Abstract
Agonistic antibodies targeting Fn14, the receptor for TWEAK, have demonstrated anti-tumor activity in xenograft models. Herein, we further explore the therapeutic potential of the humanized anti-Fn14 agonistic antibody, BIIB036, as a single agent and in combination with standard of care cancer therapeutics. Pharmacokinetic studies of BIIB036 in tumor-bearing mice revealed a half-life of approximately three days suggesting twice a week dosing would be necessary to maintain efficacy. However, in multiple xenograft models, BIIB036 treatment resulted in extended tumor growth inhibition up to 40–50 d following cessation of dosing, suggesting that frequent administration of BIIB036 may not be necessary to maintain prolonged anti-tumor activity. Subsequent xenograft studies revealed that maximal efficacy was achieved with BIIB036 dosing once every two weeks, by either intraperitoneal or subcutaneous administration. Xenograft tumors that were initially treated with BIBI036 and then re-grew up to 1000 mm3 following cessation of the first cycle of treatment remained sensitive to a second cycle of treatment. BIIB036 was also evaluated in patient derived primary colon tumor models, where efficacy compared favorably with a standard of care agent. Lastly, BIIB036 enhanced the efficacy of several standard of care chemotherapeutics, including paclitaxel in MDA-MBA-231 breast tumor xenografts, paclitaxel or carboplatin in HOP62 non-small cell lung xenografts, and 5-FU in NCI-N87 gastric xenografts, with no overlapping toxicities. These studies thus establish BIIB036 as a promising therapeutic agent with durable anti-tumor activity in human xenografts as well as patient derived primary tumor models, and enhanced activity and tolerability in combination with standard of care chemotherapeutics. Taken together, the data presented herein suggest that BIIB036 warrants evaluation in the clinic.
Introduction
The approach to cancer treatment has undergone a revolutionary change with the development of targeted therapeutics. One exciting avenue that has emerged is targeting death domain containing receptors and related family members with agonistic agents as a means to directly induce tumor cell death. Among the most promising therapeutics in this category are those targeting the TNF-related apoptosis inducing ligand (TRAIL/Apo2L) pathway, in the form of agonistic antibodies to the TRAIL receptors (TRAIL-R1 (DR4) and TRAIL-R2 (DR5)) and soluble TRAIL, all of which are currently undergoing clinical evaluation (For a review, see ref. Citation1).
More recently, the TNF superfamily member FGF-inducible molecule 14 (Fn14), the receptor for TNF-like weak inducer of apoptosis (TWEAK), has emerged as a target for agonistic antibody therapy. Since Fn14 is found at relatively low levels on normal tissues, and its expression is upregulated on many human tumor types, including breast, lung, pancreatic, esophageal and glioma,Citation2-Citation7 this may offer an attractive therapeutic window for targeting Fn14. Fn14 expression has also been detected in bone and liver metastases.Citation2 In addition, several studies have also shown that Fn14 levels in multiple tumor types correlate with tumor grade and poor prognosis.Citation6-Citation8 TWEAK ligand or agonistic antibodies to Fn14 can induce tumor cell death in vitro and in vivo.Citation2,Citation9-Citation15 Interestingly, despite lacking a death domain, Fn14 can mediate tumor cell death, similar to other non-death domain containing TNF family member receptors, such as lymphotoxin-β receptor (LTβR), CD30 and CD40.Citation15-Citation17
We recently described development of a humanized IgG1 anti-Fn14 agonistic antibody, BIIB036.Citation10 BIIB036 mimics many of the activities of the endogenous ligand, TWEAK, such as induction of NFκB signaling and tumor cell killing. Significantly, BIIB036 is efficacious in inhibiting tumor growth in multiple xenograft models. While BIIB036 can directly induce cell killing on tumor cells in vitro, maximal activity of BIIB036 in vivo requires Fc effector function.
Herein, we expand our investigation of BIIB036 to further explore its therapeutic potential for the treatment of cancer. Specifically, we evaluated the pharmacokinetic properties of BIIB036 and determined the optimal dosing schedule. Although the PK studies demonstrated that BIIB036 had a half-life of three days, extended efficacy was observed in xenograft models well beyond the dosing period. Every other week dosing schedule was determined to be optimal for maximal efficacy in these xenograft models. Furthermore, BIIB036 was efficacious in inhibiting the growth of large (~500 mm3) tumors, and tumors that received a first cycle of BIIB036 treatment maintained sensitivity upon re-treatment. Importantly, BIIB036 was also efficacious in inhibiting the growth of patient derived primary colorectal tumors grown in SCID mice. Finally, in combination, BIIB036 was able to enhance efficacy of several standard of care chemotherapeutic agents.
Results
Pharmacokinetic analysis of BIIB036
In order to determine the pharmacokinetic properties of BIIB036, WiDr colon tumor bearing mice were treated with a single IP dose (6.4 mg/kg) of BIIB036, and serum was collected at various time points following dosing. Pharmacokinetic analysis of BIIB036 levels in the serum revealed a serum half-life of 3.5 d (). A second arm of the pharmacokinetic study was designed to model the dosing schedule previously shown to be efficacious in xenograft models,Citation10 and accordingly WiDr tumor bearing mice received six weekly 6.4 mg/kg doses of BIIB036. Pharmacokinetic analysis revealed a similar half-life (3.1 d), with no evidence of accumulation of BIIB036 over time with subsequent doses. Additional single dose pharmacokinetic studies revealed a relatively similar pharmacokinetic profile in non-tumor bearing mice, and dose proportional exposure to BIIB036 at a range of doses (data not shown).
