Abstract
Sustaining proliferative signaling is one of the hallmarks of cancer. Granulocyte-colony stimulating factor (G-CSF), a hematopoietic growth factor that controls proliferation and differentiation of neural stem cells, may contribute to the development of glioma. In the present study, we evaluated the expression of G-CSF and its receptor (G-CSFR) in various grades of glioma samples and primary cell culture derived from a glioblastoma patient as well as different human glioma cell lines. We showed that G-CSF and G-CSFR were widely expressed by glioma samples, primary glioma cell culture and glioma cell lines. The expression levels of G-CSF and G-CSFR were not significantly different between different grades of glioma. G-CSF promoted proliferation, migration and invasion of glioma cells. Neutralization of G-CSFR with its antibody inhibited growth and metastasis of glioma cells in vitro. We also showed that activation of signal transducer and activator transcription 3 (STAT 3) as well as expression of several of its downstream effectors was regulated by G-CSF. Taken together, the present results suggest that G-CSF contributes to glioma progression that may be linked to glioma genesis and recurrence.
Introduction
Glioma is the most common primary brain tumor. According to the histopathological and clinical features, gliomas are divided into four grades—World Health Orgnization (WHO) grades I–IV.Citation1 The highest grade glioma, i.e., glioblastoma (WHO grade IV), is among the most lethal human cancer with a 5-year survival rate of less than 3%.Citation2 Though low-grade, i.e., grade I and II, gliomas are less malignant, unfortunately, they would inevitably progress toward malignant high-grade gliomas, i.e., grade III and IV.Citation3 Therefore, new therapeutic strategies are desirable requiring insights into the biological and molecular mechanisms driving the tumor growth. In this connection, we surmised that granulocyte-colony stimulating factor (G-CSF), a potent mitogen for many cell types, might contribute to the development of glioma for reasons to be described presently.
G-CSF was first identified as a granulocyte stimulator and leukemic differentiation factor,Citation4 regulating the proliferation, maturation and survival of granulocytes, macrophages, and hematopoietic progenitor cells.Citation5,Citation6 However, there is accumulating evidence indicating that G-CSF together with its receptor is expressed in many tumors, such as melanoma,Citation7 non-small cell lung cancer,Citation8 bladder cancerCitation9 and prostate cancer,Citation10 as well as brain cancers.Citation11,Citation12 Additionally, it has been reported that G-CSF can increase proliferation and invasion of many types of cancer cells in vitro.Citation11-Citation14 Furthermore, aberrant production of G-CSF has been tightly linked to tumor generation and progression in many tumor types.Citation7-Citation15 Recent studies have found that G-CSF regulates proliferation and differentiation of neural stem cells (NSCs),Citation16,Citation17 a cell type in the CNS that has the potentiality to transform into tumor cells.Citation18,Citation19 In the light of these findings, the possibility that G-CSF may play an essential role in glioma generation and progression is considered.
G-CSF is a cytokine that is secreted into extracellular spaces and act through binding to a specific cell surface receptor G-CSF receptor (G-CSFR, also called CSF3R, colony-stimulating factor receptor 3).Citation20 Upon receptor activation, intracellular signaling is propagated by many factors, among which are Src and Janus kinase (JAK) tyrosine kinase family members, leading to the activation of transcription factors of signal transducers and activators of transcription (STAT) family, particularly STAT 3.Citation21 STAT 3 pathway is activated in glioma and has been identified recently as one of 5 core pathways altered in human glioblastoma.Citation22 These data led us to hypothesize that G-CSF may activate STAT 3 in glioma to contribute to its biological behaviors.
In the present study, we explored the potential role of G-CSF on glioma cells in vitro. First, we determined the relationship between G-CSF/G-CSFR and WHO glioma grades. Second, we examined the effect of G-CSF on proliferation, migration and invasion of glioma cells in the hope that the underlying molecular mechanisms may be unraveled. We report here that G-CSF induces STAT 3 activation and downstream genes expression. The present results suggest that G-CSF signaling participates in the progression of glioma, and that it plays an important role in the generation and recurrence of glioma.
Results
G-CSF and its receptor were highly expressed in human glioma samples
To evaluate the potential contribution of G-CSF to glioma, we measured expression of G-CSF and its receptor (G-CSFR) in glioma samples and normal brain tissue. G-CSF mRNA was expressed in all four grades of gliomas (). G-CSFR mRNA and protein were also detectable in all four grades of glioma samples by RT-PCR and western blot, respectively (). G-CSF and G-CSFR were co-expressed in 7/8 grade I/II gliomas, 8/9 grade III gliomas, 12/12 grade IV gliomas. The differential expression of G-CSF and G-CSFR in four grades of gliomas was not statistically different (p > 0.05) (). Remarkably, neither G-CSF nor G-CSFR was detected in the normal brain tissues by RT-PCR or western blot (). Consistent with this result, all four grades of gliomas contained diffusely distributed G-CSFR-positive cells as detected by immunohistochemistry assay, G-CSFR immunopositive cells were undetected in normal brain tissue ().
Figure 1. Expression of G-CSF and G-CSFR in human glioma samples. (A) RT-PCR results revealed G-CSF and G-CSFR mRNA expression in human glioma samples; mRNA expression level of G-CSF and G-CSFR was undetected in normal brain tissue (NB). (B) Relative mRNA levels in grade I/II gliomas (n = 7), grade III gliomas (n = 8), grade IV gliomas (n = 12), and normal brain tissue (NB, n = 5). (C) Western blot assay shows expression of G-CSFR in glioma samples. Expression of G-CSFR was undetected in normal brain tissue (NB). (D) Prevalence of G-CSF and G-CSFR expression in grade I/II gliomas (7/8), grade III gliomas (8/9), grade IV gliomas (12/12), and normal brain tissue (NB, 0/5).
