Abstract
Several proteins have been suggested in promoting tumor formation in numerous human tissues by inactivating the tumor suppressor p53. This has generated interest in the development of small molecules to block these inhibitors of p53 and to regain p53 activity. Recently, we identified a small molecule, Inauhzin, which can inhibit SIRT1 activity and activate p53. SIRT1 is a deacetylase that deacetylates p53 and facilitates Mdm2 mediated p53 destabilization. In this study, we tested if combining Inauhzin with Nutlin-3, an inhibitor of MDM2-p53 binding, might synergistically activate p53 to suppress tumor growth. Indeed, at lower doses, combination of Inauhzin and Nutlin-3 exhibited a synergistic effect on inhibiting cell growth and promoting apoptosis in human colon and lung cancer cell lines in a p53-dependent fashion. Minimal effects were observed with treatment of either compound alone. Using a xenograft tumor model, we also showed a synergistic effect with both compounds. Thus, to fully regain p53 activity, targeting its multiple inhibitory proteins might be a better approach. Our study provides evidence supporting this concept for achieving better therapeutic efficacy in tumors that possess wild type p53.
Introduction
The tumor suppressor p53 is essentially important for preventing mammalian cells from undergoing neoplasia and tumorigenesis primarily due to its ability to activate the transcription of numerous genes and some miRNAs responsible for executing p53-dependent apoptosis, autophagy, senescence, and DNA repair as well as suppression of cell proliferation, growth, migration and angiogenesis,1-Citation3 though it can also induce apoptosis independently of its transcriptional activity.Citation4 In order to ensure that p53 is not overly active under normal conditions, cells also employ MDM2 (murine double minute 2 and also called HDM2 in human) and MDMX (an MDM2 analog) to control p53’s cytotoxic functions. MDM2 and MDMX often form a complex and work together to inactivate p53 by either directly inhibiting its transcriptional activity or mediating its rapid proteasomal turnover via an ubiquitin-dependent mechanism.Citation5-Citation11 This negative regulation is in a feedback fashion as the gene encoding MDM2 is a transcriptional target of p53Citation10-Citation14 and often accounts for inactivation of p53 in a number of human cancers harboring wild type p53.Citation15
MDM2 negative regulation of p53 is implicated in the development of tumors in wild type p53-containing cells. This has led to intensive studies over the past to identify small molecules that could specifically target individual protein molecules of this loop for developing a better molecule-targeting anti-cancer therapy.Citation16 Indeed, several small molecules and peptides have been reported to activate p53 by either blocking its binding to MDM2, such as Nutlin-3,Citation17 Rita,Citation18 or MI-219,Citation19 or inhibiting MDM2 E3 ubiquin ligase activity,Citation20 or inhibiting MDMX-p53 binding.Citation21 Recently a different strategy was also reported to activate p53 by targeting deacetylase(s). It has been shown that p53 acetylation by p300/CBP and ubiquitination by MDM2 are mutually exclusive,Citation22-Citation26 and deacetylation could facilitate MDM2-mediated p53 degradation.Citation23,Citation25,Citation26 Therefore, inhibition of SIRT1-catalyzed p53 deacetylation would prevent MDM2-mediated p53 degradation and lead to p53 activation. Several inhibitors of this kind have been identified, such as TenovinsCitation27 or Inauhzin by our group.Citation28 Targeting SIRT1 is biologically significant because this protein is highly expressed in human cancers frequently due to the inactivation of HIC-1 (hyper methylated in cancer) via promoter methylation,Citation29-Citation31 which encodes a transcriptional repressor that specifically inhibits the expression of SIRT1.Citation31 Therefore, indirectly blocking the MDM2-p53 loop by enhancing p53 acetylation via inhibition of its deacetylase SIRT1 could serve as an alternative strategy for the development of an anti-cancer therapy.
Since p53 deacetylation mediated by SIRT1 appears to be a prerequisite for MDM2-mediated ubiquitination and degradation of p53,Citation26 we hypothesized that dual inhibition of SIRT1 and MDM2 activities might synergize the activation of p53. In this study, we tested this idea by combining Inauhzin, a SIRT1 inhibitor,Citation28 with Nutlin-3, an inhibitor of the MDM2-p53 interaction,Citation17 using cell-based assays and a xenograft tumor model. As a result, we found that indeed these two compounds at much lower doses can synergistically activate p53 and induce its proapoptotic activity in human lung and colon cancer cell lines. We also observed growth suppression of xenograft tumors with combination of Inuahzin and Nutlin3. Hence, our studies as detailed below suggest a dual-targeting strategy by simultaneously inactivating two p53-inhibitory molecules for the development of a possibly more effective anti-cancer therapy.