Table 1. Pharmacokinetic Parameters of BIIB036 Administered Intraperitoneally (6.4 mg/kg) in Adult Female (Nude) Tumor Bearing Mice
Extended efficacy of BIIB036 following termination of dosing
The pharmacokinetic studies suggested optimal dosing of BIIB036 for efficacy in tumor-bearing mice to be twice weekly. However, in early studies with BIIB036 we had determined that dosing of BIIB036 either on a twice a week or once a week dosing schedule resulted in similar efficacy in xenograft models (data not shown). Therefore, in the current studies we evaluated efficacy of BIIB036 administered intraperitoneally (IP) on a once per week (QW) schedule for six weeks in multiple xenograft models at doses ranging from 3.2 to 25.6 mg/kg, with the aim of determining efficacy over the course of the dosing period as well as beyond termination of dosing. Xenograft models established from several Fn14-positive tumor cell linesCitation10 were selected for this study. Statistically significant (p < 0.0002) efficacy compared with Ig treated controls was observed at all doses over the course of the dosing period (), confirming our previous observations.Citation10
Figure 1. BIIB036 inhibits tumor growth in xenograft models during and beyond the period of dosing. (A) Efficacy of BIIB036 treatment in WiDr colon carcinoma xenograft model is shown. Tumor volume as a function of time is plotted. Nude mice implanted with WiDr tumor cells were treated with varying doses of BIIB036 (25.6, 12.8, 6.4 or 3.2 mg/kg) or human Ig control (25.6 mg/kg) QW for 6 weeks. Data are mean ± SEM of n = 10 mice per group. Significant efficacy is evident at all doses relative to control group (p < 0.002 on day 83 for all dose groups). Extended efficacy > 50 d beyond the dosing period is observed. (B) Efficacy of BIIB036 treatment in MDA-MB-231 breast carcinoma xenograft model is shown. SCID mice implanted with MDA-MB-231 tumor cells were treated with varying doses of BIIB036 (25.6, 12.8 or 6.4 mg/kg) or human Ig control (25.6 mg/kg) QW for 6 weeks. Data are mean ± SEM of n = 9 mice per group. Significant efficacy is evident at all doses relative to control group (p < 0.002 on day 78 for all dose groups). Extended efficacy up to 40 d beyond the dosing period is observed. (C) Efficacy of BIIB036 treatment in NCI-N87 gastric carcinoma xenograft model is shown. SCID mice implanted with NCI-N87 tumor cells were treated with varying doses of BIIB036 (12.8, 6.4 or 3.2 mg/kg) or human Ig control (12.8 mg/kg) QW for 6 weeks. Data are mean ± SEM of n = 10 mice per group. Significant efficacy is evident at all doses relative to control group (p < 0.002 on day 78 for all dose groups). Extended efficacy > 50 d beyond the dosing period is observed.
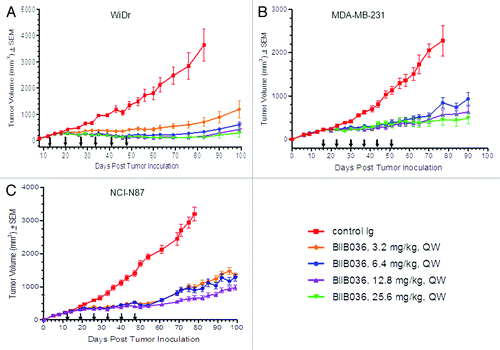
Remarkably, in all three xenograft models extended efficacy was observed well beyond the period of dosing. In the WiDr xenograft model (), the control Ig treated mice required euthanasia due to tumor burden on day 83. On that day, more than 4 weeks after the final dose of BIIB036 had been administered, mice treated with BIIB036 (at doses 6.4 to 25.6 mg/kg) showed > 90% inhibition compared with the control mice (p < 0.0001). Similarly, in the MDA-MB-231 () and NCI-N87 () models, at day 78, statistically significant (p < 10−8) tumor growth inhibition was observed in all BIIB036 treated groups relative to control Ig treated mice, as shown in . Even at 40–50 d after the final dose of BIIB036 was administered, tumors continued to exhibit substantial growth inhibition. Interestingly, in both the MDA-MB-231 and NCI-N87 models, although equivalent efficacy was maintained at all doses during the dosing period, after termination of dosing tumors re-grew slightly faster in mice receiving lower doses of BIIB036.
In the pharmacokinetic study described above, average serum concentrations of BIIB036 at day 14 following administration of the sixth dose (6.4 mg/kg) were approximately 500 ng/ml, and by extrapolation expected to be < 2 ng/ml by day 40 post dosing. Thus, the extended efficacy observed beyond the dosing period in multiple xenograft models is particularly remarkable given the negligible levels of BIIB036 expected to be present at the late time points.
BIIB036 treated tumors remain sensitive to a second cycle of treatment
Previous studiesCitation10 and the studies described above revealed that treatment of established tumors (~250 mm3) with BIIB036 resulted in robust anti-tumor activity. We next evaluated whether even larger tumors (~500 mm3) would respond to BIIB036, and whether previously treated tumors that had re-grown to nearly 1000 mm3 would be sensitive to a second cycle of BIIB036 treatment.
