mouse myeloma fusion partner. Antibody production from clones was identified by an ELISA and the antibodies were affinity purified using a Pierce Sulfolink column (ThermoFisher) and isotyped with a kit from Biomeda.
A polyclonal murine antibody was also generated to the CCKBR with a 17 amino acid KLH-conjugated peptide corresponding to amino acids 5 through 21 with a proline linker (KLNRSVQGTGPGPGASLPPPPC). Specificity for the monoclonal CCKCR antibody for the CCKCR over the CCKBR was evaluated by immunocytochemistry and Western analysis in overexpressing HEK-293 cells with pcDNA3.1 (vector-only) or pcDNA3.1 containing a COOH-terminal, 6X-Histidine epitopetagged receptor cDNA for CCKBR or CCKCR
Localization of the CCKCR in cancer cells was assessed by confocal fluorescent microscopy and electron microscopy in over-expressing COS-1 cells transiently transfected either with a human CCKBR or CCKCR cDNA construct using the selective antibody.
Evaluation of pancreatic tumors and normal pancreas tissues for CCKCR expression.
Tissue sections from human pancreatic cancer specimens, normal pancreas tissue and other pancreatic lesions were prepared from formalin-fixed paraffin embedded specimens, mounted on slides and subjected to antigen retrieval in 10 mM citrate buffer (pH = 6.0). Specimens were blocked with 3% normal goat serum, and then reacted with the CCKCR antibody (1:200) for 60 min at room temperature. Immunoreactivity was identified using Envision + labeled polymer/HRP (Dako Cytomation), visualized with DAB (Dako), and counterstained with Meyers hematoxylin. Pancreatic cancer cells were grown on glass coverslips, fixed, and reacted with the CCKCR antibody (1:100) and an anti-murine IgG AlexaFluor 488 secondary antibody (1:2,000) and visualized on a Leica TCS SP2 AOBS confocal microscope.
Effects of the CCKCR expression on cancer cell growth in vitro.
Human pancreatic cancer cells were stably transfected with a pCAGEN vector (Addgene) containing either the human CCKCR or CCKBR cDNA construct. Positive clones were selected by G418 resistance and CCKCR and CCKBR transcripts and expression were confirmed by qRT-PCR and by flow cytometry with the CCKCR antibody with wild-type and vector-only transfected cells as controls. The effect of overexpression of the CCKBR and CCKCR on cell growth was tested in vitro by the MTS proliferation assay and BrdU uptake in overexpressing cells compared with control cells.
Confirmation of CCKCR protein expression by flow cytometry.
Cells were harvested using Cellstripper (Cellgro) and collected by centrifugation. The supernatant was aspirated and cells were resuspended in 2% paraformaldehyde in 1xPBS at a concentration of 5 x 105 cells/ml. Cells were incubated for an hour at 4°C, followed by centrifugation to pellet fixed cells. Supernatant was aspirated and a 0.2% Tween-20 in 1x PBS solution was added at the same cell to volume concentration and incubated for 30 min at 37°C. Cells were again collected by centrifugation, supernatant aspirated, and a blocking buffer was added (10% FBS, 0.1% Tween-20 in 1x PBS; 30 min at room temperature). Cells were centrifuged and supernatant was aspirated, and the cells were incubated with primary antibody (either CCKCR or a non-specific mouse IgG1, Sigma M9269, 1:200) in buffer (5% FBS, 0.1% Tween-20 in 1x PBS) in the dark for 4 h at 4°C. Cells were collected by centrifugation, washed twice with 0.2% Tween-20 in 1xPBS, and incubated with an AlexaFluor 488-labeled goat anti-mouse secondary antibody (1:1,000) for one hour at 4°C. After washing, the cells were resuspended in 1x PBS and analyzed on a Becton Dickinson LSR II Flow Cytometer using the appropriate laser and filter set.
Statistical analysis.