Results
Inauhzin synergizes the potential of Nutlin-3 in p53 activation
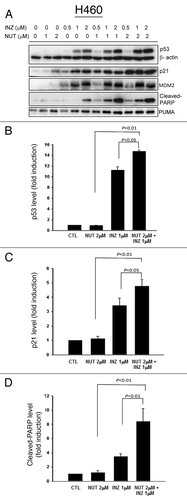
Previous studies suggesting that p53 deacetylation by SIRT1 is likely a prerequisite for MDM2-mediated p53 ubiquitination and degradation prompted us to test the idea of whether inhibition of SIRT1 activity by Inauhzin would potentiate the ability of Nutlin-3 to induce p53 more effectively by inhibiting MDM2-p53 binding. To this end, we treated p53-positive or null human colon cancer HCT116 cells with different doses of Inauhzin or Nutlin-3 alone or of their mixtures as shown in and Figure S3A for 18 h and harvested for western blot (WB) analyses. As expected,Citation17,Citation28 Inauhzin or Nutlin-3 by itself induced p53 level and activity (as measured by the level of p21, PUMA and cleaved PARP) in a dose dependent fashion (). Of note, either Nutlin-3 at 2 μM or Inauhzin at 0.5 μM was barely effective in activation of p53, as merely a trace amount of p53 or cleaved PARP was detected after treatment with the low dose of either of the compounds (). However, interestingly, the level of p53, p21, PUMA or cleaved PARP was dramatically induced when combining the two compounds at the same lower doses. In particular, co-treatment of p53-positive HCT116 cells with 1 μM of Inauhzin and 2 μM of Nutlin-3 more significantly activated p53 as measured by its protein level as well as the level of its target p21, PUMA or cleaved PARP as indication of apoptosis (), and hence, this dose set was also used in some of the following studies. These results were reproducible as shown in . This synergistic effect was not only observed in colon cancer cells, as it was also evident in human non-small lung cancer p53-containing H460 () and A549 (Fig. S1). This synergistic effect was specific to p53, as co-treatment of p53 null HCT116 (Fig. S4A) or H1299 (Fig. S4B) cells with the same combination of the compounds did not show any effect on these protein levels. Taken together, these results demonstrate that simultaneous inhibition of SIRT1 by Inauhzin and of MDM2 by Nutlin-3 can synergistically activate p53 in human cancer cells.
Figure 1. Inauhzin and Nutlin-3 significantly enhance the expression level and activity of p53 in HCT116 p53+/+ cells in a dose-dependent manner. HCT116 p53+/+ cells were treated with Inauhzin or Nutlin-3 at the indicated concentrations for 18 h and harvested for WB analysis. 50 μg protein was loaded in each lane, and an anti-α-tublin antibody was used as an loading control. Densitometric analysis of immunoreactive bands of p53, p21 and cleaved-PARP was expressed as fold change relative to the control group.
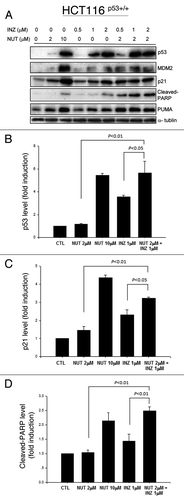
Figure 2. Inauhzin and Nutlin-3 significantly enhance the expression level and activity of p53 in H460 cells in a dose-dependent manner. H460 Cells were treated with Inauhzin or Nutlin-3 at the indicated concentrations for 18 h and harvested for WB analysis. 50 μg protein was loaded in each lane, and an anti-β-actin antibody was used as an loading control. Densitometric analysis of immunoreactive bands of p53, p21 and cleaved-PARP was expressed as fold change relative to the control group.
Inauhzin and Nutlin-3 synergistically inhibit p53-dependent cell growth
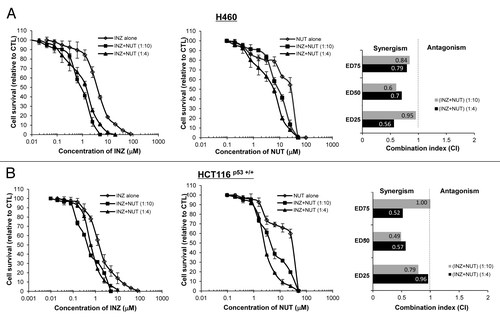
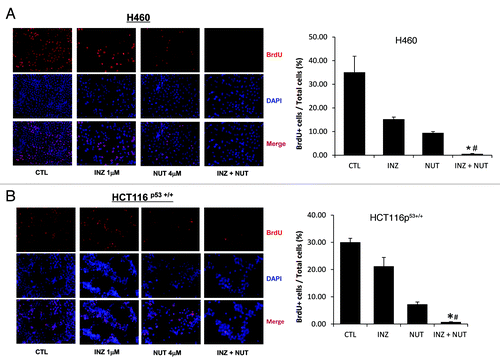
To substantially demonstrate that the co-effect of Inauhzin and Nutlin-3 on the p53 pathway is indeed synergistic, we performed a set of cell survival assays by treating either H460 or HCT116 cells with different combinations of the two compounds in various molar ratios (INZ: NUT = 1:4 or 1:10) as indicated in . The survival rates of cells after treatment with Inauhzin and Nutlin-3 in different combinations were compared with that for each individual compound alone as shown in . The values for combination index (CI), which indicates either synergism (less than 1) or antagonism (greater than 1), were calculated as described in the Materials and Methods when the two compounds reduced cellular survival by 25% (effective dose 25, ED25), 50% (ED50) or 75% (ED75) and plotted in the right panels of . In line with the results as shown in and , Inauhzin and Nutlin-3 displayed apparent synergism in suppressing the growth of these two tumor cell lines, as the CI values at a range of effective doses were clearly less than 1, particularly when a lower dose of Nutlin-3 was used (). This synergism of the two compounds was not observed in p53-null cells (Fig. S4), suggesting that the synergistic suppression of cell growth by the two compounds is p53-dependent. Consistently, this combination of the two compounds (molar ratios INZ: NUT = 1:4 or 1:2) also more significantly suppressed cell proliferation as assessed by BrdU staining in both H460 and HCT116 cells (Fig. Four and S2). These results demonstrate that Inauhzin and Nutlin-3 can synergistically repress the growth of human cancer cell lines in a p53-dependent manner.