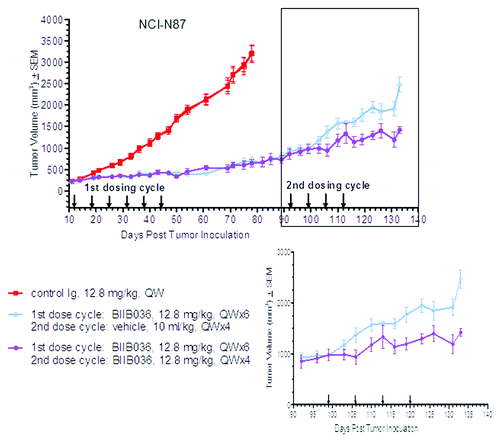
In this study, NCI-N87 tumor bearing mice received BIIB036 (12.8 mg/kg) IP, QW for 6 weeks, or an Ig control. The BIIB036 treated mice demonstrated significant anti-tumor activity compared with the control groups (p < 10−8 at day 78), as expected. Tumors in the BIIB036 treated group continued to be monitored following termination of treatment until the average tumor volume reached approximately 975 mm3 (day 92). At that point, half of the BIIB036 treated mice were administered a vehicle control while the other half of the group received a second cycle of BIIB036 (12.8 mg/kg) IP, QW for 4 weeks. As shown in , significant tumor inhibition (43%) was observed in the BIIB036 re-treated group relative to the vehicle control group (p < 0.0006 at day 133), indicating that large tumors previously treated with BIIB036 remained sensitive upon re-treatment.
Figure 2. BIIB036 inhibits growth of large tumors upon second cycle of treatment. NCI-N87 tumor cells were implanted into CB17 SCID mice. When tumors reached ~500 mm3, one group of mice (n = 10) received a first cycle of treatment with BIIB036 (12.8 mg/kg) and one group received control Ig, administered IP QW for 6 weeks. Tumors in the BIIB036 treated group were then allowed to regrow until they reached an average size of ~975 mm3 (day 52). In a second cycle of treatment, half of the BIIB036-treated group (n = 5) was re-treated with BIIB036 (12.8 mg/kg) whereas the other half of the BIIB036-treated group (n = 5) received vehicle control, QW for 4 weeks. Data are mean ± SEM. Significant efficacy is observed in the BIIB036 treated mice following a second cycle of treatment (p < 0.0006 at day 133).
Optimization of BIIB036 dosing schedule and route
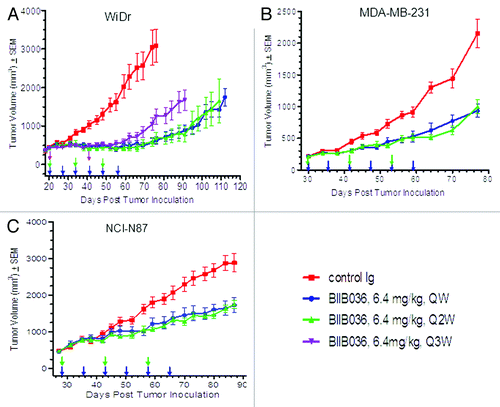
The extended efficacy observed in xenograft studies () led us to consider the possibility that BIIB036 could be administered on a less frequent dosing schedule. To that end, we compared efficacy of BIIB036 in WiDr tumor-bearing mice when dosed at 6.4 mg/kg for six weeks on a once per week (QW), once every other week (Q2W) or once every third week (Q3W) dosing schedule. As shown in , equivalent anti-tumor efficacy was observed at all the dosing schedules over the course of the dosing period. However, following termination of dosing, tumors in the Q3W treated group grew back more rapidly compared with tumors in the QW and Q2W treated groups. In contrast, the Q2W treated group maintained equivalent efficacy as the QW group throughout the duration of the study (no statistically significant difference). Similar results were observed in studies comparing QW, Q2W and Q3W dosing at 12.8 mg/kg in the same WiDr model (data not shown). These results led us to conclude that dosing every other week provides equivalent and sustained efficacy comparable to dosing every week. These findings were confirmed in two additional xenograft models (MDA-MB-231 and NCI-N87) where equivalent efficacy of BIIB036 (6.4 mg/kg) was observed on a Q2W as compared with QW dosing schedule both during the dosing period and beyond (). Similar results were obtained in the NCI-N87 model where BIIB036 administered at lower doses (0.9, 1.8 and 3.2 mg/kg) showed equivalent efficacy when dosed Q2W as compared with QW (data not shown). Thus, in multiple xenograft models and at a range of doses it is evident that dosing on an alternate week schedule results in equivalent efficacy to dosing every week.
Figure 3. Maximal efficacy of BIIB036 is achieved with a Q2W dosing schedule. (A) Efficacy of BIIB036 (6.4 mg/kg, IP) administered QW, Q2W or Q3W in WiDr colon tumor xenografts is compared. Statistically equivalent efficacy throughout the study is maintained with dosing QW or Q2W, whereas tumors in the Q3W treatment group grew back more rapidly following termination of dosing. BIIB036 administered on a QW vs. Q2W is compared in the (B) MDA-MB-231 and (C) NCI-N87 tumor models, and no statistical difference in efficacy is observed. Data are mean ± SEM of n = 10 mice per group.
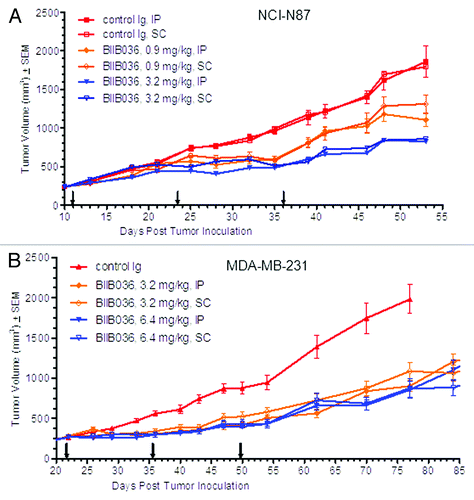
In all of the studies described above, BIIB036 was administered by the IP route. We had previously observed that intravenous administration of the murine version of BIIB036 (3.2 or 6.4 mg/kg) in the MDA-MB-231 xenograft model resulted in equivalent efficacy as compared with intraperitoneal (IP) administration (data not shown). We next considered whether subcutaneous administration of BIIB036 could provide similar efficacy in xenograft models. In the next series of studies, we assessed efficacy of BIIB036 when administered SC compared with the IP route. To that end, NCI-N87 tumor-bearing mice were dosed with BIIB036 administered either IP or SC on a Q2W dosing schedule. Equivalent efficacy with no statistical difference was observed at 3.2 mg/kg regardless of the route of administration, as shown in . This observation was confirmed with administration Q2W at suboptimal doses of 0.9 mg/kg () or 1.8 mg/kg (data not shown). These results were further validated in an additional xenograft model, MDA-MB-231, where statistically significant efficacy was observed with BIIB036 (6.4 or 3.2 mg/kg) administered Q2W, either SC or IP (). Pharmacokinetic analysis revealed relatively similar exposures when BIIB036 was administered SC or IP (data not shown), thus likely accounting for the equivalent efficacy observed in the xenograft models.
