Abstract
Neuroblastoma is the most common extracranial solid tumor in the pediatric population. Sorafenib (Nexavar), a multikinase inhibitor, blocks cell proliferation and induces apoptosis in certain types of cancers. Here, we tested antitumor effects of sorafenib (≤ 10 µM) on four human neuroblastoma cell lines, CHLA255, CHLA171, CHLA90 and SK-N-AS. Sorafenib inhibited cell proliferation and induced apoptosis of neuroblastoma tumor cells in a dose-dependent manner. Sorafenib inhibited phosphorylation of Signal Transducer and Activator of Transcription 3 (STAT3) proteins at Tyr705 in these cells, associated with inhibition of phosphorylated JAK2, an upstream kinase that mediates STAT3 phosphorylation. Expression of a constitutively-activated STAT3 mutant (pSTAT3-C) partially blocked the antitumor effects of sorafenib on neuroblastoma cells. Sorafenib also inhibited the phosphorylation of STAT3 induced by IL-6 and sphingosine-1-phosphate (S1P), a recently identified regulator for STAT3, in these tumor cells. Moreover, sorafenib downregulated phosphorylation of MAPK (p44/42) in neuroblastoma cells, consistent with inhibition of their upstream regulators MEK1/2. Sorafenib inhibited expression of cyclin E, cyclin D1/D2/D3, key regulators for cell cycle, and the antiapoptotic proteins Mcl-1 and survivin. Finally, sorafenib suppressed the growth of human neuroblastoma cells in a mouse xenograft model. Taken together, these findings suggest the potential use of sorafenib for the treatment of pediatric neuroblastomas.
Introduction
Neuroblastoma is the most common extracranial malignant tumor in infants and children and represents 8–10% of all childhood tumors.Citation1,Citation2 These tumors are derived from progenitor cells of the sympathetic nervous system. However, the mechanisms causing persistence of embryonal cells that later give rise to neuroblastic tumors are mainly unknown. A hallmark of neuroblastoma is cellular heterogeneity. Despite the advances in treatment options, clinical prognosis of aggressive neuroblastomas, especially in older patients (> 1 year) or with amplification of MYCN, remains dismal.Citation3 Combined chemotherapy failed to effectively eradicate the disease for advanced-stage neuroblastoma. Therefore, there is a critical need to find new drugs that are less toxic and target cell signaling pathways implicated as key mediators in the formation of neuroblastoma.
Sorafenib (Nexavar), a multi-kinase inhibitor, was originally developed for its inhibitory effect on Raf and receptor tyrosine kinase (RTK) signaling.Citation4 Recent findings showed that sorafenib inhibited tumor growth and angiogenesis, and induced apoptosis through either Raf-MEK-MAPK dependent or independent pathways, depending on the type of tumors being investigated.Citation5,Citation6 Sorafenib induces apoptosis in imatinib mesylate-resistant Bcr/Abl human leukemia cells in association with STAT5 inhibition.Citation7 We previously reported that sorafenib induces apoptosis and inhibits cell proliferation associated with the inhibition of STAT3 signaling in medulloblastomas and glioblastomas.Citation8,Citation9 Evaluation of sorafenib from Phase I and II clinical trials on several forms of advanced solid tumors showed favorable tolerability and promising clinical antitumor activity.Citation10-Citation12
The activity of STAT proteins, particularly STAT3, is frequently elevated in a wide variety of solid tumors and hematological malignancies, and is associated with proliferation and maintenance of tumors.Citation13,Citation14 Thus, STAT3 has emerged as a promising molecular target for cancer therapy.Citation15 STAT3 is activated when tyrosine residue 705 is phosphorylated by Janus tyrosine kinases (JAKs) or the proto-oncogene tyrosine protein kinase Src associated with cytokine receptors, such as these for interleukins and interferons.Citation14 Interleukin-6 (IL-6) is an important activator for STAT3 signaling pathway in normal and tumor cells.Citation16,Citation17 Various types of cancers metastasize to the bone, including neuroblastoma. IL-6 helps bone-homing cancer cells in facilitating bone invasion and growth of metastatic lesions.Citation18 Notably, IL-6 in bone marrow microenvironment promotes the growth and survival of neuroblastoma cells.Citation19 Currently, sphingosine-1-phosphate receptor-1 (S1PR1), a G protein-coupled receptor for sphingosine-1-phosphate (S1P), has been reported to upregulate STAT3 activaty in tumors via increasing JAK2 tyrosine kinase activity.Citation20 S1P-S1PR1-induced STAT3 activation is persistent, in contrast to transient STAT3 activation by IL-6. Interestingly, S1PR1 is elevated in STAT3-positive tumors, a positive feedback loop for persistent STAT3 activation.