The Kaplan-Meier method was used to estimate the survival curve for patients with pancreatic cancer with an A-allele (A/A or A/C) compared with survival of those pancreatic cancer patients with the C/C genotype by examining the 95% confidence interval for median survival of patients.Citation32 The log-rank test was used to compare the difference between the two groups using ASA Proc LIFETEST. The effect of various tumor characteristics and patient demographics as predictors on survival (genotype, lymph node status, histologic tumor grade, surgical margins, age at diagnosis, gender, adjuvant chemotherapy, smoking history, diabetes and family history of pancreatic cancer) were assessed using the Cox proportional hazards regression both individually and in combination with the genotype using Proc PHREG. The allelic frequencies and probability of development of adenocarcinoma was modeled on the A-allele vs. no A-allele condition using logistic regression analysis.
Differences in proliferation (MTS assay and BrdU assay) between control cells and CCKCR overexpressing cancer cells were analyzed with t-test and Bonferroni correction for multiple comparisons to control.
Disclosure of Potential Conflicts of Interest
J.S., J.H. and G.M. have intellectual property and a provisional patent for the CCKCR antibody. None have received financial rewards for this invention.
Abbreviations
| CCK-BR | = | cholecystokinin-B receptor |
| CCK-CR | = | cholecystokinin-C receptor |
| GWAS | = | genome-wide association study |
| SNP | = | single nucleotide polymorphism |
| HCG | = | human chorionic gonadotropin |
| MTS | = | 3-(4,5-dimethylthiazol-2-yl)-5-(3carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt |
| qRT-PCR | = | quantitative reverse transcription polymerase chain reaction |
| IVS4 | = | intervening sequence 4 or intron 4 |
Figures and Tables
Figure 1 Specificity of the CCKCR antibody. (A) Protein gel blot with protein extracts of HEK-293 cells that were transiently transfected with pcDNA3.1 (vector-only) or 6xHis epitope-tagged receptor cDNA for the CCKBR or the CCKCR (left panel). Both CCKBR and CCKCR extracts react to the 6xHis antibody whereas the vector-extract only does not. The same HEK-293 cell extracts were reacted with the monoclonal antibody specific to the CCKCR (right panel). Only the cells expressing the CCKCR demonstrated reactivity whereas the antibody did not cross react with the cells expressing only the CCKBR. (B) COS1 cells transiently transfected with either the cDNA for the CCKCR or the CCKBR are shown. The CCKCR antibody shows specificity to the cells expressing this receptor whereas the anti-CCKBR antibody reacts to both the CCKCR and the CCKBR cells.
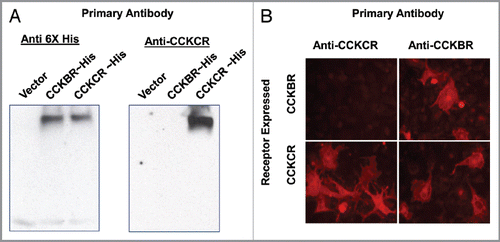
Figure 2 Localization of the CCKCR. (A) Confocal micrograph image of permeabilized CCKCR expressing cells reacted with the CCKCR monoclonal antibody and an Alexafluor-488 labeled anti-mouse secondary antibody showing the binding to the cytoplasmic membrane. (B) The same cells that are not permeabilized lack immunoreactivity. Nuclei are visualized with DAPI. (C) An electronmicrograph (EM) image of the CCKCR monoclonal antibody in a CCKCR expressing pancreatic cancer cell with secondary gold labeling, shows the receptor location to intracytoplasmic membrane. (D) EM image of CCKCR positive cells reacted with the secondary antibody only (negative control).