Figure 4. Equivalent efficacy of BIIB036 is observed with administration IP vs. SC Mice bearing. (A) NCI-N87 or (B) MDA-MB-231 xenograft tumors were administered BIIB036 either IP or SC on a Q2W schedule at various doses as indicated. Data are mean ± SEM of n = 10 mice per group. No statistical difference between IP and SC treatment groups at any given dose is observed.
BIIB036 is efficacious in inhibiting growth of patient-derived primary tumors grown in athymic nude mice
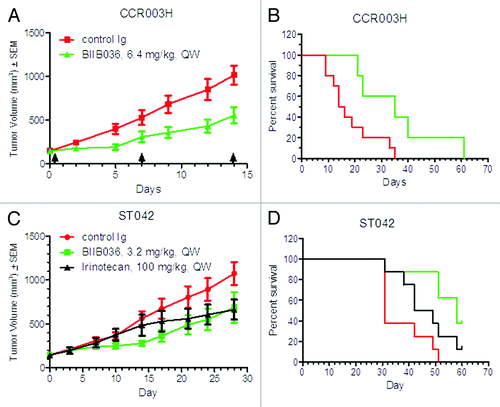
Having demonstrated that BIIB036 is effective in multiple xenograft models, we next assessed whether BIIB036 would be efficacious in patient derived primary tumors grown in mice. Low passage human colorectal tumors were implanted into athymic nude mice, and when tumors reached approximately 150 mm3, treatment with BIIB036 or control Ig administered IP, QW was initiated. In two of nine patient derived primary models tested, BIIB036 exhibited significant anti-tumor efficacy compared with control Ig. As shown in , in the CCR003H patient derived model, BIIB036 (6.4 mg/kg) treatment resulted in a 45% reduction in tumor growth relative mice treated with a control Ig. In addition, an increased mean time to progression (defined as time for treated tumors to reach 1000 mm3) was observed. The mean time to progression was 35 d for the BIIB036 treated group as compared with 15 d for the control treated group (p < 0.02), as shown in . Similarly, BIIB036 at 3.2 mg/kg demonstrated tumor growth inhibition (36%) in the STO42 patient derived tumor model (), with an increase in time to progression of 49 d as compared with 26 d in mice treated with control Ig (p < 0.02) (). In addition, in the STO42 model, efficacy of BIIB036 was compared with the standard of care agent, irinotecan (IP, QW for 3 weeks). BIIB036 treatment compared favorably to treatment with irinotecan where the time to progression was 42 d. Taken together, these studies demonstrate that BIIB036 effectively inhibits tumor growth of patient derived primary tumor models, suggesting that BIIB036 may be efficacious for the treatment of human cancers.
Figure 5. BIIB036 inhibits growth of patient derived primary tumors. Low passage colorectal tumor fragments derived from patients and passaged in mice were implanted into nude mice and administered treatment IP on a QW schedule. In (A) and (B), the CCR003H tumor model was tested with treatment of BIIB036 (n = 5) or control Ig (n = 10) at 6.4 mg/kg. In (C) and (D), the ST042 tumor model was tested with treatment of BIIB036 (n = 5) or control Ig (n = 10) at 3.2 mg/kg or irinotecan (n = 5; 100 mg/kg, QW). In (A) and (C), tumor growth is shown as mean ± SEM. In (B) and (D), time to progression (i.e., tumor reaches1000 mm3) is plotted as % survival.
BIIB036 enhances efficacy of standard of care chemotherapeutics in xenograft models
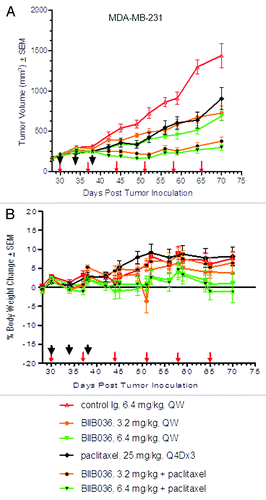
We next assessed whether BIIB036 could enhance efficacy of standard of care chemotherapeutic agents. Paclitaxel is a mitotic inhibitor widely used as a chemotherapeutic for the treatment of breast, lung and other cancers. To determine whether BIIB036 would enhance the efficacy of paclitaxel, MDA-MB-231 triple-negative breast tumor-bearing mice were treated with BIIB036 (3.2 or 6.4 mg/kg, QW), paclitaxel, or the combination of paclitaxel and BIIB036. Paclitaxel was administered in these studies at the maximum tolerated dose (25 mg/kg, Q4Dx3), as previously determined (data not shown). As shown in , as single agents BIIB036 and paclitaxel each significantly inhibited growth of the MD-MB-231 tumors compared with the control group. Repeated measure analysis followed by the Tukey’s test of the combination data from days 30–70 revealed that the combination of paclitaxel and BIIB036 (6.4 mg/kg) was significantly superior compared with either paclitaxel or BIIB036 (6.4 mg/kg) single agents alone (p < 0.01 for BIIB036 + paclitaxel relative to paclitaxel or BIIB036). Similar results were observed when paclitaxel was combined with BIIB036 at 3.2 mg/kg (p < 0.001 for BIIB036 + paclitaxel relative to paclitaxel or BIIB036). As shown in , in the combination dose groups, no overlapping toxicities were observed, as evidenced by a lack body weight loss over the course of the study. In fact, mice in all treatment groups gained weight over the duration of the study. A similar experiment was performed using the MDA-MB-231 breast cancer model, where BIIB036 was dosed at 3.2 or 6.4 mg/kg on a Q2W dosing schedule, alone or in combination with paclitaxel. This study confirmed the results of the study described above where BIB036 was dosed on a QW schedule, as the combination groups similarly showed statistically significant superior efficacy compared with the single agents (data not shown).