In the present study, we show that sorafenib suppresses cell proliferation and induces apoptosis in four human neuroblastoma cell lines. Sorafenib inhibits phosphorylation of STAT3 at Tyr705 in these tumor cells, associated with inhibition of phosphorylated JAK2. Sorafenib also inhibits STAT3 phosphorylation induced by IL-6 and S1P. Sorafenib downregulates phosphorylation of MAPK (p44/42) and MEK1/2. Sorafenib inhibits the expression of cyclin E and cyclin D1/D2/D3, and antiapoptotic proteins, Mcl-1 and survivin. Finally, sorafenib blocks the growth of human neuroblastoma cells in a mouse xenograft model.
Results
Sorafenib inhibits proliferation and induces apoptosis in four human neuroblastoma cell lines
To investigate the effects of sorafenib on cell proliferation in neuroblastomas, we performed dose-response and time-course studies in four human neuroblastoma cell lines, SK-N-AS (), CHLA255 (), CHLA171 () and CHLA90 (). Cells were treated with increasing concentrations of sorafenib (1.5, 2.5, 5 µM) for 24 hours and 48 hours. Control cells were treated with the vehicle (DMSO) only. Because previous studies suggest that sorafenib binds to serum proteins,Citation21 all treatments with sorafenib were performed in 1% serum to reduce the effect of serum. Sorafenib markedly inhibited cell proliferation of all four cell lines in a dose- and time-dependent manner. We next investigated whether sorafenib could induce apoptosis in these tumor cells. After treatment with increasing concentrations of sorafenib (1.5, 2.5, 5, 10 µM) for 48 hours, cells were analyzed by Annexin V/propidium iodide (PI) staining and flow cytometry. Apoptotic cells shown in included both early apoptotic cells (Annexin V-positive) and late apoptotic cells (Annexin V- and PI-positive). Sorafenib inhibited survival of these tumor cells in a dose-dependent manner. These results show that proliferation and survival was greatly reduced for human neuroblastoma cells exposed to ≤ 10 µM sorafenib, which has been shown to be a therapeutically achievable concentration in clinical trials with doses of 400 mg sorafenib twice daily.Citation22
Figure 1. Sorafenib inhibits cell proliferation of human neuroblastoma cells. SK-N-AS (A), CHLA255 (B), CHLA171 (C) and CHLA90 (D) cells were treated with 0, 1.0, 2.5 or 5 µM sorafenib for 24 hours and 48 hours. Cell proliferation was evaluated by MTS assay. Sorafenib inhibits cell growth in all four human neuroblastoma cell lines. Each experiment was performed in triplicate or duplicate and repeated twice independently. Each bar graph represents the mean, and the error bars represent ± SD.
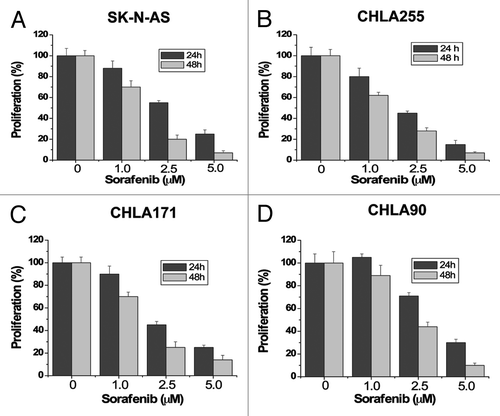
Figure 2. Sorafenib induces apoptosis of human neuroblastoma cells. SK-N-AS (A), CHLA255 (B), CHLA171 (C) and CHLA90 (D) cells were treated with 0, 1.0, 2.5, 5, or 10 µM sorafenib for 48 hours. Apoptotic cells represented Annexin V-FITC positive (early stage of apoptosis) or PI and Annexin V-FITC double-positive (late stage of apoptosis) cells as determined by flow cytometry. Each experiment was performed in triplicate or duplicate and repeated twice independently. Each bar graph represents the mean, and the error bars represent ± SD.