Figure 3. Inauhzin and Nutlin-3 demonstrate synergistic cytotoxicity in the Nutlin-3 low-sensitive cells. H460 (A) and HCT116 p53+/+ (B) cells were plated in 96-well plates and treated with Inauhzin or Nutlin-3 individually at serial dilutions, or both simultaneously at fixed molar drug ratios (INZ: NUT = 1:4 or 1:10) for 72 h. Cell viability was determined using a WST cell growth assay. The data here represent the mean of three independent experiments ± SEM. CI values presented indicate the interaction of Inauhzin with Nutlin-3 when the combined treatment inhibits cell growth by 25% (ED25), 50% (ED50) or 75% (ED75), compared with the control for their individual treatment alone.
Inauhzin and Nutlin-3 synergistically induce p53-dependent apoptosis
The results from , and Figure S1 showing the increased amount of cleaved PARP by o-treatment of p53-possessing H460, HCT116 and A549 cells with Inauhzin and Nutlin-3 suggest that this combinational treatment might also induce p53-dependent apoptosis. To test this, we conducted FACS analyses of H460 or HCT116 cells after treatment with 4 or 10 μM of Nutlin-3 for 48 h in the absence or presence of 1 μM of Inauhzin. As shown in , Nutlin-3 was not so effective in induction of apoptosis in both of these cell lines even at 10 μM (as less than 2% of apoptotic HCT116 or 1% of apoptotic H460 cells were detected), whereas 1 μM of Inauhzin induced approximately ~5–8% of apoptotic cells. However, combinational treatment of these cells with 1 μM of Inauhzin with 4 or 10 μM of Nutlin-3 significantly raised the apoptotic rate to 13% or 17%. This apoptotic effect of these compounds was not seen in p53 null cells (data not shown).Citation28 Clearly, Inauhzin and Nutlin-3 potentiate each other’s ability to induce p53-dependent apoptosis. Intriguingly, when analyzing the effect of these two compounds on the cell cycle progression, we observed that the treatment of HCT116 p53 +/+ with either Nutlin-3 or Inauhzin induced G1 or G2 arrest individually (Fig. S3). This result was repeated in H460 cells as well (data not shown). Because Nutlin-3 and Inauhzin affects G1 and G2 phases, respectively (Fig. S3), their synergistic effect on either of these two individual phases was not evident (Fig. S3); however, the number of S phase cells was significantly reduced when the cells were co-treated with the two compounds at the same lower doses as that used for individual treatment (Fig. S3). Together with the results in – and Figure S3, these data demonstrate that these two p53 activators can synergistically suppress cell survival by inducing apoptosis and inhibiting cell proliferation. Of note, it would be interesting to investigate in the future why Inauhzin induces G2 arrest while Nutlin-3 induces G1 arrest even though both of the compounds elevate the levels of p53 and p21.
Figure 5. INZ significantly enhances the pro-apoptotic effect of Nutlin-3 on H460 and HCT116 p53+/+ cells. H460 (A) and HCT116 p53+/+ (B) were plated in 6cm-diameter dishes, and treated with Inauhzin and Nutlin-3 at the indicated concentrations for 48 h. Cells were stained with propidium iodide, and subjected to flow cytometry to determine the DNA content. The apoptotic cells, identified by sub-G1 DNA content, were presented as the M1 population. The data was expressed as the mean ± SEM (n = 4). *p < 0.01 vs. INZ 1μM. #p < 0.01 vs. NUT 4μM. &p < 0.01 vs. NUT 10μM.
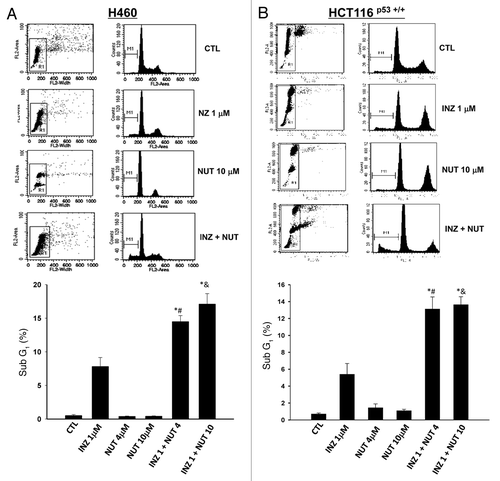
Figure 4. Inauhzin significantly enhances the inhibition of cell proliferation induced by low dose of Nutlin-3 in H460 and HCT116 p53+/+ cells. H460 (A) and HCT116 p53+/+ (B) were plated in 6cm-diameter dishes, and treated with Inauhzin and Nutlin-3 at the indicated concentrations for 48 h. Immunofluorescence was performed to determine the amount of BrdU incorporation. Representative images of BrdU immunostaining were shown (Magnification 200 × ), and quantitative analysis was performed. The data was presented as the mean of the ratios of BrdU positive cells and total cells ± SEM (n = 3). *p < 0.01 vs. INZ 1μM. #p = 0.05 vs. NUT 4μM (A) or p = 0.1 vs. NUT 4μM (B).