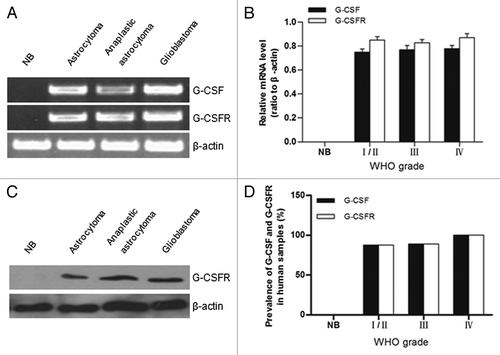
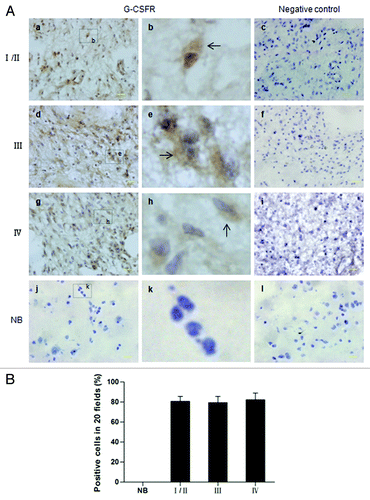
Figure 2. Immunohistochemical analysis of G-CSFR expression in human glioma samples and normal brain tissue. (A) Immunohistochemistry shows G-CSFR expression is mainly localized in the cytoplasm (b, e, and h, brown; arrow) of glioma cells. G-CSFR is widely expressed in glioma cells (a, d, and g), but not in the normal brain tissue (NB, j). c, f, i, and l are negative controls for grade I/II glioma, grade III glioma, grade IV glioma, and normal brain tissue (NB), respectively. For negative controls, cells were similarly processed as the tested groups but without the primary antibody. (B) Average G-CSFR-positive cells in grade I/II gliomas (80.71% ± 5.06%), grade III gliomas (79.50% ± 6.07%), grade IV gliomas (82.36% ± 6.68%), and normal brain tissue (0%). The difference between grades was not statistically significant (p > 0.05).
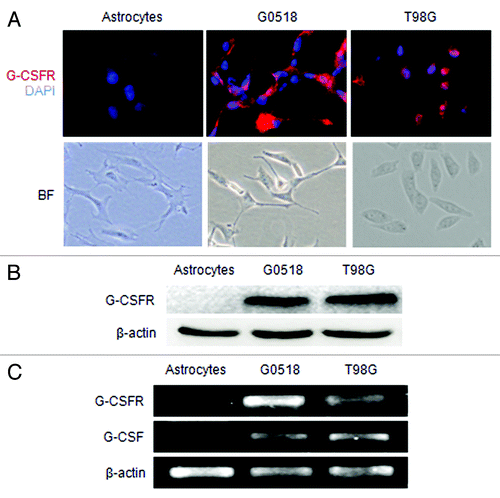
Both immunofluorescence labeling and western blot assay showed that G-CSFR protein was expressed by the primary cell culture G0518 derived from a glioblastoma patient and glioma cell lines T98G, U251 and U87 (; data not shown). RT-PCR assay revealed that G-CSF and G-CSFR mRNA was also expressed in these cells (; and data not shown). However, neither G-CSF nor its receptor was detected in the primary cell culture of astrocytes (). These findings indicated that G-CSF and G-CSFR expression was induced and augmented in gliomas as opposed to the normal brain tissue lacking the expression.
Figure 3. Expression of G-CSF and G-CSFR in glioma cell cultures. (A) Shows G-CSFR immunofluorescence (red) in glioma cell culture derived from glioma sample (G0518) and glioma cell lines (T98G, U251, and U87), but not in primary cultured astrocytes. Cell nuclei were counterstained with DAPI (blue). Magnification: 200 × scale: 100 μm. (B) Western blot shows G-CSFR protein expression in glioma cells and primary astrocytes. (C) RT-PCR shows mRNA expression levels of G-CSF and G-CSFR in glioma cells: G0518, and T98G cells, but not in primary astrocytes.
G-CSF promoted cell viability and proliferation of glioma cells
To study the effect of G-CSF on glioma cells, recombinant human G-CSF was added to the culture medium of glioma cells. Four media with different but increasing concentrations (10, 50, 100, 200 ng/ml) of G-CSF and medium without G-CSF were used for experimental and control groups, respectively. G-CSF increased glioma cell viability over time in a dose dependent manner compared with the control groups (p < 0.05 or p < 0.01) (). The effect of G-CSF on cell viability was progressively increased with concentration till 100 ng/ml. G-CSF at 200 ng/ml did not exert a greater effect; rather it appeared to decline (p > 0.05) (). In view of this, we have used 100 ng/ml for all subsequent experimental analyses.
Figure 4. Effect of G-CSF on cell viability and proliferation of glioma cells. (A and B) Cell viability was determined by MTT assay. (A) Showing dose dependent effect of G-CSF on glioma cells; (B) Showing time dependent effect of G-CSF on glioma cells. Data are expressed as ratio to the control value (without G-CSF). (C, D) BrdU assay shows that the percentage of BrdU positive cells is significantly increased in G-CSF treated groups compared with groups without G-CSF treatment. (E, F) Colony formation assay shows that the G-CSF treated cells formed more colonies compared with those without G-CSF treatment. (*p < 0.05, #p < 0.01; each experimental point was performed with three wells, and values represent mean ± SD of three independent experiments).
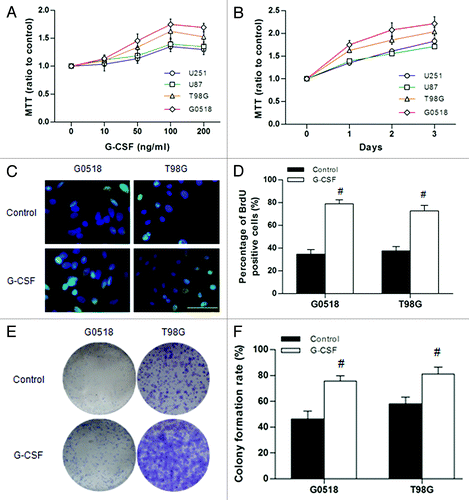
To further ascertain that G-CSF could stimulate proliferation of glioma cells, BrdU incorporation assay and colony formation assay were performed. The results showed that G-CSF significantly increased cellular proliferation as manifested by an increase in frequency (G0518:78.93% ± 3.52%; T98G: 72.67% ± 2.88%, both p < 0.01) of BrdU positive cells following G-CSF treatment (). Colony formation assay also demonstrated that G-CSF increased proliferation of glioma cells as colony formation rate increased significantly with G-CSF treatment compared with those without treatment (G0518: 75.67% ± 4.04% vs. 46.33% ± 5.11%; T98G: 81.00% ± 5.57% vs. 58.00% ± 5.29%, both p < 0.01) (). These results have shown that G-CSF promotes growth of glioma cells.