Figure 6. BIIB036 enhances efficacy of paclitaxel without overlapping toxicities. (A) MDA-MB-231 tumor bearing mice were administered BIIB036 (3.2 or 6.4 mg/kg), paclitaxel (25 mg/kg Q4X3), or a combination of BIIB036 (3.2 or 6.4 mg/kg) plus paclitaxel. Data are mean ± SEM of n = 10 mice per group. Statistically superior tumor inhibition is observed in the combination treatment as compared with single agent groups (repeated measure ANOVA, p < 0.01 for BIIB036 (6.4 mg/kg) combination relative to BIIB036 (6.4 mg/kg) or paclitaxel alone; p < 0.001 for BIIB036 (3.2 mg/kg) combination relative to BIIB036 (3.2 mg/kg) or paclitaxel alone). (B) Percent change in body weight of each group of mice from the MDA-MB-231 xenograft experiment in (A) is plotted as a function of time. No reduction in body weight is observed in any of the treatment groups.
We next tested the ability of BIIB036 to enhance the activity of paclitaxel in the HOP62 non-small cell lung xenograft model. As shown in , significantly enhanced activity of BIIB036 in combination with paclitaxel was observed as compared with the single agent treatment groups (repeated measure ANOVA days 19–50: p < 0.0001 BIIB036 + paclitaxel relative to BIIB036, p < 0.05 BIIB036+ paclitaxel relative to paclitaxel), confirming the results in the breast cancer model. Notably, all of the tumors in the combination treatment group regressed, whereas none of the tumors in the BIIB036 treated group regressed, and only 30% of the tumors in the paclitaxel treated group showed a slight regression followed by re-growth. At termination of the study (day 57), 80% of the tumors in the combination group were either not palpable or too small to be measured, whereas all of the tumors in the single agent groups were palpable and measurable. Thus, in multiple xenograft models we observed enhanced efficacy when BIIB036 was added to paclitaxel therapy
Figure 7. BIIB036 enhances efficacy of several standard of care chemotherapeutics. (A) HOP62 lung tumor bearing mice were administered paclitaxel (25 mg/kg Q4DX3), BIIB036 (6.4 mg/kg) or a combination of paclitaxel plus BIIB036 (6.4 mg/kg). Data are mean ± SEM of n = 10 mice per group. Statistically superior tumor inhibition is observed in the BIIB036 combination treatment as compared with single agent groups (repeated measure ANOVA, p < 0.0001). (B) HOP62 lung tumor bearing mice were administered carboplatin (50 mg/kg, Q4DX4), BIIB036 (6.4 mg/kg) or a combination of carboplatin plus BIIB036 (6.4 mg/kg). Data are mean ± SEM of n = 10 mice per group. Statistically superior tumor inhibition is observed in the BIIB036 combination treatment as compared with single agent groups (repeated measure ANOVA p < 0.05). (C) NCI-N87 tumor bearing mice were administered 5-FU (50 mg/kg, Q2DX6), BIIB036 (6.4 mg/kg) or a combination of 5-FU plus BIIB036 (6.4 mg/kg). Data are mean ± SEM of n = 9 mice per group. Statistically superior tumor inhibition is observed in the BIIB036 combination treatment as compared with single agent groups (repeated measure ANOVA, p < 0.001).
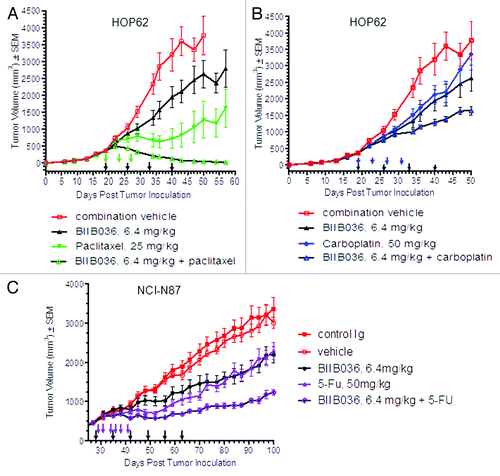
The alkylating agent carboplatin is another chemotherapeutic frequently used for the treatment of non-small cell lung cancer (NSCL) cancer. Therefore, we assessed whether BIIB036 could enhance the efficacy of carboplatin in the HOP62 NSCL tumor-bearing mice. As shown in , efficacy of the BIIB036 + carboplatin combination was significantly superior compared with either single agent therapy (repeated measure ANOVA days 19–50: p < 0.05). Thus, BIIB036 is also effective in enhancing efficacy of carboplatin therapy.