Effects of sorafenib on expression of regulatory proteins important for cell growth in neuroblastoma cells
Sorafenib inhibited cell growth of human neuroblastoma (). To investigate whether sorafenib affected the expression of regulatory proteins involved in cell cycle, immunoblotting analyses were performed for cyclin E and cyclin D1/D2/D3, the positive regulators for cell cycle, and p21Cip1 and p27Kip1, the negative regulators for cell cycle. showed that expression of cyclin E, cyclin D1, cyclin D2 and cyclin D3 was downregulated by sorafenib, while expression of p21Cip1 and p27Kip1 was not affected in CHLA255 and SK-N-AS cells. These results correlated with the inhibition of cell proliferation induced by sorafenib in neuroblastoma cells.
Figure 3. Effects of sorafenib on the expression of cell cycle and apoptosis regulatory proteins. (A and B) The role of sorafenib in the regulation of cell cycle key proteins. SK-N-AS and CHLA255 cells were treated with sorafenib (0, 2.5, 5 µM) for 24 hours. Sorafenib inhibited expression of cyclin E and cyclin D1/D2/D3 in these cells. (C and D) Sorafenib inhibited expression of anti-apoptotic proteins, Mcl-1 and survivin, in human neuroblastoma cells. Expression of four anti-apoptotic proteins, Mcl-1, Bcl-2, Bcl-xL and survivin, and three pro-apoptotic proteins, Bim, Bax and Bak, was detected in SK-N-AS and CHLA255 cells after 24 hours sorafenib treatment.
Effects of sorafenib on the expression of pro-apoptotic and anti-apoptotic genes in human neuroblastoma cells
Bcl-2 family proteins have a critical role in survival of normal and tumor cells.Citation23 The expression of three anti-apoptotic proteins, Mcl-1, Bcl-2 and Bcl-xL, and three pro-apoptotic proteins, Bak, Bax and Bim from the Bcl-2 family was further investigated in neuroblastoma tumor cells after sorafenib treatment. Though Bcl-2, Bcl-xL, Bax and Bak were not affected by sorafenib, decreased Mcl-1 protein level was observed after sorafenib treatment in SK-N-AS and CHLA255 cells (). Bim was increased in CHLA255 cells.
The anti-apoptotic protein survivin is a negative prognostic marker in neurooblastoma patients and knockdown of survivin causes apoptosis of neuroblastoma cells.Citation24 We therefore investigated whether its expression was inhibited by sorafenib in these tumor cells. Expression of survivin was reduced by sorafenib in SK-N-AS and CHLA255 cells by immunoblotting assays (). Suppression of Mcl-1 and survivin function may promote cytochrome c release, resulting in induction of apoptosis by the intrinsic pathway.
Sorafenib reduces phosphorylation of STAT3 and MAPK (p44/42) in human neuroblastoma cells
We investigated the levels of total and phosphorylated STAT3, AKT and MAPK (p44/42) proteins in SK-N-AS and CHLA255 cells after sorafenib treatment. Cells were treated with 5 µM sorafenib for 0, 1, 5, 15, 30 or 45 min respectively. Total protein levels of STAT3, AKT, and MAPK were not significantly changed after sorafenib treatment (). Inhibition of phosphorylated STAT3 at Tyr705 was detected between 5 and 15 min following sorafenib treatment. Phosphorylation of STAT3 at Ser727 was also inhibited by sorafenib treatment, but starting time of inhibition about 15 min later than one of Tyr705. The inhibition of phosphorylated STAT3 was time-dependent. Phosphorylation of MAPK (p44/42) was strongly decreased after sorafenib treatments in these cells. However, phosphorylation of AKT did not change substantially. These results indicate that inhibition of STAT3 and MAPK (p44/42) signaling is an early response to sorafenib treatment in neuroblastoma cells.
Figure 4. Phosphorylation of STAT3 and JAK2 is inhibited by sorafenib and constitutively activated STAT3 mutant partially blocks the inhibiting effects of sorafenib. (A) Sorafenib inhibited phosphorylated STAT3 and MAPK(p44/42) in SK-N-AS and CHLA255 human neuroblastoma cells. Cells were treated with 5 µM sorafenib for 0, 1, 5, 15, 30 and 45 minutes. Then total proteins were prepared and immunoblottings were performed with specific antibodies. β-actin works as an internal control. (B) SK-N-AS cells were stably transfected with a constitutively activated STAT3 mutant, pSTAT3-C, and the success of transfection was confirmed by immunobloting assay with flag-antibody (left panel). SK-N-AS cells transfected with pSTAT3-C or vector only were treated with 5 µM sorafenib for 24 hours and 48 hours, and cell proliferation was evaluated by MTS assay. (C) Effects of sorafenib on phosphorylation of JAK2 and Src. After 5 µM sorafenib treatment with different times as indicated in the figure, results from immunoblottings showed that sorafenib inhibited activation of JAK2.