G-CSFR antibody decreased growth and survival of glioma cells
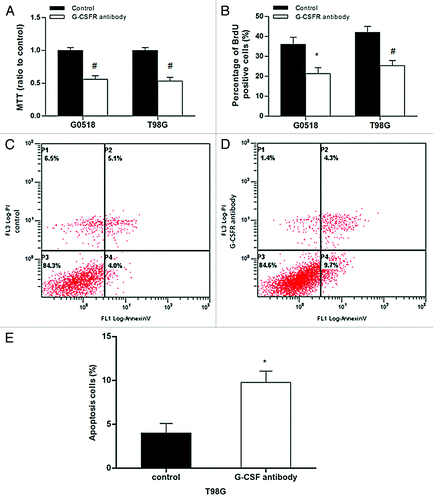
To further determine the effect of G-CSF on glioma, G-CSFR antibody was used to block the G-CSF signaling. BrdU incorporation assay and colony formation assay showed that the frequency of BrdU positive cells and colony formation rate in G-CSFR antibody treated glioma cells decreased significantly compared with those without treatment (p < 0.05 or p < 0.01) (). Additionally, G-CSFR antibody treatment significantly induced apoptosis in T98G glioma cells as evidenced by elevated annexin V-positive cells compared with those without treatment (9.77% ± 1.30% vs. 4.00% ± 1.10%, p < 0.05) ().
Figure 5. Effects of G-CSFR antibody in glioma cells. (A, B) MTT and BrdU incorporation assays show G-CSFR antibody decreases cell viability and growth, respectively. (C, D, and E) Flow cytometry assay revealed G-CSFR antibody significantly increased Annexin-V-positive cells in glioma cells, indicating that G-CSFR antibody induced apoptosis. (*p < 0.05, #p < 0.01; each experimental point was performed with three wells, and values represent mean ± SD of three independent experiments).
G-CSF increased migration and invasion of glioma cells
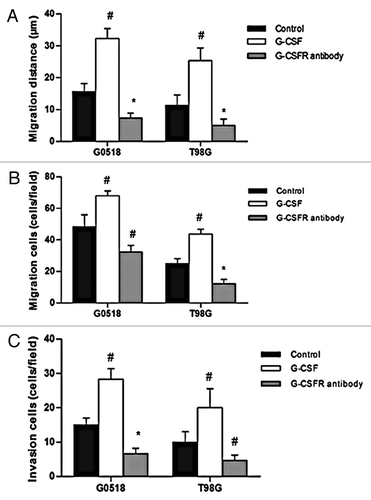
We next evaluated whether G-CSF plays a role on migration and invasion of glioma cells. In wound-healing assay, G-CSF treated glioma cells migrated a significantly further distance into the “denuded” area compared with those without treatment (p < 0.01) (). Transwell migration test showed that G-CSF drastically promoted migration of G0518 and T98G glioma cells as manifested by an increase of cells on the bottom side of the membranes (p < 0.01) (). In contrast to the promotion effect of G-CSF on migration, G-CSFR antibody significantly decreased migration ability of gliomas both on migration distance and migration cells (p < 0.01 or p < 0.01) (). Moreover, matrigel coated transwell invasion test showed that G-CSF significantly increased invasion of glioma cells, while G-CSFR antibody obviously decreased invasion of glioma cells (p < 0.05 or p < 0.01) (). These results demonstrated that G-CSF can increase migration and invasion of glioma cells and blocking of G-CSF signaling can decrease migration and invasion of glioma cells.
Figure 6. Effects of G-CSF on migration and invasion of glioma cells. (A, B) G-CSF treatment significantly increased migration ability of G0518 and T98G, as demonstrated by increases in migration distance (A) and migration cells (B). G-CSFR antibody inhibited migration ability of glioma cell. (C) Invasion ability of G-CSF treated group is higher than that of untreated group, while that of G-CSFR antibody treated group is decreased. (*p < 0.05, #p < 0.01. Each experimental point was performed with three wells, and values represent mean ± SD of three independent experiments).
G-CSF regulated phosphorylation of STAT 3 in glioma cells
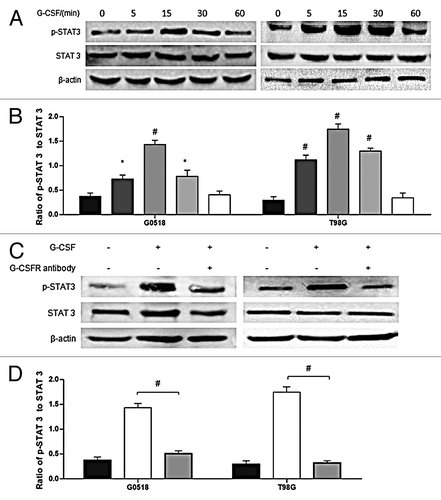
It was reported that G-CSF functions through STAT 3 pathway.Citation23,Citation24 Thus, to find out the underlying mechanisms of G-CSF on glioma cells, we checked the activation of STAT 3 which was found to be increased with G-CSF stimulation (). Phosphorylation of STAT 3 was elicited from 5 min following G-CSF stimulation, peaking at about 15 min; it returned to the basal level at about 1 h (). To further determine if phosphorylation of STAT 3 by G-CSF was through combination of G-CSF and G-CSF receptor, glioma cells were incubated with G-CSF receptor antibody (1 μg/ml) for 6 h, and then stimulated with G-CSF. G-CSF induced STAT 3 activation was inhibited by G-CSF receptor antibody (). These results suggest that STAT 3 is involved in G-CSF signaling in glioma cells.
Figure 7. Phosphorylation of STAT 3 is regulated by G-CSF. (A, B) western blot analysis shows time-dependent effects of G-CSF on activation of STAT 3 in G0518 and T98G cells. (C, D) western blot analysis shows that G-CSFR antibody inhibits G-CSF induced elevation of p-STAT 3. (*p < 0.05, #p < 0.01, Each experimental point was performed with three wells, and values represent mean ± SD of three independent experiments).