Finally, we tested the ability of BIIB036 to enhance efficacy of the pyrimidine analog fluorouracil (5-FU), which is widely used in the treatment of gastrointestinal tumors. In the NCI-N87 gastric xenograft model, we observed significantly enhanced tumor growth inhibition in mice treated with a combination of BIIB036 + 5-FU as compared with mice treated with either single agent therapy (repeated measure ANOVA days 27–100, p < 0.001) (). Remarkably, as shown in , treatment was initiated when tumors were relatively large (> 500 mm3), and yet tumors regressed upon treatment with the combination of BIIB036 and 5-FU, although not BIIB036 or the standard of care agent alone. Thus, taken together, BIIB036 can enhance the efficacy of a variety of chemotherapeutics, including paclitaxel, carboplatin and 5-FU, in multiple xenograft models.
Discussion
Studies described in this manuscript explored the therapeutic potential of the anti-Fn14 agonistic antibody, BIIB036, using pharmacokinetic studies, xenograft and patient-derived primary tumor model efficacy studies, as single agents and in combination with standard of care chemotherapeutics. Pharmacokinetic analysis revealed an approximately three day half-life for BIIB036 administered IP in tumor bearing mice. Despite the relatively short half-life, maximal efficacy in xenograft models was achieved with BIIB036, administered either IP or SC, once every two weeks. Moreover, extended tumor growth inhibition was observed even up to 40–50 d following termination of BIIB036 dosing. Additionally, even large tumors (nearly 1000 mm3), that had previously responded to BIIB036 treatment, remained sensitive to a second cycle of treatment with BIIB036. Significantly, BIIB036 enhanced the sensitivity to several chemotherapeutic agents in multiple xenograft models with no overlapping toxicities, supporting the utility of BIIB036 in the combination setting. In patient derived primary colorectal tumor models, which more closely mimic human tumors, BIIB036 showed efficacy that was at least comparable to the standard chemotherapeutic agent irinotecan. Taken together, these studies highlight the robust efficacy of BIIB036 both as a single agent therapeutic and in combination with standard of care agents.
The pharmacokinetic studies suggested that twice weekly dosing would be an optimal schedule for administration of BIIB036. However, we found that maximal efficacy of BIIB036 was achieved upon dosing once every other week, i.e., equivalent to at least four half-lives. We also observed remarkable extended efficacy of BIIB036 well beyond the dosing period. Taken together, these results demonstrate the potent and prolonged anti-tumor activity, and suggest the possibility of infrequent or pulse dosing of BIIB036 in the clinic. Given that the half-life of a typical humanized antibody in man is 14–21 d, relatively infrequent dosing of BIIB036 in the clinic is likely. Importantly, infrequent dosing would enable ease of administration for patients and may also minimize toxicities.
The extended efficacy observed upon cessation of dosing is remarkable, although the mechanism by which BIIB036 induces such a prolonged effect is not well understood. We previously reportedCitation10 that BIIB036 mediates its anti-tumor effect in vivo by direct killing of tumor cells marked by caspase cleavage as well by Fc-mediated effects involving cross-linking of Fn14 receptors and/or antibody-dependent cell-mediated cytotoxicity. However, neither of these mechanisms would seemingly account for prolonged activity of the antibody. One possible explanation is that although the half-life of BIIB036 in serum is only approximately three days, perhaps BIIB036 has a prolonged half-life in tumor tissue, and this could account for the prolonged anti-tumor activity observed in vivo. An alternate yet intriguing possibility is that Fn14 may be expressed, or preferentially expressed, on cancer stem cells (CSCs), and so by eliminating the CSC population a durable effect is observed. Notably, the TRAIL-R2 receptor DR5 was shown to be enriched in pancreatic CSCs compared with the bulk of tumor cells, and this is thought to account for the particularly potent effect of the anti-DR5 antibody tigatuzimab in combination with gemcitabine.Citation18 Thus, it is possible that Fn14, too, may be preferentially expressed on CSCs, thus providing an explanation for its prolonged and profound anti-tumor activity.
It is significant that BIIB036 can enhance the activity of several standard of care chemotherapeutics in multiple in vivo xenograft models. Despite the emergence of exciting and effective targeted agents in oncology, in reality most patients continue to receive these new agents in combination with standard chemo- and/or radiation therapies. Thus, the fact that we observed enhanced activity of BIIB036 as a combination therapy with no added toxicity is particularly compelling. Moreover, while clinical trials with TRAIL agonists have demonstrated acceptable safety and tolerability, there has been little evidence to-date of single agent efficacy.Citation1 However, in numerous ongoing studies, anti-TRAIL receptor agonists and soluble TRAIL are currently being evaluated in combination with standard of care therapies, and the expectation, largely based on a wealth of pre-clinical data, is that the TRAIL therapies may show efficacy in the combination setting. Thus, by analogy, it is significant that BIIB036 has demonstrated enhanced efficacy when administered in combination with a variety of standard of care agents, since BIIB036 may also require combination dosing to reveal its full potential as an anti-cancer therapeutic in the clinic.
BIIB036 exhibited anti-tumor activity in two patient derived primary colon tumor models, with efficacy comparable to that of a standard of care agent. The data from the patient derived primary tumor models provides important validation for BIIB036. In contrast to traditional xenograft models, primary tumor models are not passaged in cell culture and undergo only a limited number of passages in mice. The primary tumor models thereby more closely reflect the genetic features of the original cancer,Citation19 and therefore positive outcomes in primary tumor models are more likely to predict success in the clinic. Thus, the demonstration of efficacy in primary tumor models represents a high hurdle and is a promising indication of the potential for BIIB036 to be efficacious in man. Taken together, the data presented herein suggests that BIIB036 warrants clinical evaluation.