Inauhzin and Nutlin-3 synergistically suppress xenograft tumor growth
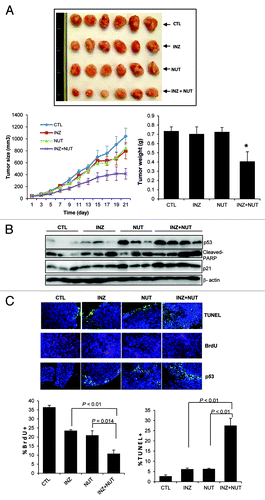
Although Nutlin-3 can suppress the growth of xenograft tumors derived from human osteosarcoma or leukemia cells,Citation17,Citation32 the anti-tumor activity of Nutlin-3 even at the dose of 200 mg/Kg per oral administration was marginal in an HCT116-derived xenograft tumor model.Citation32 Our previous study showed that Inauhzin is quite effective in suppressing the growth of HCT116 xenograft tumors.Citation28 Hence, to translate the aforementioned synergism of these compounds in HCT116 cells into a xenograft model, we tested if Inauhzin and Nutlin-3 could also synergistically repress the growth of this type of xenograft tumors. Since Inauhzin at 30 mg/Kg via intraperitoneal (IP) injection reduced the HCT116 tumor volume by ~70%,Citation28 we decided to use the dose of Inauhzin at 15 mg/Kg for the synergistic study with 150 mg/Kg of Nutlin-3 using an xenograft tumor model derived from HCT116 cells as detailed in the Materials and Methods. At the doses as mentioned above, each of the compounds could individually reduce the average tumor volume by less than 20% after treatment for 21 d (). However, when combining the two treatments at the same doses, the final average volume of tumors was decreased by 60%, which was also statistically significant (). This result again demonstrates that Inauhzin and Nutlin-3 indeed can synergistically suppress the growth of HCT116-derived xenograft tumors ex vivo. Consistent with this result, the two compounds synergistically induced p53 level and activity () as well as remarkably suppressed cell proliferation and induced apoptosis (). These results demonstrate that Inauhzin and Nutlin-3 can activate p53 and suppress xenograft tumor growth in animals as well.
Figure 6. Combination of low doses of Inauhzin and Nutlin-3 demonstrates a significant synergy on p53 induction, apoptosis and tumor suppression of HCT116 p53+/+ xenografts. (A) Combination of Inauhzin and Nutlin-3 presented a remarkable suppression on the growth of xenograft tumors derived from HCT116 p53+/+ cells. Mice bearing HCT116 p53+/+ xenografts were treated with INZ (15 mg/Kg) intraperitoneally every day, NUT orally (150 mg/Kg twice per day, 4 continuous days followed by 2 d off), or vehicles for 21 d. Images of tumors isolated are presented at the left. The tumor growth is shown by the mean tumor volumes ± SEM (middle), and the final tumor weight is shown in columns (right) (n = 6 mice per group; *p < 0.05). (B) WB analysis of proteins extracted from HCT116p53+/+ tumor samples in panel A with antibodies as indicated on right. (C) Representative images of TUNEL staining, BrdU and p53 immunostaining of xenograft tumor sections are presented. Quantification of BrdU and TUNEL staining is expressed as Mean ± SEM (n = 4 mice per group).
Discussion
Although Nutlin-3 is able to activate the p53 pathway and to inhibit cell growth in several wild type p53-harboring tumor cells, including SJSA osteosarcoma cells, MHM osteosarcoma cells, and MV4-11 leukemia cells,Citation17,Citation32 single treatment with Nutlin-3 is often less efficient in altering the growth or promoting apoptosis of cancer cells, such as HCT116, H460 or A549, in xenograft tumor models even though they also contain wild type p53.Citation17,Citation32 This suggests that other pathways might also be involved to abrogate p53 function. Thus, identifying other small molecules that could work with Nutlin3 may increase the efficacy to this compound.