G-CSF modulated expression of STAT 3 downstream genes in glioma cells
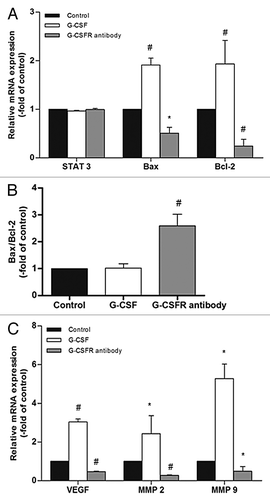
We next checked the expression of STAT 3 and downstream genes following G-CSF or G-CSFR antibody treatment by real time RT-PCR. Neither G-CSF nor G-CSFR antibody affected STAT 3 expression in glioma cells (p > 0.05) (). G-CSF treatment significantly increased mRNA expression level of STAT 3 downstream genes Bax (1.90 ± 0.10 fold over control, p < 0.01), Bcl-2 (1.87 ± 0.12 fold over control, p < 0.05), VEGF (3.03 ± 0.15 fold over control, p < 0.01), MMP2 (2.43 ± 0.93 fold over control, p < 0.05) and MMP9 (5.28 ± 0.75 fold over control, p < 0.05), while G-CSFR antibody decreased mRNA expression levels of these genes (p < 0.05 or p < 0.01)(). Furthermore, ratio of Bax to Bcl-2 was significantly increased following G-CSFR antibody treatment (2.59 ± 0.43 fold over control, p < 0.05) (). Together, these results have shown that G-CSF regulates expression of STAT 3 downstream genes.
Figure 8. STAT 3 downstream genes are modulated by G-CSF. (A, C) Real time RT-PCR shows neither G-CSF nor G-CSFR antibody affects STAT 3 expression. On the other hand, G-CSF upregulates expression of STAT 3 downstream genes including Bax, Bcl-2, VEGF, MMP 2, and MMP 9. Neutralization of G-CSFR with its antibody inhibits expression of these genes. (B) Ratio of Bax to Bcl-2 is not affected by G-CSF, but is significantly elevated by G-CSFR antibody. (*p < 0.05, #p < 0.01). Each experimental point was performed with three wells, and values represent mean ± SD of three independent experiments).
Discussion
This study sought to investigate the role of G-CSF, notably the underlying regulatory molecular mechanism, in human glioma cells. First, our results showed that both G-CSF and G-CSFR were widely expressed in glioma samples and primary cell culture derived from glioma samples as well as glioma cell lines. Second, exogenous application of G-CSF significantly promoted the proliferation, migration and invasion of glioma cells in vitro, while blockade of G-CSF signaling by G-CSFR antibody exerted an opposing effect. The expression levels of phosphorylated STAT 3 and STAT 3 downstream genes were elevated by G-CSF; however, they were suppressed by G-CSFR antibody. These results suggest that G-CSF may contribute to glioma development and relapse.
The present results have shown that G-CSF or G-CSFR expression was not detected in the normal brain cortex or primary cultured astrocytes, although it was reported to be expressed by cultured astrocytes by others;Citation25 furthermore, expression of G-CSF and G-CSFR is upregulated in ischemic brain.Citation26 A possible explanation for this may be that G-CSF and G-CSFR expression in the normal brain does exist but is at too low level to be detected by the present methods. Nonetheless, it can be confidently concluded that expression of G-CSF and G-CSFR is induced and upregulated in glioma cells as shown in this study. Indeed, both G-CSF and G-CSFR were highly expressed by gliomas, a feature which is in accord with previous findings.Citation11,Citation12 Very interestingly, G-CSF and G-CSFR expression was not significantly different between different grades of gliomas. The co-expression of G-CSF and G-CSFR in glioma suggests that it may facilitate both autocrine and paracrine mode of stimulation. It is further suggested that such autocrine/paracrine signaling may play an important role in the establishment and maintenance of these tumors. The broad range of human gliomas displaying altered G-CSF expression strongly suggests that G-CSF signaling axis plays a pivotal role in the events underlying gliomagenesis. However, to further clarify the relationship between G-CSF signaling and gliomagenesis, in vivo overexpression of G-CSF in normal brain would be essential.
G-CSF is a potent mitogen for many cell types, such as neural progenitors and neural stem cells (NSCs) in the central nervous system (CNS).Citation16,Citation17 Here, we showed that G-CSF not only increased cell growth but also colony formation rate of glioma cells. On the other hand, blocking G-CSF signaling by G-CSFR antibody decreased cell growth, which may be attributed partly to apoptosis. G-CSF signaling functionally interacts with signals evoked by other secreted molecules, such as VEGF, in the modulation of angiogenic process as reflected by the high expression of both G-CSFR and vascular endothelial growth factor receptor (VEGFR) in many human gliomas.Citation11,Citation12,Citation27,Citation28 It is further evidenced by the ability of G-CSF signaling to upregulate VEGF expression.Citation16,Citation29 All in all, it seems therefore likely that G-CSF signaling is involved in tumor expansion and survival, stimulating growth, inhibiting apoptosis and indirectly promoting nutrient supply to the tumor mass.
Signal transducer and activation transcription 3 (STAT 3) is a DNA-binding transcriptional factor that can be activated by receptor-associated kinases. Activated STAT 3 induces expression of many genes, such as Bax, Bcl-2 and VEGF.Citation30,Citation31 The Bax gene is pro-apoptotic, while Bcl-2 is important in preventing apoptosis.Citation32-Citation34 VEGF is associated with angiogenesis.Citation35 STAT 3 is constitutively activated in many human cancers, including glioma, and plays a key role in the processes of neoplastic transformation and tumor progression.Citation36,Citation37 In this study, G-CSF activated STAT 3 in a time-dependent manner; G-CSFR antibody inhibited G-CSF-induced activation of STAT 3. Moreover, G-CSF evoked expression of STAT 3 downstream genes, while G-CSFR antibody inhibited expression of these genes. These findings indicate that STAT 3 pathway is involved in the G-CSF function in glioma cells.