Overexpression of constitutively activated STAT3 partially reverses the effects of sorafenib
To further confirm that inhibition of STAT3 activity is critical for the biological effects of sorafenib on neuroblastoma tumor cells, a constitutively-activated STAT3 mutant (pSTAT3-C)Citation25 was transfected into SK-N-AS cells. Stable cell lines were established by antibiotic selection (G418) and confirmed by immunoblotting analysis (). pRC was used as a control vector in the transfection and subsequent analyses. Cells containing either pSTAT3-C or control vector were treated with 5 µM sorafenib for 24 hours and 48 hours, and then proliferation assays were performed. Expression of constitutively-activated STAT3 increased the resistance of SK-N-AS cells to sorafenib compared with control transfected cells ().
Phosphorylated JAK2 is inhibited by sorafenib in human neuroblastoma cells
Phosphorylation of STAT3 at Tyr705 is usually mediated by receptor-associated tyrosine kinases (JAKs) or non-receptor tyrosine kinases (Src).Citation13 Inhibiting the phosphorylation of STAT3 (Tyr705) by sorafenib was detected at 5 min in SK-N-AS cells and 15 min in CHLA255 cells after treatment (). To elucidate how sorafenib rapidly inhibited phosphorylated STAT3 at Tyr705, total and phosphorylated proteins of JAK2, and Src were examined after sorafenib treatment in SK-N-AS and CHLA255 cells. Inhibiting phosphorylated JAK2 by sorafenib was detected after 5 min treatment (), which was associated with the inhibition of phosphorylated STAT3 at Tyr705.
Sorafenib inhibits phosphorylation of STAT3 induced by IL-6 and S1P in human neuroblastoma cells
IL-6 is a critical cytokine to activate JAK2-STAT3 signaling in normal and cancer cells. It has been reported that stromal-derived IL-6 contributes to the formation of a bone marrow microenvironment favorable to the progression of metastatic neuroblastoma.Citation19 Therefore, we investigate whether sorafenib inhibits the phosphorylation of STAT3 induced by IL-6 in human neuroblastoma cells. SK-N-AS and CHLA255 cells were pre-treated with sorafenib (5 µM) or vehicle (DMSO) for 20 min, and then IL-6 (10 ng/ml) was added to these cells for 10 min. Immunoblotting analyses showed that IL-6 increased phosphorylation of STAT3 at Tyr705, not Ser727 (). Sorafenib treatment blocked the effect of IL-6 on the induction of STAT3 phosphorylation ().
Figure 5. Sorafenib inhibits the phosphorylation of STAT3 induced by IL-6 and S1P in neuroblastoma cells. (A) Sorafenib inhibited the phosphorylation of STAT3 at Tyr705 induced by IL-6. SK-N-AS and CHLA255 neoroblastoma cells were treated with 5 µM sorafenib for 20 minutes, and then cells were added 10 ng/ml IL-6 for another 10 minutes. Phosphorylation of STAT3 was detected by immunoblottings. (B) Expression of S1PR1 in human neuroblastoma cells was detected by immunoblottings. (C) S1P induced phosphorylation of STAT3 at Tyr705. CHLA255 and CHLA171 cells were treated with 100 nM S1P for one hour. IL-6 treatment works as positive control. (D) Sorafenib blocked the induction of phosphorylated STAT3 by S1P. Before S1P treatment (100 nM), CHLA255 and CHLA171 cells were treated with 5 µM sorafenib for 20 minutes. Then immunobloting analyses were performed to detect the level of phosphorylated STAT3.
Recently, S1P-S1PR1 signaling has emerged as a new regulator for STAT3 activation via JAK2 in cancer cells.Citation20 In order to clarify the role of S1P in human neuroblastoma cells, we first investigate whether these cells express the receptors for the S1P. showed expression of S1PR1 in all four cell lines we tested. S1P treatment (100 nM, 1 h) increased phosphorylation of STAT3 at Tyr705 in neuroblastoma cells (), and IL-6 treatment worked as a positive control in these tumor cells. Interestingly, pre-treatment of 5 µM sorafenib for 20 minutes strongly blocked the phosphorylation of STAT3 induced by S1P ().