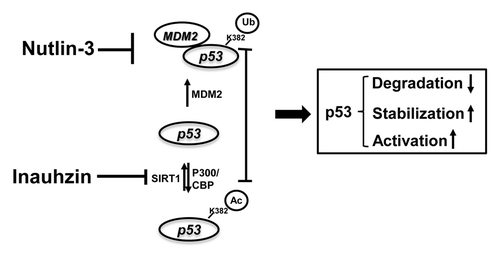
As reported previously, SIRT-1 deacetylates p53 at acetylated lysine 382, which is also the target residue for ubiquitination by MDM2, therefore facilitates the MDM2-dependent ubiquitination and degradation of p53.Citation26 Consistent with this notion, SIRT-1 is often highly expressed in several cancers due to the promoter silence of its repressor, HIC-1, via DNA hypermethylation.Citation29-Citation31 Hence, predictably, inactivation of SIRT-1 in such cancers would improve the therapeutic potency of Nutlin-3. Here the SIRT-1 inhibitor, Inauhzin, comes to play a role, as it is quite potent in stimulating p53 level and p53 activity, by preventing p53 from deacetylation and therefore stabilizing p53.Citation28 Indeed, as expected, in both p53-containing H460 and HCT116 cells, p53 level and activity (indicated by the p21 and PUMA levels) exhibited a dramatic augment after combined treatment with Inauhzin and Nutlin-3, even at relatively lower doses, compared with either single regimen ( and ). On one hand, Nutlin-3 blocks the binding of MDM2 to p53, inhibiting p53 degradation and releasing more p53 molecules for acetylation, and facilitates the acetylation of p53 at its C-teminus. On the other hand, Inauhzin induces p53 acetylation by inhibiting SIRT1-mediated deacetylation, further prevents the association of MDM2 with p53,Citation33 blocks the ubiquitination sites, and induces p53 conformation change to attenuate the MDM2-mediated ubiquitination of other lysine residues,Citation25 consequently activating p53. The sum outcome of this double treatment is, therefore, the more remarkable induction of p53 acetylation, level and activity (; Fig S5). Our subsequent finding further confirms the p53-dependent synergy between Inauhzin and Nutlin-3 on cell growth inhibition, which might be attributed to the augment of apoptotic response and cell growth arrest, in p53-wild type cells, but not in p53-null cells (, and ; Fig. S4). Previously, Nutlin-3 resistant cells were found to be unable to undergo apoptosis, although obvious induction of p53 and p21 as well as G1 arrest caused by this compound was observed. It was evident that other pathways, such as E2F/p73 response to Nutlin-3, were limited.Citation32 SIRT-1 binds to p73 and deacetylate p73, therefore inhibiting p73 activity.Citation34 Indeed, Inauhzin has also been shown to activate p73 by inhibiting SIRT1 activity.Citation28 Hence, it is anticipated that Inauhzin may be useful in combination with Nutlin-3 for the treatment of these Nutlin-3 resistant cancer cells through the activation of p73. We will investigate the possible combinatorial role of Nutlin and Inauhzin in p73 activation in the future.
Figure 7. The model for the mechanisms underlying the p53-dependent synergistic anti-tumor effect of Inauhzin and Nutlin-3. As a blocker of the binding of MDM2 to p53, Nutlin-3 inhibits MDM2-dependent ubiquitination and degradation of p53, releasing more p53 molecules for acetylation at its C-terminus. In comparison with Nutlin-3, Inauhzin induces p53 acetylation by inhibiting SIRT1-mediated deacetylation at K382. Thereby this posttranslational modification further prevents the association of MDM2 with p53, blocks the ubiquitination sites from MDM2 and attenuates the MDM2-mediated ubiquitination of these lysine residues within p53, consequently activating p53. The sum outcome of the co-treatment of p53-contaning cancerous cells with two compounds is, therefore, the more remarkable induction of p53 acetylation, level and activity, consequently suppressing tumor growth in a p53-dependent fashion. Arrows indicate activation or increase, while bars denote inactivation.
Although originally identified as a non-genotoxic agent, Nutlin-3 has been recently shown to induce a DNA damage response and p53 SerCitation15 phosphorylation at a relatively high effective dosage (≥ 10 μM) in some tumor cells,Citation35,Citation36 suggesting a potential toxicity of Nutlin-3 on some highly-proliferative normal cells harboring wild type p53 (such as stem cells). The previous data also presented a low IC50 value of Nutlin-3 on human and mouse normal diploid fibroblasts.Citation17 Unlike most of the current anti-tumor drugs, Inauhzin bypasses DNA damage, while retaining anti-tumor activity, and exerts a minimal effect on the p53 pathway and cell viability in normal human cells (such as WI-38 and NHF-1) at the equipotent dose as Nutlin-3 to induce p53 ().Citation28 Even though more studies are needed to further elucidate the low toxicity of Inauhzin in normal cells and tissues, this feature could offer a good advantage for developing into an effective anti-cancer drug with minimum side effects on patients. This advantage would be more apparent when combining it with Nutlin-3 for cancer therapy, as even much lower doses of both of the small molecules are needed to achieve a better and synergistic anti-cancer effect (). Of note, the accumulative concentration of Inauhzin in the xenografted tumor tissues after 21-d-treatment was relatively lower (Fig. S6), implying possibly faster metabolism and less accumulation of Inauhzin in the body, which might be one of the reasons for why Inauhzin is less toxic to normal tissues. However, this low concentration of Inauhzin did enhance the anti-tumor effect of Nutlin-3, which was accumulated at a much higher concentration in tumor tissues (; Fig. S6), confirming the higher sensitivity of Nutlin-resistant cancer cells to Inauhzin and demonstrating the advantage of this combination therapy with these two small molecules. Further pharmacokinetic and pharmacodynamic analyses of Inauhzin will be necessarily conducted in the future.
Combination chemotherapy has been used in the clinic for many years for chemotherapeutic drugs that induce various types of DNA damage. The new approach will be to identify small molecules that target multiple pathways to circumvent the secondary pathway to engage to increase resistance. The combination of Nutlin-3 with Inauhzin as described in this study presents a novel combinatory therapy that may increase the efficacy for patients with tumors that maintain wild type p53, particularly those at their early stage without p53 mutation.