Diffused infiltration into surrounding normal brain is a characteristic feature of glioma. The invasion process of glioma cells can be driven by many factors, among which, MMP2 and MMP9 were suggested to be key enzymes in the invasion of glioma cells.Citation38 MMP 2 and MMP 9 are members of MMPs family, which can catalyze extracellular proteolysis and play an essential role in cell migration and tissue remodeling. Here, we showed that G-CSF promoted migration and invasion of glioma cells; G-CSFR antibody inhibited these processes. Furthermore, expression of MMP2 and MMP9 was increased following stimulation of G-CSF, but it was decreased by G-CSFR antibody. These results suggest that G-CSF regulates invasion of glioma cells through modulation of MMP 2 and MMP 9. It is relevant to note that MMP 2 and MMP 9 are involved not only in invasion, but also in proliferation, angiogenesis and recurrence.Citation39,Citation40
An important issue arose from this study was whether G-CSF is involved in glioma recurrence. Recurrence of glioblastoma is related to many biological mechanisms, such as proliferation, migration, invasion and angiogenesis, all can be promoted by G-CSF, indicating that G-CSF may play a role in glioma relapse. In the present study, we have used 100 ng/ml G-CSF for all experimental analyses. Clinically, a median G-CSF serum concentration reaches 100 ng/ml within a few hours after a single subcutaneous injection at the dose of 100 μg/kg; such a level is maintained at about 100 ng/ml for many hours.Citation41 In the present results, STAT 3 activation reached its peak at about 15 min following G-CSF stimulation. These data indicate that G-CSF used in clinic can reach its optimal concentration to act on glioma cells. However, it needs to be cautioned that long-term follow-ups should be performed in glioma patients taking G-CSF to better define the relationship between G-CSF and glioma relapse. This takes into consideration of the present results that G-CSF and its receptor may result in undesirable glioma recurrence.
Materials and Methods
Human samples
All human samples in the present study were collected from the Neurosurgery Institute of Qi Lu Hospital (Jinan, China) according to the National Regulation of Clinical Sampling in China. Gliomas were verified by pathological analysis and classified according to the World Health Organization classification standard. The adult normal brain samples were collected from surgical resection of five trauma patients. Human glioma and normal tissue used for reversed transcription polymerase chain reaction (RT-PCR) were quickly frozen in liquid nitrogen after surgical resection and conserved at −80°C until use.
Isolation of glioma cells and astrocytes and cell culture
Glioma tumor samples and normal brain tissue were obtained from recent surgical resections in accordance with a Shandong University Institutional Review Board approved protocol concurrent with national regulatory standards and with the patients giving informed consent. Briefly, tumor samples or normal brain tissue were dissociated into single cell suspension using enzymatic and mechanical methods, and plated in Dulbecco’s modified Eagle’s medium (DMEM)/High Glucose (Hyclone) supplemented with 10% fetal bovine serum (FBS; Gibco), penicillin (100 U/ml) and streptomycin (100 μg/ml). All cells were maintained at 37°C in a humidified incubator with 5% CO2. The culture of human glioma cell lines U251, U87 and T98G was performed as described previously.Citation35,Citation42
Cell viability assays
Glioma cells were plated into 96-well culture plates at an optimal density of 5 × 103 cells/ml with 200 μl culture medium per well. The cells were allowed to recover and stabilize for 6 h, and then incubated with different concentrations of G-CSF for 24 or 48 h. Cell viability was tested by MTT (3-[4, 5- dimethylthiazol-2-yl]-2, 5-diphenyltetrazolium bromide) assay as described previously.Citation17 At least three independent experiments were conducted.
Bromodeoxyuridine (BrdU) incorporation assay
Glioma cells were plated into 12-well plates with poly-l-lysine coated coverslips, and treated with 0 or 100 ng/ml G-CSF for 24 h. Two hours before harvesting the cells, BrdU (Sigma) (10 μmol/l) was added into the medium. BrdU labeling was detected following the method described previously.Citation17 At least three independent experiments were conducted.
Colony formation assay
Glioma cells were dissociated into single cells and seeded on six-well culture plates at 1,000 cells/well in triplicate wells. The cells were allowed to recover for 6h before 0 or 100 ng/ml G-CSF was added into the medium. Incubation was for 7 d, after which the cells were fixed with methyl hydrate for 20 min, and then stained with Giemsa for 20 min. The numbers of colonies containing more than 50 cells were counted. The colony formation rate was obtained based on the colony numbers/the plated cells. All experiments were performed in triplicate.
Annexin V/propidium iodide (PI) staining assay
Early and late apoptotic changes in glioma cells were determined using an Annexin V/PI Apoptosis Detection Kit (Abcam). After incubation with G-CSFR antibody for 24 h, cells were washed once with PBS and resuspended with 100 μl binding buffer, then, 5 μl FITC-conjugated annexin V and 5 μl PI were added to the binding buffer and incubated for 20 min. After staining, 400 μl of buffer was added to the mixture and samples were stored at 4°C until data acquisition by flow cytometry (FC500, Beckman Coulter). Measurements were completed within 1 h. All experiments were performed in triplicate.
Invasion and migration assay
Invasion assay was measured in a Transwell culture chamber system. The filter membrane with 8-μm pores was coated with Matrigel (BD Biosciences). Cells were starved for 6 h, then suspended in serum-free DMEM with or without G-CSF at a density of 1 × 105/ml. 200 μl suspension was seeded into the upper chambers. Six hundred microliters of FBS containing medium was added to the lower chamber. Incubation was for 24 h. The upper chamber was removed and the adherent cells were gently detached from the upper surface of the membrane. The cells on the other side of the membrane were stained with 0.1% crystal violet in methanol for 10 min. The cells were viewed under a light microscope and photographed.
Transwell migration assay was employed to evaluate the protease-independent migration ability. No Matrigel coating was performed on the 8 μm pore membrane.
Wound-healing assay (WHA) method was also used to evaluate cell migration ability. 1 × 106 cells were seeded into a well of 6-well plate. When cells grew to about 80% confluency, the medium was changed to serum-free medium and incubated for 6 h. A 10-μl pipette was used to create a wound line. Cells were washed three times, and then incubated with 0 or 100 ng/ml G-CSF. Photographs were taken at 0 and 24 h at the same site, and the migration distance was measured.