Phosphorylation of MEK1/2 is suppressed by sorafenib in human neuroblastoma cells
Since phosphorylated MAPK (p44/42) was quickly inhibited by sorafenib (), we further investigated whether the activities of B-Raf, C-Raf and MEK1/2, the upstream regulators of MAPK (p44/42), were affected by sorafenib treatment in human neuroblastoma cells. Phosphorylated B-Raf and C-Raf was transiently inhibited in SK-N-AS cells (), between 1 to 30 minutes after 5 µM sorafenib treatment. In CHLA255 cells, phosphorylated C-Raf was inhibited in time-dependent manner, but phosphorylated B-Raf was not affected (). However, the phosphorylation of MEK1/2 was markedly suppressed with a time-dependent manner in both SK-N-AS and CHLA255 cells (). The inhibition of phosphorylated MEK1/2 is associated with downregulation of MAPK (p44/42) activities.
Figure 6. Effects of sorafenib on the activation of B-Raf, C-Raf and MEK1/2, and expression of cell cycle regulatory proteins. (A) SK-N-AS and CHLA255 cells were treated with 5 µM sorafenib for 0, 1, 5, 15, 30 and 45 minutes. Then, total cell proteins were assayed for expression of phosphorylated B-Raf, C-Raf and MEK1/2 with specific antibodies. (B) Sorafenib suppressed tumor growth of neuroblastoma in a mice xenograft model. SK-N-AS neuroblastoma tumor cells (5 x 106 per animal) were impanted s.c. in female NOD/SCID/IL2Rγ null mice (n = 5). Sorafenib and the vehicle were administered at 60 mg/kg, once daily by oral gavage.
Sorafenib inhibits growth of human neuroblastoma tumor xenografts in mice
To examine the effect of sorafenib on growth of neuroblastoma cells in vivo, NOD/SCID/IL2Rgamma null mice were inoculated with SK-N-AS cells subcutaneously and were administered sorafenib by oral gavage (60 mg/kg, daily). At this dose, no lethal toxicity or weight loss was observed among treated animals. Sorafenib treatment started when tumors become palpable (about 80 mm3). After eight days treatment, sorafenib strongly inhibited the growth of neuroobastomas in mice as shown in compared with vehicle group.
Discussion
Neuroblastoma, a neoplasm of the sympathetic nervous system, predominantly affects children under 5 y of age. In the past 20 y, therapeutic advances have failed to significantly increase the 5-y survival rates of children with aggressive, advanced-stage neuroblastomas.Citation26 Thus, neuroblastoma continues to remain a clinical challenge. In this study, we report that sorafenib inhibits cell proliferation and induces apoptosis of human neuroblastoma cells, associated with suppression of JAK2/STAT3 and MEK(1/2)/MAPK(p44/42) signaling pathways.
STAT3 regulates basic biologic processes important in tumorigenesis including cell-cycle progression, apoptosis, tumor angiogenesis, and tumor-cell evasion of the immune system.Citation13,Citation14 STAT3 activation requires Tyr705 phosphorylation via JAKs or Src, resulting in dimerization, nuclear translocation, DNA binding and transcriptional activation of target genes.Citation27 Inhibition of phosphorylated STAT3 at Tyr705 by sorafenib in neuroblastoma cells can be detected between 5 to 15 minutes after treatment, which is correlated with the inhibition of phosphorylated JAK2, the upstream regulator of STAT3. Phosphorylated STAT3 at Ser727 is also reduced by sorafenib. Since phoshorylation of STAT3 at Ser727 further enhances transcriptional activation of STAT3 target genes,Citation27 this inhibition on Ser727 will increase the antitumor effects of sorafenib on these cells. Cytokine IL-6 is a critical activator for JAKs/STAT3 signaling in normal and tumor cells. Stromal-derived IL-6 in bone marrow microenvironment promotes the growth and survival of neuroblastoma cells.Citation19 Almost half of all patients with neuroblastoma have disease dissemination at diagnosis. Bone marrow is a common place for neuroblastoma metastasis. Our data showed that sorafenib inhibits the phosphorylation of STAT3 at Tyr705 induced by IL-6 in human neuroblastoma cells. Recently, it has been reported that S1P-S1PR1 signaling induces the persistent activation of STAT3 at Tyr705 in tumor cells via increasing JAK2 tyrosine kinase activity.Citation20 Our study demonstrates that sorafenib inhibits phosphorylation of STAT3 induced by S1P. Therefore, these findings may be beneficial for the treatment of neuroblastoma, especially metastatic neuroblastoma.