Materials and Methods
Chemicals and antibodies
Inauhzin (INZ) was purchased from ChemDiv or ChemBridge and dissolved with DMSO to a concentration of 50mM and stored at -20°C. Nutlin-3 (NUT) was synthesized by the IUPUI chemical core facility, and dissolved with DMSO to a concentration of 100mM, stored at -20°C. Cell counting kit was purchase from Dojindo Molecular Technologies Inc. Fluorescein in situ apoptosis detection kit was from Roche, Swiss. Antibodies for immunoblotting were mouse monoclonal anti-p53 (DO-1), rabbit polyclonal anti-p53 (FL-393), mouse monoclonal anti-Ac-p53 (K382) (2525S, Cell signaling), rabbit polyclonal anti-p21 (M19), rabbit polyclonal anti-cleaved PARP (9542, Cell signaling), mouse Ac-α-tubulin (K40), mouse anti-β-actin. 2A10 monoclonal anti-MDM2 was generated as described previously. Antibodies used for immunostaining were Mouse monoclonal anti-p53 (1C12, Cell signaling), mouse monoclonal anti-BrdU (IIB5).
Cell culture
Human non-small-cell lung carcinoma wild type p53-containing H460 and A549, human non-small-cell lung carcinoma p53-null H1299, human colon cancer HCT116 (p53+/+ and p53 −/−) cells were maintained in DMEM medium (Gibco) supplemented with 10% fetal bovine serum, PSN antibiotic mixture (10 ml/L) (Gibco BRL) at 37°C in an incubator containing humidified air with 5% CO2.
Western blot analysis
H460, A549, H1299 and HCT116 cells cultured in six-well culture plates were grown to 80% confluence, then treated with various concentrations of INZ or NUT for indicated time periods. After being washed with ice-cold PBS three times, cells were lysed with 70 μL ice-cold lysis buffer containing 150 mM NaCl, 50 mM TRIS-HCl (PH7.5), 5 mM EDTA, 0.5% NP-40, 1 mM DTT, 0.2mM PMSF, and 0.25 μg of pepstatin A/ml and 20mM leupeptin. Tumor tissues from the control and treatment groups saved in liquid nitrogen were rinsed in precooled PBS, minced and homogenized on ice with a Dounce homogenizer in RIPA lysis buffer (150 mM NaCl, 1.0% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 8.0). After incubated at 4°C for 30 min, cell or tumor lysates were obtained by centrifugation at 18,000 × g at 4°C for 15 min, and the concentration of total protein was determined by the BCC method. An equal amount (50 μg per lane) of proteins obtained as described above were loaded to each lane of an SDS-PAGE gel, and transferred onto a nitrocellulose membrane (Bio-Rad) using a semidry transfer apparatus (Bio-Rad). After blocked in 5% non-fat dry milk in 1 × TBS containing 0.1% Tween 20 (TBST) at RT for 1h, membranes were incubated with primary antibodies in the blocking buffer at 4°C for overnight, followed by three washes in TBST and incubation with secondary HRP conjugated antibody (Bio-Rad) at RT for 1 h. After three washes with TBST, blots were detected with Supersignal West Pico chemiluminescent substrate (Pierce Biotechnology Inc.) and developed using Kodak Biomax film (PerkinElmer). All blots were normalized against β-actin or β-actin as a loading control.
Cell viability assay and assessment of combined drug effect
To assess cell growth, the cell counting kit (Dojindo Molecular Technologies Inc.) was used according to manufacturer’s instructions. Briefly, Cells (5,000/100ul per well) were plated in 96-well flat bottom plates, incubated overnight at 37°C, then treated with each compound individually at serial dilutions, or both compounds simultaneously at various fixed molar drug ratios (Inauhzin: Nutlin-3 = 1: 4 or 1: 10). After continuous treatment for 72 h, WST-8 was added to each well at a final concentration of 10%, and the absorbance of the samples was recorded at 450 nm on a Microplate Reader (Molecular Device, SpecrtraMax M5e) after 3hrs of incubation at 37°C. The dose-effect curves were generated for each kind of treatment to estimate the individual fractional survival (f).
To determine the synergy of Inauhzin and Nutlin-3, the CI (combination index) values were calculated based on the dose-effect curves mentioned above, according to the following formula as previously developed:Citation38
CI = (D)1/(Df)1 + (D)2/(Df)2 + a (D)1(D)2/(Df)1(Df)2
In this formula, (D)1 and (D)2 are the concentrations of combination required to produce f, (Df)1 and (Df)2 are the concentrations of individuals required to produce f, and α = 1 or 0 depending on whether the drugs are assumed to be mutually nonexclusive or mutually exclusive, respectively. In this method, CI < 1, = 1 or > 1 indicate synergism, additivity or antagonism respectively.Citation38
Cell cycle and apoptosis analysis
Cells (1.5 × 105) were plated into 6-well plates, and incubated at 37°C overnight. After treatment of Inauhzin and Nutlin-3 at the indicated concentrations for 48 h, cells were harvested, fixed in 70% ice-cold ethanol overnight at -20°C, resuspended in propidium iodide-solution (50 µg/ml PI, 0.1 mg/ml RNase A, 0.05% Tritin X-100 in PBS) for 40min at 37°C, then analyzed for DNA content using a flow cytometer (FACS Calibur, Becton Dickinson) and proprietary software (FlowJo software and ModFit LT, Verity Software House).