Immunohistochemistry
Frozen sections of human glioma specimens on a glass slide were treated with 0.3% H2O2 for 15 min, and blocked with 5% goat serum for 30 min at room temperature. They were incubated with anti-G-CSFR antibody (1:100, Santa Cruz) overnight at 4°C. After washing with PBS, they were incubated with biotin conjugated secondary antibody for 30 min at room temperature. After further washing, they were treated with streptavidin ± biotin peroxidase complex for 30 min at room temperature. Diaminobenzidine tetrahydrochloride was used as the chromogen, and the cells were lightly counterstained with hematoxylin. For negative controls, cells were similarly processed without the primary antibody.
Immunofluorescence labeling
Glioma cells were plated on coverslips (5 × 104 cells/well) coated with poly-l-lysine. They were then fixed in 4% paraformaldehyde (PFA) and permeabilized with 0.1% Triton X-100. Subsequently, cells were incubated with primary antibody: anti-G-CSFR (1:100; rabbit polyclonal, Santa Cruz). TRITC-conjugated anti-rabbit IgG (Chemicon) was used as secondary antibodies. Cell nuclei were counterstained with 4′, 6′-diamidino-2-phenylindole (DAPI). Coverslips were mounted in 90% glycerol/PBS and analyzed using a fluorescence microscope (IX71, Olympus).
Western blots analysis
Equal amounts of proteins were loaded on 10% SDS-polyacrylamide gel. After electrophoresis, the proteins were transferred to Polyvinylidene fluoride (PVDF) membranes, and the blots were subsequently probed with the following antibodies: G-CSFR (1:500, Santa Cruz), STAT 3 (1:500, Santa Cruz), p-STAT 3 (1:1,000; CST) and β–actin (1:1,000, Abcam). For detection, horseradish peroxidase-conjugated secondary antibodies were used (1:1,000) followed by enhanced chemiluminescence development (Millipore). Normalization of the results was done by running parallel western blots using β-actin as control. The optical density was quantified using AlphaEase FC Version4 analysis software (Alphalmager HP, Alpha Innotech).
RNA extraction, Semiquantitative RT–PCR and Quantitative real time RT-PCR
Total RNA was extracted from glioma cells with 1 × 106 cells using the TRIZOL (Gibco, Invitrogen) method. The amount of RNA obtained was determined by spectrophotometric measurements at 260 nm. The sample was incubated for 10 min at 70°C followed by addition of 4 μl of 5 × transcription buffer, 2 μl of 0.1 M DTT, 1 μl of 10 mM dNTPs, and 200 U of Superscript II (Gibco, Invitrogen). The mixture was incubated for 60 min at 42°C. The final volume of the reaction was 20 μl. For PCR amplification, different amounts of the synthesized complement DNA (cDNA) (diluted 1:10 in water) were analyzed to evaluate the linearity of the reaction. Then polymerase reaction was performed in a solution contained 1.5 mM MgCl2, 200 μM of each nucleotide in PCR buffer, 10 pmol of the primers, and 0.25 U platinum Taq DNA polymerase (Gibco, Invitrogen). Primers used in RT-PCR reactions are shown in . The PCR products separated on 1.5% agarose/TAE gels were visualized by staining with ethidium bromide and semiquantified using AlphaEase FC Version4 analysis software (Alphalmager HP, Alpha Innotech). The densitometric analysis of the data was normalized to the β-actin. Results were mean ± SD from three separate experiments for each group.
Table 1. Primers for RT-PCR and real time RT-PCR.
For quantitive real-time PCR, the LightCycler 2.0 instrument (Roche Applied Science) and the LightCycler FastStart DNA Master SYBR Green I Reagent Kit (Roche Molecular Biochemicals) were used according to the protocol provided by the manufacturer. The primers are shown in . Melting curve analysis was used to confirm amplification specificity. The quantification data were analyzed with LightCycler analysis software version 4.0 (Roche Applied Science). The relative expression was normalized on the basis of β-actin. At least three independent experiments for each condition were conducted.
Statistical analysis
Quantitative data were presented as the mean ± SD of at least three independent experiments. Statistical analysis of data was performed by Student’s t-test or by one-way ANOVA using Dunnett’s test in multiple comparisons of means. Differences were considered statistically significant if the p value was < 0.05.
| Abbreviations: | ||
| Bax | = | Bcl-2 associated x protein |
| Bcl-2 | = | B cell lymphoma 2 |
| BrdU | = | Bromodeoxyuridine |
| CSF3R | = | colony-stimulating factor receptor 3 |
| DAPI | = | 4′, 6′-diamidino-2-phenylindole |
| DMEM | = | Dulbecco’s modified Eagle’s medium |
| FBS | = | fetal bovine serum |
| G-CSF | = | Granulocyte-colony stimulating factor |
| G-CSFR | = | G-CSF receptor |
| JAK | = | Janus kinase tyrosine kinase |
| MMP 2 | = | matrix metalloproteinase 2 |
| MMP 9 | = | matrix metalloproteinase 9 |
| MTT | = | 3-[4, 5- dimethylthiazol-2-yl]-2, 5-diphenyltetrazolium bromide |
| PFA | = | Paraformaldehyde |
| p-STAT 3 | = | phosphorylated signal transducer and activator transcription 3 |
| RT-PCR | = | reversed transcription polymerase chain reaction |
| STAT 3 | = | signal transducer and activator transcription 3 |
| WHA | = | wound-healing assay |
| 3 | = | |
Acknowledgments