Materials and Methods
Xenograft studies
Nu/nu (Crl:NU-Foxn1nu) and CB17 SCID female mice 6–8 weeks of age with an average weight of ~25 g purchased from Charles River Laboratories were housed in ventilated cage racks, with food and water provided ad libitum. Mice were acclimated for 72 h before going on study. All routine and experimental animal work was conducted in compliance with the Institutional Animal Care and Use Committee.
WiDr (colon), MDA-MB-231 (breast) and NCI-N87 (gastric) tumor xenografts were grown in female nu/nu mice (Crl:NU-Foxn1nu) or CB17 SCID mice as described previously.Citation10 HOP62 non-small cell tumor cell were obtained from NCI and were grown as serially transplanted tumor fragments (3 mm3) in female CB17 SCID mice.
BIIB036 was administered either intraperitoneally (IP) or subcutaneously (SC) at various doses as indicated, either on a once a week (QW), every other week (Q2W) or every third week (Q3W) schedule. For the combination studies, paclitaxel, (Bedford Laboratories) was diluted in saline and administered at 25 mg/kg, intraperitoneally (IP) on Q4Dx3 dosing schedule. Carboplatin and 5-fluorouracil (Sicor Pharmaceuticals) were diluted in saline and administered at 50 mg/kg, on a Q4Dx4 and Q2Dx6 dosing schedules, respectively. The number of mice per group was typically n = 10 in the xenograft studies, except where otherwise stated in the figure legends.
Tumor measurements were monitored twice weekly as described previously.Citation10 When the majority of tumors reached a predetermined size, mice were assigned to treatment and control groups. Tumors were size matched across all groups. Animals were monitored on a daily basis and body weight was measured twice weekly using a digital scale.
Anti-tumor activity was evaluated by comparing the tumor volume of the control groups with the tumor volume of the treated groups. Percent tumor growth inhibition is calculated by the change in mean treated tumor volume divided by the change in mean control tumor volume multiplied by 100 and subtracted from 100%. Statistical significance was determined using the unpaired Student’s t-test or ANOVA where appropriate.
Continuous tumor measurements were reported as mean ± standard error of the mean and plotted over time for xenografts studies. Statistical significance of observed differences in growth curves between treatment and control groups during and after dosing periods were evaluated by repeated measures ANOVA followed by Dunnett’s multiple range post comparison test. Differences between treatment groups were evaluated by paired repeated measures ANOVA followed by Tukey’s post-test. GraphPad Prism© software was used for statistical analysis.
Patient derived primary tumor models
Low passage Biomerk Tumorgraft™ models (Champions Biotechnology), propagated as tumor fragments in mice, were implanted into the right flank of female athymic nu/nu mice (Harlan) at age 4–6 weeks. Tumors were measured and volumes calculated as described above. When tumors reached approximately 150 mm3, animals were matched by tumor volume into treatment (n = 5) and control (n = 10) groups, and dosing was initiated (Day 0).
For tumor growth delay analysis, individual mice reaching the designated study endpoint (an estimated tumor volume of approximately 1000 mm3) were assigned a time to endpoint (TTE) value corresponding to that day; the tumor growth delay study was concluded once all mice reached the study endpoint or 60 d following study initiation. Tumor bearing animals not reaching the designated volume endpoint by study completion were assigned a TTE value corresponding to the final study day for statistical calculations. Statistical differences in median TTE values between control and each treatment group were compared using an unpaired, two-tailed student t-test.
PK studies
WiDr colon tumor-bearing female mice received a single IP dose of BIIB036 at 6.4 mg/kg. Blood was collected via cardiac puncture from three mice at each of the following time points after the dose: 1, 6, 24, 48, 96, 168 and 336 h, and from four untreated mice. In a second arm of the study, mice were dosed with BIIB036 at 6.4 mg/kg once a week for 6 weeks. After the last dose, blood was collected via cardiac puncture from three mice at each of the following time points: 6, 24, 48, 96, 168 and 336 h. ELISA was performed to assess serum BIIB036 concentrations using mFn14-mFc coated on 96-well microtiter plates, and detection with an HRP-conjugated F(ab’)2 goat-anti-human IgG (H+L). The mean plasma concentrations of the test articles were used to calculate the pharmacokinetic parameters by a non-compartmental analysis (NCA) extravascular input model (Model 200, WinNonlin Professional Version 5.2; Pharsight Inc., Mountain View, CA). The parameters determined in this analysis are defined as follows: Maximum serum concentration values (Cmax) were defined as the highest observed serum concentrations, and the minimum serum concentration values (Cmin) were defined as the serum concentration 7 d after dosing. AUC is the area–under-the-concentration-time-curve. The estimation of area under the concentration-time vs. time curves (AUC) was based upon log trapezoidal rule. The terminal rate constant (λ) was derived from the slope of the terminal log-linear phase of serum concentration-time curves. The apparent terminal half-lives (t½) were calculated as 0.693/λ.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We acknowledge Keli Perron for growing all tumor cell lines for the in vivo studies, and Qin Wang for PK analysis.