BrdU labeling and immunofluorescence
To determine the synergistic effects of Inauhzin and Nutlin-3 on the cell proliferation, logarithmically growing cells were treated with Inauhzin and Nutlin-3 for 48 h, followed by incubation with 10 μM BrdU for 6 h. Then cells were fixed in precooled methanol, and treated with HCl (2 N HCl, 1% Triton X-100). A mouse anti-BrdU antibody (Santa Cruz) and a Rhodamine-conjugated anti-mouse IgG (Vector Laboratories) were used for BrdU detection, and DAPI was added to cells for counterstaining. The BrdU positive cells were counted under a fluorescence microscopy. At least nine randomly chosen fields of view were examined with a minimum number of 1000 cells scored for each condition.
In vivo studies
Five-week-old female SCID mice were purchased from In Vivo Therapeutics (IVT) Core, Indiana University Simon Cancer Center. Mice were subcutaneously inoculated with 3 × 106 HCT116 p53+/+ cells in the right flank and tumor growth was monitored with calipers. After the mean tumor volume reached 50–100 mm3, animals were administered Inauhzin intraperitoneally (IP), Nutlin-3 orally, or vehicles (4% DMSO for Inauhzin, EtOH: Tween: 5% Glucose = 5:5:90 for Nutlin-3) as described previously.Citation17,Citation28,Citation32 Tumor volume was measured every other day, and inhibition of tumor growth (T/C) was calculated on the last day of treatment.
To determine p53 induction in vivo, tumors were harvested and homogenized in RIPA buffer with a protease inhibitor mixture (Sigma). Tumor homogenates were analyzed by western blot.
Cell proliferation in tumors was assessed by BrdU labeling and immunostaining. 200 mg/kg body weight of BrdU was administrated to mice via intraperitoneal injection 2 h before the mice were sacrificed. In situ p53 expression was detected by immunofluorescence. Briefly, tumors were fixed in 4% paraformaldehyde overnight at 4°C, embedded in paraffin, and cut into 6mM-thick sections. Slides were boiled in 10 mM Sodium Citrate (pH 6.0) in a microwave for 15 min for antigen retrieval, cooled for 30 min at room temperature, and washed with PBST (PBS, 0.1% Tween 20). Tissue permeabilization was achieved by incubating the slides in 1% Triton X-100 in PBS for 10 min. After blocked for 1 h in blocking buffer (PBS containing 1.5% BSA and 0.2% Triton X-100), sections were incubated overnight at 4°C in a humidity chamber with a mouse anti-BrdU monoclonal antibody, or a rabbit anti-p53 polyclonal antibody (FL393) diluted 1:100 in the blocking buffer, followed by incubation with a rhodamine-conjugated or a fluorescein-conjugated IgG. Apoptosis was determined by in situ TUNEL staining, using the Fluorescein In situ cell death detection kit (Roche) according to manufacturer's instructions.
All animal experiments were conducted in accordance with the National Institutes Health “Guide for the Care and Use of Laboratory Animals” and were approved by the Institutional Animal Care and Use Committee at Indiana University School of Medicine.
Statistics
Data were reported as means ± SEM with N being the sample size. Comparisons among groups were analyzed by using One-way ANOVA. Probability values of p < 0.05 were considered statistically significant.
Additional material
Download Zip (1.3 MB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank the members in the Lu lab for active discussion. This work was partially supported by NIH-NCI grants CA127724, CA095441 and CA129828 to H.L.
Supplemental Material
Supplemental material can be downloaded here:
Lane D, Levine A. p53 Research: the past thirty years and the next thirty years. Cold Spring Harb Perspect Biol 2010; 2:a000893; http://dx.doi.org/10.1101/cshperspect.a000893; PMID: 20463001
References
- Leung AK, Sharp PA. MicroRNA functions in stress responses. Mol Cell 2010; 40:205 - 15; http://dx.doi.org/10.1016/j.molcel.2010.09.027; PMID: 20965416
- Boominathan L. The guardians of the genome (p53, TA-p73, and TA-p63) are regulators of tumor suppressor miRNAs network. Cancer Metastasis Rev 2010; 29:613 - 39; http://dx.doi.org/10.1007/s10555-010-9257-9; PMID: 20922462
- Speidel D. Transcription-independent p53 apoptosis: an alternative route to death. Trends Cell Biol 2010; 20:14 - 24; http://dx.doi.org/10.1016/j.tcb.2009.10.002; PMID: 19879762
- Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992; 69:1237 - 45; http://dx.doi.org/10.1016/0092-8674(92)90644-R; PMID: 1535557
- Oliner JD, Pietenpol JA, Thiagalingam S, Gyuris J, Kinzler KW, Vogelstein B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature 1993; 362:857 - 60; http://dx.doi.org/10.1038/362857a0; PMID: 8479525