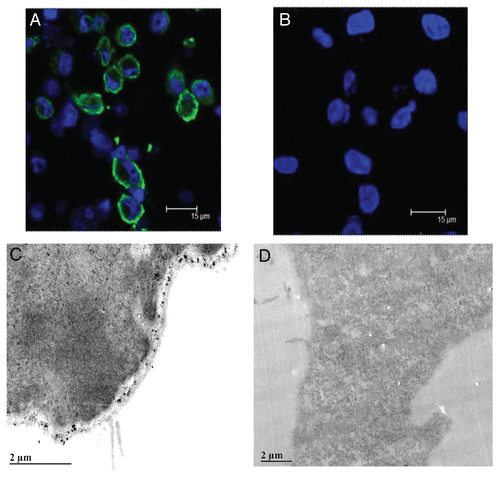
Figure 3 CCKCR phenotype is associated with the presence of the SNP A/A or A/C genotype. (A) CCKCR immunoreactivity is found in malignant cells of a pancreatic cancer of a patient having genotype A/A. CCKCR immunoreactivity is identified in a nerve invaded by cancer cells (arrow). The surrounding connective tissue (*CT) is void of CCKCR staining. (B) CCKCR positive immunoreactivity is seen from a pancreatic tumor of a patient with genotype A/C (arrows point to cancer cells). Again note the fibrous connective tissue surrounding the cancer cells is void of receptors. (C) A pancreatic cancer specimen from a subject with genotype C/C shows lack of CCKCR immunoreactivity. Arrow is pointing to malignant glands where cancer cells do not have the CCKCR. (D) A tissue specimen from a procured normal pancreas with genotype A/A showing the lack of CCKCR immunoreactivity in normal benign tissue. (E) PANC-1 cancer cells which have the A/C genotype show immunoreactivity to the CCKCR antibody by confocal microscopy. (F) AsPC-1 pancreatic cancer cells which have the C/C genotype do not react with the CCKCR antibody. Nuclei are stained with DAPI.
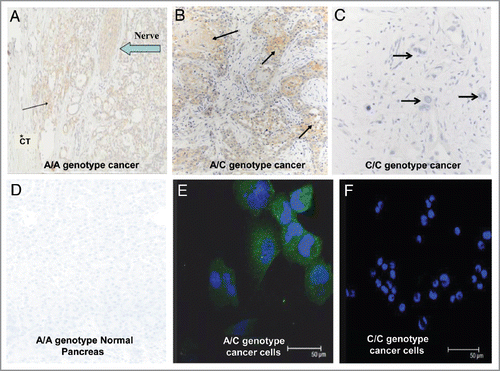
Figure 4 The SNP genotype correlates with survival. Kaplan Meier survival curve shows significantly prolonged survival in patients with pancreatic cancer who have the C/C genotype compared with those with either the A/A or the A/C genotype (p = 0.0001). Patients still alive at the last censored date is shown by a + on the survival curve. The number of patients surviving at different time intervals is displayed below the curve.
Figure 5 Overexpression of the CCKCR and not CCKBR in pancreatic cancer cells stimulates growth. (A) MTS cell growth assay was performed in AsPC-1 wild type C/C cells, vector transfected AsPC-1 cells, or AsPC-1 cells transfected to overexpress the either the CCKBR or CCKCR. Growth was significantly increased in the CCKCR over-expressing cancer cells compared with vector and wild type controls and also compared with CCKBR overexpressing cells suggesting that the CCKCR receptor expression provides a growth benefit to the cancer (***p = 2.7 x 10–11). The assay was performed after 48 h of growth and done three times. (B) BrdU incorporation was also assessed with AsPC-1 wild-type, vector transfected, CCKBR or CCKCR transfected cells. BrdU incorporation was markedly increased (***p = 5.8 x 10-7) in CCKCR transfected cancer cells compared with control cells.
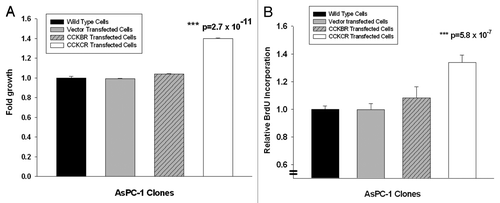
Figure 6 Overexpression of the CCK-CR in human pancreatic cancer cells is confirmed by flow cytometry. Overexpressing cancer cells were reacted with either no antibody for background control (A) or with a nonspecific IgG antibody control (B). Cancer cells that overexpress the CCKCR show increased immunoreactivity to the selective CCKCR monoclonal antibody (C). AB, antibody; mAB, monoclonal antibody.