Cyclin D1, a critical gene in cell cycle control, is regulated by STAT3.Citation13 Therefore, inhibition of STAT3 activation by sorafenib is likely to contribute to inhibition of cell proliferation. Our data show that D-type and E-type cyclins are common targets for sorafenib in human neuroblastomas. Expression of Mcl-1 and survivin, the antiapoptotic proteins, is also regulated by STAT3 signaling pathway.Citation13 Sorafenib inhibits expression of both Mcl-1 and survivin in human neuroblastomas. These results are consistent with inhibiting proliferation and inducing apoptosis by sorafenib in neuroblastoma cells.
The Raf-MEK-MAPK signaling pathway promotes cell proliferation, cell survival and metastasis, along with the overwhelming frequency in which this pathway is aberrantly activated in cancer.Citation28 Sorafenib inhibits activation of MEK1/2-MAPK(p44/42) signaling in human neuroblastoma cells. This action should enhance the inhibitory effects of sorafenib on these tumor cells. Sorafenib exhibits strong antiangiogenic activity via inhibition of vascular endothelial growth factor receptors (VEGFRs) and platelet-derived growth factor receptor (PDGFR).Citation21 We demonstrate that sorafenib inhibits the expression of VEGFR and PDGFR in neuroblastoma cells (data not shown). In summary, sorafenib is potentially a promising drug for the treatment of pediatric neuroblastomas.
Materials and Methods
Reagents and antibodies
Sorafenib was purchased from LC Laboratories. Anti-cyclin D1, and anti-cyclin D3 were obtained from Calbiochem. Anti-cyclin E was obtained from BD Biosciences. Anti-cyclin D2 and anti-Mcl-1 were obtained from Santa Cruz. Horseradish peroxidase-labeled anti-mouse and anti-rabbit secondary antibodies were from GE Healthcare. All other antibodies were purchased from Cell Signaling.
Cell culture
The human neuroblastoma cell line SK-N-AS was from the American Type Culture Collection. CHLA255, CHLA171 and CHLA90 human neuroblastoma cells were obtained from Dr. C. Patrick Reynolds (Children Hospital Los Angeles). Human neuroblastoma cells were maintained in Iscove’s Modified Dulbecco’s Medium supplemented with 10% FBS and 1% antibiotic-antimycotic. All cultured cells were grown in a humidified atmosphere of 5% CO2 at 37°C.
Proliferation assays
Cell proliferation assays were performed with CellTiter 96 Aqueous One Solution Cell proliferation Assay from Promega which contains 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS). Each well of 96-well plate was seeded with 5000 cells in culture medium. After overnight culture (16 hours) the cells were treated with different concentrations of sorafenib and controls were treated with vehicle (DMSO). After 24 hours or 48 hours treatment, MTS was added to the cells according to the supplier’s protocol and absorbance was measured at 490 nm using an automated ELISA plate reader.
Apoptosis assay
SK-N-AS, CHLA255, CHLA171 and CHLA90 cells (2 x 105) were seeded in 60 mm culture dishes in culture medium. The following day the cells were treated with indicated concentrations of sorafenib for 48 hours. After treatment, all cells including both detached and attached cells were collected, and the apoptotic cells were detected by Annexin V-FITC Apoptosis Detection Kit (BD Biosciences). The cells were stained with Annexin V-FITC and propidium iodide (PI) according to the supplier’s instructions. Viable and apoptotic cells were detected by flow cytometry in the Analytical Cytometry Core at City of Hope National Medical Center. Apoptotic cells include both the early apoptotic portion (Annexin V-positive) and the late apoptotic portion (Annexin V- and PI-positive).
Immunoblotting analysis
Twenty µg total proteins were resolved in a 4–15% gradient TRIS-HCl gel from BIO-RAD. After gel electrophoresis, the proteins were transferred to Hybond-C membranes (Amersham). The membranes were blocked for 1 h at room temperature (RT) in 10% non-fat dry milk in PBST (1 X PBS with 0.1% Tween-20), followed by an overnight incubation at 4°C with primary antibodies in PBST with 2% non-fat dry milk. Horseradish peroxidase labeled anti-mouse or anti-rabbit secondary antibodies were incubated 1 h at RT. Immunoreactivity was detected with SuperSignal West Pico substrate (Pierce).