This work was supported by funding from the National Natural Science Foundation of China (No. 30872645, 81071057), and the Natural Science Foundation of Shandong Province (No. Y2008C57, JQ200823).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 2007; 114:97 - 109; http://dx.doi.org/10.1007/s00401-007-0243-4; PMID: 17618441
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al, European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups, National Cancer Institute of Canada Clinical Trials Group. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005; 352:987 - 96; http://dx.doi.org/10.1056/NEJMoa043330; PMID: 15758009
- Kleihues P, Soylemezoglu F, Schäuble B, Scheithauer BW, Burger PC. Histopathology, classification, and grading of gliomas. Glia 1995; 15:211 - 21; http://dx.doi.org/10.1002/glia.440150303; PMID: 8586458
- Morstyn G, Burgess AW. Hemopoietic growth factors: a review. Cancer Res 1988; 48:5624 - 37; PMID: 2458827
- Solaroglu I, Cahill J, Jadhav V, Zhang JH. A novel neuroprotectant granulocyte-colony stimulating factor. Stroke 2006; 37:1123 - 8; http://dx.doi.org/10.1161/01.STR.0000208205.26253.96; PMID: 16514095
- Lee ST, Park JE, Kim DH, Kim S, Im WS, Kang L, et al. Granulocyte-colony stimulating factor attenuates striatal degeneration with activating survival pathways in 3-nitropropionic acid model of Huntington’s disease. Brain Res 2008; 1194:130 - 7; http://dx.doi.org/10.1016/j.brainres.2007.11.058; PMID: 18166168
- Mattei S, Colombo MP, Melani C, Silvani A, Parmiani G, Herlyn M. Expression of cytokine/growth factors and their receptors in human melanoma and melanocytes. Int J Cancer 1994; 56:853 - 7; http://dx.doi.org/10.1002/ijc.2910560617; PMID: 7509778
- Nakata H, Uemura Y, Kobayashi M, Harada R, Taguchi H. Cyclooxygenase-2 inhibitor NS-398 suppresses cell growth and constitutive production of granulocyte-colony stimulating factor and granulocyte macrophage-colony stimulating factor in lung cancer cells. Cancer Sci 2003; 94:173 - 80; http://dx.doi.org/10.1111/j.1349-7006.2003.tb01415.x; PMID: 12708493
- Chakraborty A, White SM, Lerner SP. Granulocyte colony-stimulating factor receptor signals for beta1-integrin expression and adhesion in bladder cancer. Urology 2004; 63:177 - 83; http://dx.doi.org/10.1016/S0090-4295(03)00786-6; PMID: 14751388
- Pirtskhalaishvili G, Nelson JB. Endothelium-derived factors as paracrine mediators of prostate cancer progression. Prostate 2000; 44:77 - 87; http://dx.doi.org/10.1002/1097-0045(20000615)44:1<77::AID-PROS10>3.0.CO;2-G; PMID: 10861760
- Nitta T, Sato K, Allegretta M, Brocke S, Lim M, Mitchell DJ, et al. Expression of granulocyte colony stimulating factor and granulocyte-macrophage colony stimulating factor genes in human astrocytoma cell lines and in glioma specimens. Brain Res 1992; 571:19 - 25; http://dx.doi.org/10.1016/0006-8993(92)90505-4; PMID: 1377084
- Mueller MM, Herold-Mende CC, Riede D, Lange M, Steiner HH, Fusenig NE. Autocrine growth regulation by granulocyte colony-stimulating factor and granulocyte macrophage colony-stimulating factor in human gliomas with tumor progression. Am J Pathol 1999; 155:1557 - 67; http://dx.doi.org/10.1016/S0002-9440(10)65472-7; PMID: 10550313
- Mueller MM, Peter W, Mappes M, Huelsen A, Steinbauer H, Boukamp P, et al. Tumor progression of skin carcinoma cells in vivo promoted by clonal selection, mutagenesis, and autocrine growth regulation by granulocyte colony-stimulating factor and granulocyte-macrophage colony-stimulating factor. Am J Pathol 2001; 159:1567 - 79; http://dx.doi.org/10.1016/S0002-9440(10)62541-2; PMID: 11583982
- Mueller MM, Fusenig NE. Constitutive expression of G-CSF and GM-CSF in human skin carcinoma cells with functional consequence for tumor progression. Int J Cancer 1999; 83:780 - 9; http://dx.doi.org/10.1002/(SICI)1097-0215(19991210)83:6<780::AID-IJC14>3.0.CO;2-C; PMID: 10597195
- Eaves C, Jiang X, Eisterer W, Chalandon Y, Porada G, Zanjani E, et al. New models to investigate mechanisms of disease genesis from primitive BCR-ABL(+) hematopoietic cells. Ann N Y Acad Sci 2003; 996:1 - 9; http://dx.doi.org/10.1111/j.1749-6632.2003.tb03226.x; PMID: 12799276
- Jung KH, Chu K, Lee ST, Kim SJ, Sinn DI, Kim SU, et al. Granulocyte colony-stimulating factor stimulates neurogenesis via vascular endothelial growth factor with STAT activation. Brain Res 2006; 1073-1074:190 - 201; http://dx.doi.org/10.1016/j.brainres.2005.12.037; PMID: 16423324
- Liu H, Jia D, Fu J, Zhao S, He G, Ling EA, et al. Effects of granulocyte colony-stimulating factor on the proliferation and cell-fate specification of neural stem cells. Neuroscience 2009; 164:1521 - 30; http://dx.doi.org/10.1016/j.neuroscience.2009.09.045; PMID: 19782730
- Zheng H, Ying H, Yan H, Kimmelman AC, Hiller DJ, Chen AJ, et al. Pten and p53 converge on c-Myc to control differentiation, self-renewal, and transformation of normal and neoplastic stem cells in glioblastoma. Cold Spring Harb Symp Quant Biol 2008; 73:427 - 37; http://dx.doi.org/10.1101/sqb.2008.73.047; PMID: 19150964
- Wang Y, Yang J, Zheng H, Tomasek GJ, Zhang P, McKeever PE, et al. Expression of mutant p53 proteins implicates a lineage relationship between neural stem cells and malignant astrocytic glioma in a murine model. Cancer Cell 2009; 15:514 - 26; http://dx.doi.org/10.1016/j.ccr.2009.04.001; PMID: 19477430
- Nicola NA. Hemopoietic cell growth factors and their receptors. Annu Rev Biochem 1989; 58:45 - 77; http://dx.doi.org/10.1146/annurev.bi.58.070189.000401; PMID: 2549855