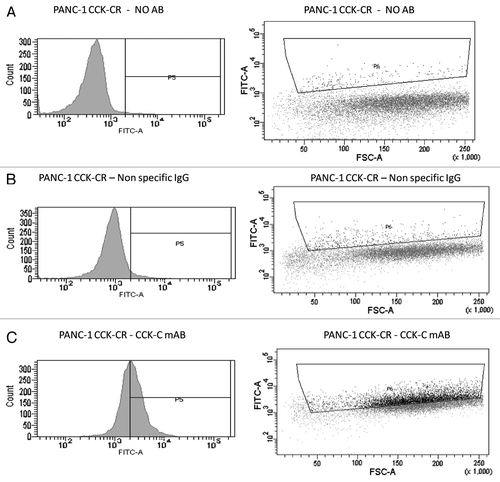
Table 1 Genotype frequency according to histology
Table 2 Patient and tumor characteristics as predictors of survival
Acknowledgments
The research was supported by the National Institutes of Health, National Cancer Institute grant (R01 CA117926 to J.P.S.); and an American Cancer Society post-doctoral fellowship award (PF-04-104-01-CSM to J.F.H.) Additional funding was provided through an educational grant from the National Institutes of Health (R25 DK078381 to G.L.M.) for a student in the STEP-UP program. We acknowledge the technical assistance of David Stanford and Nate Shaeffer in the Cell Science Flow Cytometry Core, Joe Bednarczyk in the DNA Sequencing Core, and Rob Brucklacher and Georgina Bixler of the Functional Genomics Core of the Section of Research Resources. We appreciate the technical assistance of Calpurnia Jayakumar (Department of Medicine) and Richard Bruggeman (The Morphologic and Molecular Pathology Research Lab of the Department of Pathology) from the Pennsylvania State University College of Medicine for immunohistochemical staining of tissue specimens. We acknowledge Daniel Beard from the Tumor Bank from the Penn State Cancer Institute for providing additional cancer samples and tumor registry information.
References
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, et al. Cancer statistics, 2008. CA Cancer J Clin 2008; 58:71 - 96; PMID: 18287387; http://dx.doi.org/10.3322/CA.2007.0010
- Rocha Lima CM, Centeno B. Update on pancreatic cancer. Curr Opin Oncol 2002; 14:424 - 430; PMID: 12130928; http://dx.doi.org/10.1097/00001622200207000-00010
- Goldstein D, Carroll S, Apte M, Keogh G. Modern management of pancreatic carcinoma. Intern Med J 2004; 34:475 - 481; PMID: 15317546; http://dx.doi.org/10.1111/j.1444-0903.2004.00658.x
- Wolff RA. Novel therapies for pancreatic cancer. Cancer J 2001; 7:349 - 258; PMID: 11561610
- Montgomery RC, Hoffman JP, Riley LB, Rogatko A, Ridge JA, Eisenberg BL. Prediction of recurrence and survival by post-resection CA 19-9 values in patients with adenocarcinoma of the pancreas. Ann Surg Oncol 1997; 4:551 - 556; PMID: 9367020; http://dx.doi.org/10.1007/BF02305535
- Louhimo J, Alfthan H, Stenman UH, Haglund C. Serum HCG beta and CA 72–4 are stronger prognostic factors than CEA, CA 19-9 and CA 242 in pancreatic cancer. Oncology 2004; 66:126 - 131; PMID: 15138364; http://dx.doi.org/10.1159/000077438
- Koopmann J, Buckhaults P, Brown DA, Zahurak ML, Sato N, Fukushima N, et al. Serum macrophage inhibitory cytokine 1 as a marker of pancreatic and other periampullary cancers. Clin Cancer Res 2004; 10:2386 - 2392; PMID: 15073115; http://dx.doi.org/10.1158/1078-0432.CCR-03-0165
- Koopmann J, Fedarko NS, Jain A, Maitra A, Iacobuzio-Donahue C, Rahman A, et al. Evaluation of osteopontin as biomarker for pancreatic adenocarcinoma. Cancer Epidemiol Biomarkers Prev 2004; 13:487 - 491; PMID: 15006928
- Garcea G, Neal CP, Pattenden CJ, Steward WP, Berry DP. Molecular prognostic markers in pancreatic cancer: a systematic review. Eur J Cancer 2005; 41:2213 - 2236; PMID: 16146690; http://dx.doi.org/10.1016/j.ejca.2005.04.044
- Goggins M. Molecular markers of early pancreatic cancer. J Clin Oncol 2005; 23:4524 - 4531; PMID: 16002843; http://dx.doi.org/10.1200/JCO.2005.19.