Plasmids transfections
The constitutively-activated STAT3 mutant plasmid (pSTAT3-C) was murine STAT3 which was cloned into pRc/CMV vector with a FLAG epitope.Citation25 pSTAT3-C was transfected into SK-N-AS cells by LipofectamineTM 2000 (Invitrogen). Stable cell line was selected by G418 and confirmed by immunoblotting analysis.
Tumor xenografts
Female NOD/SCID/IL2Rgamma null (NSG, Stock # 005557) mice at 6–8 weeks old from Jackson Laboratories (Bar Harbor, Maine) were used for the in vivo preclinical therapeutic experiment. SK-N-AS cells at 5 x 106 cells in 100 microliter serum free medium were inoculated subcutaneously into the dorsal skin of the mice. Sorafenib was first dissolved in DMSO (final 5%) and then diluted with 30% Solutol HS15 (BASF) in water. When tumors become palpable (about 80 mm3), the mice were randomly divided into two groups with 5 mice per group. Sorafenib and the vehicle were then administered at 60 mg/kg, once daily by oral gavage with the protocol approved by the IACUC of City of Hope.
Acknowledgments
Supported by ThinkCure foundation (to H.Y.) and NCI grant CA1155674 (to R.J.).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Esiashvili N, Anderson C, Katzenstein HM. Neuroblastoma. Curr Probl Cancer 2009; 33:333 - 60; http://dx.doi.org/10.1016/j.currproblcancer.2009.12.001; PMID: 20172369
- Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet 2007; 369:2106 - 20; http://dx.doi.org/10.1016/S0140-6736(07)60983-0; PMID: 17586306
- Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer 2003; 3:203 - 16; http://dx.doi.org/10.1038/nrc1014; PMID: 12612655
- Wilhelm S, Carter C, Lynch M, Lowinger T, Dumas J, Smith RA, et al. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov 2006; 5:835 - 44; http://dx.doi.org/10.1038/nrd2130; PMID: 17016424
- Liu L, Cao Y, Chen C, Zhang X, McNabola A, Wilkie D, et al. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res 2006; 66:11851 - 8; http://dx.doi.org/10.1158/0008-5472.CAN-06-1377; PMID: 17178882
- Chang YS, Adnane J, Trail PA, Levy J, Henderson A, Xue D, et al. Sorafenib (BAY 43-9006) inhibits tumor growth and vascularization and induces tumor apoptosis and hypoxia in RCC xenograft models. Cancer Chemother Pharmacol 2007; 59:561 - 74; http://dx.doi.org/10.1007/s00280-006-0393-4; PMID: 17160391
- Rahmani M, Nguyen TK, Dent P, Grant S. The multikinase inhibitor sorafenib induces apoptosis in highly imatinib mesylate-resistant bcr/abl+ human leukemia cells in association with signal transducer and activator of transcription 5 inhibition and myeloid cell leukemia-1 down-regulation. Mol Pharmacol 2007; 72:788 - 95; http://dx.doi.org/10.1124/mol.106.033308; PMID: 17595328
- Yang F, Van Meter TE, Buettner R, Hedvat M, Liang W, Kowolik CM, et al. Sorafenib inhibits signal transducer and activator of transcription 3 signaling associated with growth arrest and apoptosis of medulloblastomas. Mol Cancer Ther 2008; 7:3519 - 26; http://dx.doi.org/10.1158/1535-7163.MCT-08-0138; PMID: 19001435
- Yang F, Brown C, Buettner R, Hedvat M, Starr R, Scuto A, et al. Sorafenib induces growth arrest and apoptosis of human glioblastoma cells through the dephosphorylation of signal transducers and activators of transcription 3. Mol Cancer Ther 2010; 9:953 - 62; http://dx.doi.org/10.1158/1535-7163.MCT-09-0947; PMID: 20371721
- Abou-Alfa GK, Schwartz L, Ricci S, Amadori D, Santoro A, Figer A, et al. Phase II study of sorafenib in patients with advanced hepatocellular carcinoma. J Clin Oncol 2006; 24:4293 - 300; http://dx.doi.org/10.1200/JCO.2005.01.3441; PMID: 16908937
- Strumberg D, Clark JW, Awada A, Moore MJ, Richly H, Hendlisz A, et al. Safety, pharmacokinetics, and preliminary antitumor activity of sorafenib: a review of four phase I trials in patients with advanced refractory solid tumors. Oncologist 2007; 12:426 - 37; http://dx.doi.org/10.1634/theoncologist.12-4-426; PMID: 17470685