- Ward AC, Hermans MH, Smith L, van Aesch YM, Schelen AM, Antonissen C, et al. Tyrosine-dependent and -independent mechanisms of STAT3 activation by the human granulocyte colony-stimulating factor (G-CSF) receptor are differentially utilized depending on G-CSF concentration. Blood 1999; 93:113 - 24; PMID: 9864153
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008; 455:1061 - 8; http://dx.doi.org/10.1038/nature07385; PMID: 18772890
- de Koning JP, Dong F, Smith L, Schelen AM, Barge RM, van der Plas DC, et al. The membrane-distal cytoplasmic region of human granulocyte colony-stimulating factor receptor is required for STAT3 but not STAT1 homodimer formation. Blood 1996; 87:1335 - 42; PMID: 8608222
- Rusthoven J, Bramwell V, Stephenson B, Provincial Systemic Treatment Disease Site Group. Use of granulocyte colony-stimulating factor (G-CSF) in patients receiving myelosuppressive chemotherapy for the treatment of cancer. Cancer Prev Control 1998; 2:179 - 90; PMID: 10093631
- Sabeh F, Shimizu-Hirota R, Weiss SJ. Protease-dependent versus -independent cancer cell invasion programs: three-dimensional amoeboid movement revisited. J Cell Biol 2009; 185:11 - 9; http://dx.doi.org/10.1083/jcb.200807195; PMID: 19332889
- Malipiero UV, Frei K, Fontana A. Production of hemopoietic colony-stimulating factors by astrocytes. J Immunol 1990; 144:3816 - 21; PMID: 1692062
- Huang H, Held-Feindt J, Buhl R, Mehdorn HM, Mentlein R. Expression of VEGF and its receptors in different brain tumors. Neurol Res 2005; 27:371 - 7; http://dx.doi.org/10.1179/016164105X39833; PMID: 15949234
- Joensuu H, Puputti M, Sihto H, Tynninen O, Nupponen NN. Amplification of genes encoding KIT, PDGFRalpha and VEGFR2 receptor tyrosine kinases is frequent in glioblastoma multiforme. J Pathol 2005; 207:224 - 31; http://dx.doi.org/10.1002/path.1823; PMID: 16021678
- Ohki Y, Heissig B, Sato Y, Akiyama H, Zhu Z, Hicklin DJ, et al. Granulocyte colony-stimulating factor promotes neovascularization by releasing vascular endothelial growth factor from neutrophils. FASEB J 2005; 19:2005 - 7; http://dx.doi.org/10.1096/fj.04-3496fje; PMID: 16223785
- Haura EB, Turkson J, Jove R. Mechanisms of disease: Insights into the emerging role of signal transducers and activators of transcription in cancer. Nat Clin Pract Oncol 2005; 2:315 - 24; http://dx.doi.org/10.1038/ncponc0195; PMID: 16264989
- Brantley EC, Benveniste EN. Signal transducer and activator of transcription-3: a molecular hub for signaling pathways in gliomas. Mol Cancer Res 2008; 6:675 - 84; http://dx.doi.org/10.1158/1541-7786.MCR-07-2180; PMID: 18505913
- Yin C, Knudson CM, Korsmeyer SJ, Van Dyke T. Bax suppresses tumorigenesis and stimulates apoptosis in vivo. Nature 1997; 385:637 - 40; http://dx.doi.org/10.1038/385637a0; PMID: 9024662
- Yin XM, Oltvai ZN, Korsmeyer SJ. BH1 and BH2 domains of Bcl-2 are required for inhibition of apoptosis and heterodimerization with Bax. Nature 1994; 369:321 - 3; http://dx.doi.org/10.1038/369321a0; PMID: 8183370
- McKernan DP, Dinan TG, Cryan JF. “Killing the Blues”: a role for cellular suicide (apoptosis) in depression and the antidepressant response?. Prog Neurobiol 2009; 88:246 - 63; http://dx.doi.org/10.1016/j.pneurobio.2009.04.006; PMID: 19427352
- Niola F, Evangelisti C, Campagnolo L, Massalini S, Buè MC, Mangiola A, et al. A plasmid-encoded VEGF siRNA reduces glioblastoma angiogenesis and its combination with interleukin-4 blocks tumor growth in a xenograft mouse model. Cancer Biol Ther 2006; 5:174 - 9; http://dx.doi.org/10.4161/cbt.5.2.2317; PMID: 16340308
- Brantley EC, Benveniste EN. Signal transducer and activator of transcription-3: a molecular hub for signaling pathways in gliomas. Mol Cancer Res 2008; 6:675 - 84; http://dx.doi.org/10.1158/1541-7786.MCR-07-2180; PMID: 18505913
- Weissenberger J, Loeffler S, Kappeler A, Kopf M, Lukes A, Afanasieva TA, et al. IL-6 is required for glioma development in a mouse model. Oncogene 2004; 23:3308 - 16; http://dx.doi.org/10.1038/sj.onc.1207455; PMID: 15064729
- Sabeh F, Shimizu-Hirota R, Weiss SJ. Protease-dependent versus -independent cancer cell invasion programs: three-dimensional amoeboid movement revisited. J Cell Biol 2009; 185:11 - 9; http://dx.doi.org/10.1083/jcb.200807195; PMID: 19332889
- Komatsu K, Nakanishi Y, Nemoto N, Hori T, Sawada T, Kobayashi M. Expression and quantitative analysis of matrix metalloproteinase-2 and -9 in human gliomas. Brain Tumor Pathol 2004; 21:105 - 12; http://dx.doi.org/10.1007/BF02482184; PMID: 15696970
- Zhang Q, Chen X, Zhou J, Zhang L, Zhao Q, Chen G, et al. CD147, MMP-2, MMP-9 and MVD-CD34 are significant predictors of recurrence after liver transplantation in hepatocellular carcinoma patients. Cancer Biol Ther 2006; 5:808 - 14; doi.org/10.4161/cbt.5.7.2754 http://dx.doi.org/10.4161/cbt.5.7.2754; PMID: 16775432
- Yamamoto N, Sekine I, Nakagawa K, Takada M, Fukuoka M, Tanigawara Y, et al. A pharmacokinetic and dose escalation study of pegfilgrastim (KRN125) in lung cancer patients with chemotherapy-induced neutropenia. Jpn J Clin Oncol 2009; 39:425 - 30; http://dx.doi.org/10.1093/jjco/hyp038; PMID: 19395467
- Zhang Y, Hao H, Zhao S, Liu Q, Yuan Q, Ni S, et al. Downregulation of GRIM-19 promotes growth and migration of human glioma cells. Cancer Sci 2011; 102:1991 - 9; http://dx.doi.org/10.1111/j.1349-7006.2011.02059.x; PMID: 21827581