- Goggins M, Canto M, Hruban R. Can we screen high-risk individuals to detect early pancreatic carcinoma?. J Surg Oncol 2000; 74:243 - 248; PMID: 10962454; http://dx.doi.org/10.1002/1096-9098(200008)74:4<243::AIDJSO1>3.0.CO;2-C
- Tada M, Omata M, Kawai S, Saisho H, Ohto M, Saiki RK, et al. Detection of ras gene mutations in pancreatic juice and peripheral blood of patients with pancreatic adenocarcinoma. Cancer Res 1993; 53:2472 - 2474; PMID: 8495407
- Korc M. Driver Mutations. Cancer Biol Ther 2010; 10:588 - 591; PMID: 20716952; http://dx.doi.org/10.4161/cbt.10.6.13128
- Pisegna JR, de Weerth A, Huppi K, Wank SA. Molecular cloning of the human brain and gastric cholecystokinin receptor: structure, functional expression and chromosomal localization. Biochem Biophys Res Commun 1992; 189:296 - 303; PMID: 1280419; http://dx.doi.org/10.1016/0006-291X(92)91557-7
- Wank SA, Harkins R, Jensen RT, Shapira H, de Weerth A, Slattery T. Purification, molecular cloning, and functional expression of the cholecystokinin receptor from rat pancreas. Proc Natl Acad Sci USA 1992; 89:3125 - 3129; PMID: 1313582; http://dx.doi.org/10.1073/pnas.89.7.3125
- Wank SA, Pisegna JR, de Weerth A. Brain and gastrointestinal cholecystokinin receptor family: structure and functional expression. Proc Natl Acad Sci USA 1992; 89:8691 - 8695; PMID: 1528881; http://dx.doi.org/10.1073/pnas.89.18.8691
- Wank SA. G protein-coupled receptors in gastrointestinal physiology. I. CCK receptors: an exemplary family. Am J Physiol 1998; 274:G607 - G613; PMID: 9575840
- Smith JP, Rickabaugh CA, McLaughlin PJ, Zagon IS. Cholecystokinin receptors and PANC-1 human pancreatic cancer cells. Am J Physiol 1993; 265:G149 - G155; PMID: 8338163
- Smith JP, Liu G, Soundararajan V, McLaughlin PJ, Zagon IS. Identification and characterization of CCK-B/gastrin receptors in human pancreatic cancer cell lines. Am J Physiol 1994; 266:R277 - R283; PMID: 8304551
- Smith JP, Zagon IS. Cholecystokinin receptors and human pancreatic adenocarcinomas. Int J Pancreatol 1994; 16:243 - 246
- Weinberg DS, Ruggeri B, Barber MT, Biswas S, Miknyocki S, Waldman SA. Cholecystokinin A and B receptors are differentially expressed in normal pancreas and pancreatic adenocarcinoma. J Clin Invest 1997; 100:597 - 603; PMID: 9239407; http://dx.doi.org/10.1172/JCI119570
- Smith JP, Verderame MF, McLaughlin P, Zagon IS. Characterization of the CCK-C (cancer) receptor in human pancreatic cancer. Digestion 1999; 60:401
- Smith JP, Verderame MF, McLaughlin P, Martenis M, Ballard E, Zagon IS. Characterization of the CCK-C (cancer) receptor in human pancreatic cancer. Int J Mol Med 2002; 10:689 - 694; PMID: 12429993
- Hellmich MR, Rui XL, Hellmich HL, Fleming RY, Evers BM, Townsend CM Jr. Human colorectal cancers express a constitutively active cholecystokininB/gastrin receptor that stimulates cell growth. J Biol Chem 2000; 275:32122 - 32128; PMID: 10913157; http://dx.doi.org/10.1074/jbc.M005754200
- Ding WQ, Kuntz SM, Miller LJ. A misspliced form of the cholecystokinin-B/gastrin receptor in pancreatic carcinoma: role of reduced sellular U2AF35 and a suboptimal 3′-splicing site leading to retention of the fourth intron. Cancer Res 2002; 62:947 - 952; PMID: 11830556