Related Research Data
References
- Wiezorek J, Holland P, Graves J. Death receptor agonists as a targeted therapy for cancer. Clin Cancer Res 2010; 16:1701 - 8; http://dx.doi.org/10.1158/1078-0432.CCR-09-1692; PMID: 20197482
- Culp PA, Choi D, Zhang Y, Yin J, Seto P, Ybarra SE, et al. Antibodies to TWEAK receptor inhibit human tumor growth through dual mechanisms. Clin Cancer Res 2010; 16:497 - 508; http://dx.doi.org/10.1158/1078-0432.CCR-09-1929; PMID: 20068083
- Feng SL, Guo Y, Factor VM, Thorgeirsson SS, Bell DW, Testa JR, et al. The Fn14 immediate-early response gene is induced during liver regeneration and highly expressed in both human and murine hepatocellular carcinomas. Am J Pathol 2000; 156:1253 - 61; http://dx.doi.org/10.1016/S0002-9440(10)64996-6; PMID: 10751351
- Han H, Bearss DJ, Browne LW, Calaluce R, Nagle RB, Von Hoff DD. Identification of differentially expressed genes in pancreatic cancer cells using cDNA microarray. [erratum appears in Cancer Res 2002 Aug;62(15):4532] Cancer Res 2002; 62:2890 - 6; PMID: 12019169
- Michaelson JS, Cho S, Browning B, Zheng TS, Lincecum JM, Wang MZ, et al. Tweak induces mammary epithelial branching morphogenesis. Oncogene 2005; 24:2613 - 24; http://dx.doi.org/10.1038/sj.onc.1208208; PMID: 15735761
- Tran NL, McDonough WS, Donohue PJ, Winkles JA, Berens TJ, Ross KR, et al. The human Fn14 receptor gene is up-regulated in migrating glioma cells in vitro and overexpressed in advanced glial tumors. Am J Pathol 2003; 162:1313 - 21; http://dx.doi.org/10.1016/S0002-9440(10)63927-2; PMID: 12651623
- Wang S, Zhan M, Yin J, Abraham JM, Mori Y, Sato F, et al. Transcriptional profiling suggests that Barrett’s metaplasia is an early intermediate stage in esophageal adenocarcinogenesis. Oncogene 2006; 25:3346 - 56; http://dx.doi.org/10.1038/sj.onc.1209357; PMID: 16449976
- Willis AL, Tran NL, Chatigny JM, Charlton N, Vu H, Brown SA, et al. The fibroblast growth factor-inducible 14 receptor is highly expressed in HER2-positive breast tumors and regulates breast cancer cell invasive capacity. Mol Cancer Res 2008; 6:725 - 34; http://dx.doi.org/10.1158/1541-7786.MCR-08-0005; PMID: 18505918
- Chicheportiche Y, Bourdon PR, Xu H, Hsu YM, Scott H, Hession C, et al. TWEAK, a new secreted ligand in the tumor necrosis factor family that weakly induces apoptosis. J Biol Chem 1997; 272:32401 - 10; http://dx.doi.org/10.1074/jbc.272.51.32401; PMID: 9405449
- Michaelson JS, Amatucci A, Kelly R, Su L, Garber E, Day ES, et al. Development of an Fn14 agonistic antibody as an anti-tumor agent. MAbs 2011; 3:362 - 75; http://dx.doi.org/10.4161/mabs.3.4.16090; PMID: 21697654
- Nakayama M, Ishidoh K, Kayagaki N, Kojima Y, Yamaguchi N, Nakano H, et al. Multiple pathways of TWEAK-induced cell death. J Immunol 2002; 168:734 - 43; PMID: 11777967
- Nakayama M, Ishidoh K, Kojima Y, Harada N, Kominami E, Okumura K, et al. Fibroblast growth factor-inducible 14 mediates multiple pathways of TWEAK-induced cell death. J Immunol 2003; 170:341 - 8; PMID: 12496418
- Schneider P, Schwenzer R, Haas E, Mühlenbeck F, Schubert G, Scheurich P, et al. TWEAK can induce cell death via endogenous TNF and TNF receptor 1. Eur J Immunol 1999; 29:1785 - 92; http://dx.doi.org/10.1002/(SICI)1521-4141(199906)29:06<1785::AID-IMMU1785>3.0.CO;2-U; PMID: 10382740
- Vince JE, Chau D, Callus B, Wong WW, Hawkins CJ, Schneider P, et al. TWEAK-FN14 signaling induces lysosomal degradation of a cIAP1-TRAF2 complex to sensitize tumor cells to TNFalpha. J Cell Biol 2008; 182:171 - 84; http://dx.doi.org/10.1083/jcb.200801010; PMID: 18606850
- Wilson CA, Browning JL. Death of HT29 adenocarcinoma cells induced by TNF family receptor activation is caspase-independent and displays features of both apoptosis and necrosis. Cell Death Differ 2002; 9:1321 - 33; http://dx.doi.org/10.1038/sj.cdd.4401107; PMID: 12478469
- Lukashev M, LePage D, Wilson C, Bailly V, Garber E, Lukashin A, et al. Targeting the lymphotoxin-beta receptor with agonist antibodies as a potential cancer therapy. Cancer Res 2006; 66:9617 - 24; http://dx.doi.org/10.1158/0008-5472.CAN-06-0217; PMID: 17018619
- Younes A, Kadin ME. Emerging applications of the tumor necrosis factor family of ligands and receptors in cancer therapy. J Clin Oncol 2003; 21:3526 - 34; http://dx.doi.org/10.1200/JCO.2003.09.037; PMID: 12972530
- Rajeshkumar NV, Rasheed ZA, García-García E, López-Ríos F, Fujiwara K, Matsui WH, et al. A combination of DR5 agonistic monoclonal antibody with gemcitabine targets pancreatic cancer stem cells and results in long-term disease control in human pancreatic cancer model. Mol Cancer Ther 2010; 9:2582 - 92; http://dx.doi.org/10.1158/1535-7163.MCT-10-0370; PMID: 20660600
- Rubio-Viqueira B, Jimeno A, Cusatis G, Zhang X, Iacobuzio-Donahue C, Karikari C, et al. An in vivo platform for translational drug development in pancreatic cancer. Clin Cancer Res 2006; 12:4652 - 61; http://dx.doi.org/10.1158/1078-0432.CCR-06-0113; PMID: 16899615