- Gridelli C, Maione P, Del Gaizo F, Colantuoni G, Guerriero C, Ferrara C, et al. Sorafenib and sunitinib in the treatment of advanced non-small cell lung cancer. Oncologist 2007; 12:191 - 200; http://dx.doi.org/10.1634/theoncologist.12-2-191; PMID: 17296815
- Yu H, Jove R. The STATs of cancer--new molecular targets come of age. Nat Rev Cancer 2004; 4:97 - 105; http://dx.doi.org/10.1038/nrc1275; PMID: 14964307
- Haura EB, Turkson J, Jove R. Mechanisms of disease: Insights into the emerging role of signal transducers and activators of transcription in cancer. Nat Clin Pract Oncol 2005; 2:315 - 24; http://dx.doi.org/10.1038/ncponc0195; PMID: 16264989
- Pathak AK, Bhutani M, Nair AS, Ahn KS, Chakraborty A, Kadara H, et al. Ursolic acid inhibits STAT3 activation pathway leading to suppression of proliferation and chemosensitization of human multiple myeloma cells. Mol Cancer Res 2007; 5:943 - 55; http://dx.doi.org/10.1158/1541-7786.MCR-06-0348; PMID: 17855663
- Catlett-Falcone R, Landowski TH, Oshiro MM, Turkson J, Levitzki A, Savino R, et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity 1999; 10:105 - 15; http://dx.doi.org/10.1016/S1074-7613(00)80011-4; PMID: 10023775
- Bollrath J, Phesse TJ, von Burstin VA, Putoczki T, Bennecke M, Bateman T, et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell 2009; 15:91 - 102; http://dx.doi.org/10.1016/j.ccr.2009.01.002; PMID: 19185844
- Tawara K, Oxford JT, Jorcyk CL. Clinical significance of interleukin (IL)-6 in cancer metastasis to bone: potential of anti-IL-6 therapies. Cancer Manag Res 2011; 3:177 - 89; PMID: 21625400
- Ara T, Song L, Shimada H, Keshelava N, Russell HV, Metelitsa LS, et al. Interleukin-6 in the bone marrow microenvironment promotes the growth and survival of neuroblastoma cells. Cancer Res 2009; 69:329 - 37; http://dx.doi.org/10.1158/0008-5472.CAN-08-0613; PMID: 19118018
- Lee H, Deng J, Kujawski M, Yang C, Liu Y, Herrmann A, et al. STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat Med 2010; 16:1421 - 8; http://dx.doi.org/10.1038/nm.2250; PMID: 21102457
- Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res 2004; 64:7099 - 109; http://dx.doi.org/10.1158/0008-5472.CAN-04-1443; PMID: 15466206
- Strumberg D, Richly H, Hilger RA, Schleucher N, Korfee S, Tewes M, et al. Phase I clinical and pharmacokinetic study of the Novel Raf kinase and vascular endothelial growth factor receptor inhibitor BAY 43-9006 in patients with advanced refractory solid tumors. J Clin Oncol 2005; 23:965 - 72; http://dx.doi.org/10.1200/JCO.2005.06.124; PMID: 15613696
- Maddika S, Ande SR, Panigrahi S, Paranjothy T, Weglarczyk K, Zuse A, et al. Cell survival, cell death and cell cycle pathways are interconnected: implications for cancer therapy. Drug Resist Updat 2007; 10:13 - 29; http://dx.doi.org/10.1016/j.drup.2007.01.003; PMID: 17303468
- Lamers F, van der Ploeg I, Schild L, Ebus ME, Koster J, Hansen BR, et al. Knockdown of survivin (BIRC5) causes apoptosis in neuroblastoma via mitotic catastrophe. Endocr Relat Cancer 2011; 18:657 - 68; http://dx.doi.org/10.1530/ERC-11-0207; PMID: 21859926
- Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, et al. Stat3 as an oncogene. Cell 1999; 98:295 - 303; http://dx.doi.org/10.1016/S0092-8674(00)81959-5; PMID: 10458605
- Ishola TA, Chung DH. Neuroblastoma. Surg Oncol 2007; 16:149 - 56; http://dx.doi.org/10.1016/j.suronc.2007.09.005; PMID: 17976976
- Clevenger CV. Roles and regulation of stat family transcription factors in human breast cancer. Am J Pathol 2004; 165:1449 - 60; http://dx.doi.org/10.1016/S0002-9440(10)63403-7; PMID: 15509516
- Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007; 26:3291 - 310; http://dx.doi.org/10.1038/sj.onc.1210422; PMID: 17496923