- Biagini P, Monges G, Vuaroqueaux V, Parriaux D, Cantaloube JF, De Micco P. The human gastrin/cholecystokinin receptors: type B and type C expression in colonic tumors and cell lines. Life Sci 1997; 61:1009 - 1018; PMID: 9296339; http://dx.doi.org/10.1016/S0024-3205(97)00605-X
- Song I, Brown DR, Wiltshire RN, Gantz I, Trent JM, Yamada T. The human gastrin/cholecystokinin type B receptor gene: alternative splice donor site in exon 4 generates two variant mRNAs. Proc Natl Acad Sci USA 1993; 90:9085 - 9089; PMID: 8415658; http://dx.doi.org/10.1073/pnas.90.19.9085
- Chao C, Goluszko E, Lee YT, Kolokoltsov AA, Davey RA, Uchida T, et al. Constitutively active CCK2 receptor splice variant increases Src-dependent HIF-1 alpha expression and tumor growth. Oncogene 2007; 26:1013 - 1019; PMID: 16909104; http://dx.doi.org/10.1038/sj.onc.1209862
- Cheng ZJ, Harikumar KG, Ding WQ, Holicky EL, Miller LJ. Analysis of the cellular and molecular mechanisms of trophic action of a misspliced form of the type B cholecystokinin receptor present in colon and pancreatic cancer. Cancer Lett 2005; 222:95 - 105; PMID: 15837546; http://dx.doi.org/10.1016/j.canlet.2004.09.008
- Laverman P, Roosenburg S, Gotthardt M, Park J, Oyen WJ, de Jong M, et al. Targeting of a CCK(2) receptor splice variant with (111)In-labelled cholecystokinin-8 (CCK8) and (111)In-labelled minigastrin. Eur J Nucl Med Mol Imaging 2008; 35:386 - 392; PMID: 17934729; http://dx.doi.org/10.1007/s00259-007-0604-1
- Smith JP, Verderame MF, McLaughlin PJ, Zagon IS. Characterization of the CCK-C (cancer) receptor in human pancreatic cancer. Gastroenterology 1999; 116:A1164
- Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc 1958; 53:457 - 481; http://dx.doi.org/10.2307/2281868
- Johnson AD, O'Donnell CJ. An open access database of genome-wide association results. BMC Med Genet 2009; 10:6; PMID: 19161620; http://dx.doi.org/10.1186/1471-2350-10-6
- Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010; 467:1114 - 1117; PMID: 20981102; http://dx.doi.org/10.1038/nature09515
- Carter H, Samayoa J, Hruban RH, Karchin R. Prioritization of driver mutations in pancreatic cancer-specific high-throughput annotation of somatic mutations (CHASM). Cancer Biol Ther 2010; 10:582 - 587; PMID: 20581473; http://dx.doi.org/10.4161/cbt.10.6.12537
- Finn OJ. Cancer immunology. N Engl J Med 2008; 358:2704 - 2715; PMID: 18565863; http://dx.doi.org/10.1056/NEJMra072739
- Palucka K, Ueno H, Banchereau J. Recent developments in cancer vaccines. J Immunol 2011; 186:1325 - 1331; PMID: 21248270; http://dx.doi.org/10.4049/jimmunol.0902539
- Brinkman BM. Splice variants as cancer biomarkers. Clin Biochem 2004; 37:584 - 594; PMID: 15234240; http://dx.doi.org/10.1016/j.clinbiochem.2004.05.015
- Gardina PJ, Clark TA, Shimada B, Staples MK, Yang Q, Veitch J, et al. Alternative splicing and differential gene expression in colon cancer detected by a whole genome exon array. BMC Genomics 2006; 7:325; PMID: 17192196; http://dx.doi.org/10.1186/14712164-7-325
- Long JC, Caceres JF. The SR protein family of splicing factors: master regulators of gene expression. Biochem J 2009; 417:15 - 27; PMID: 19061484; http://dx.doi.org/10.1042/BJ20081501
- Shepard PJ, Hertel KJ. The SR protein family. Genome Biol 2009; 10:242; PMID: 19857271; http://dx.doi.org/10.1186/gb-2009-10-10-242