- Chen J, Marechal V, Levine AJ. Mapping of the p53 and mdm-2 interaction domains. Mol Cell Biol 1993; 13:4107 - 14; PMID: 7686617
- Kruse JP, Gu W. Modes of p53 regulation. Cell 2009; 137:609 - 22; http://dx.doi.org/10.1016/j.cell.2009.04.050; PMID: 19450511
- Wade M, Wang YV, Wahl GM. The p53 orchestra: Mdm2 and Mdmx set the tone. Trends Cell Biol 2010; 20:299 - 309; http://dx.doi.org/10.1016/j.tcb.2010.01.009; PMID: 20172729
- Kawai H, Lopez-Pajares V, Kim MM, Wiederschain D, Yuan ZM. RING domain-mediated interaction is a requirement for MDM2’s E3 ligase activity. Cancer Res 2007; 67:6026 - 30; http://dx.doi.org/10.1158/0008-5472.CAN-07-1313; PMID: 17616658
- Linke K, Mace PD, Smith CA, Vaux DL, Silke J, Day CL. Structure of the MDM2/MDMX RING domain heterodimer reveals dimerization is required for their ubiquitylation in trans. Cell Death Differ 2008; 15:841 - 8; http://dx.doi.org/10.1038/sj.cdd.4402309; PMID: 18219319
- Wu X, Bayle JH, Olson D, Levine AJ. The p53-mdm-2 autoregulatory feedback loop. Genes Dev 1993; 7:7A 1126 - 32; http://dx.doi.org/10.1101/gad.7.7a.1126; PMID: 8319905
- Barak Y, Juven T, Haffner R, Oren M. mdm2 expression is induced by wild type p53 activity. EMBO J 1993; 12:461 - 8; PMID: 8440237
- Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer 2009; 9:749 - 58; http://dx.doi.org/10.1038/nrc2723; PMID: 19776744
- Momand J, Jung D, Wilczynski S, Niland J. The MDM2 gene amplification database. Nucleic Acids Res 1998; 26:3453 - 9; http://dx.doi.org/10.1093/nar/26.15.3453; PMID: 9671804
- Shangary S, Wang S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: a novel approach for cancer therapy. Annu Rev Pharmacol Toxicol 2009; 49:223 - 41; http://dx.doi.org/10.1146/annurev.pharmtox.48.113006.094723; PMID: 18834305
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004; 303:844 - 8; http://dx.doi.org/10.1126/science.1092472; PMID: 14704432
- Issaeva N, Bozko P, Enge M, Protopopova M, Verhoef LG, Masucci M, et al. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat Med 2004; 10:1321 - 8; http://dx.doi.org/10.1038/nm1146; PMID: 15558054
- Shangary S, Qin D, McEachern D, Liu M, Miller RS, Qiu S, et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc Natl Acad Sci U S A 2008; 105:3933 - 8; http://dx.doi.org/10.1073/pnas.0708917105; PMID: 18316739
- Yang Y, Ludwig RL, Jensen JP, Pierre SA, Medaglia MV, Davydov IV, et al. Small molecule inhibitors of HDM2 ubiquitin ligase activity stabilize and activate p53 in cells. Cancer Cell 2005; 7:547 - 59; http://dx.doi.org/10.1016/j.ccr.2005.04.029; PMID: 15950904
- Reed D, Shen Y, Shelat AA, Arnold LA, Ferreira AM, Zhu F, et al. Identification and characterization of the first small molecule inhibitor of MDMX. J Biol Chem 2010; 285:10786 - 96; http://dx.doi.org/10.1074/jbc.M109.056747; PMID: 20080970
- Kobet E, Zeng X, Zhu Y, Keller D, Lu H. MDM2 inhibits p300-mediated p53 acetylation and activation by forming a ternary complex with the two proteins. Proc Natl Acad Sci U S A 2000; 97:12547 - 52; http://dx.doi.org/10.1073/pnas.97.23.12547; PMID: 11070080
- Ito A, Lai CH, Zhao X, Saito S, Hamilton MH, Appella E, et al. p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J 2001; 20:1331 - 40; http://dx.doi.org/10.1093/emboj/20.6.1331; PMID: 11250899
- Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 1997; 90:595 - 606; http://dx.doi.org/10.1016/S0092-8674(00)80521-8; PMID: 9288740
- Li M, Luo J, Brooks CL, Gu W. Acetylation of p53 inhibits its ubiquitination by Mdm2. J Biol Chem 2002; 277:50607 - 11; http://dx.doi.org/10.1074/jbc.C200578200; PMID: 12421820
- Ito A, Kawaguchi Y, Lai CH, Kovacs JJ, Higashimoto Y, Appella E, et al. MDM2-HDAC1-mediated deacetylation of p53 is required for its degradation. EMBO J 2002; 21:6236 - 45; http://dx.doi.org/10.1093/emboj/cdf616; PMID: 12426395
- Lain S, Hollick JJ, Campbell J, Staples OD, Higgins M, Aoubala M, et al. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell 2008; 13:454 - 63; http://dx.doi.org/10.1016/j.ccr.2008.03.004; PMID: 18455128
- Zhang Q, Zeng SX, Zhang Y, Zhang Y, Ding D, Ye Q, et al. A small molecule Inauhzin inhibits SIRT1 activity and suppresses tumour growth through activation of p53. EMBO Mol Med 2012; 4:298 - 312; http://dx.doi.org/10.1002/emmm.201100211; PMID: 22331558
- Huffman DM, Grizzle WE, Bamman MM, Kim JS, Eltoum IA, Elgavish A, et al. SIRT1 is significantly elevated in mouse and human prostate cancer. Cancer Res 2007; 67:6612 - 8; http://dx.doi.org/10.1158/0008-5472.CAN-07-0085; PMID: 17638871
- Jung-Hynes B, Nihal M, Zhong W, Ahmad N. Role of sirtuin histone deacetylase SIRT1 in prostate cancer. A target for prostate cancer management via its inhibition?. J Biol Chem 2009; 284:3823 - 32; http://dx.doi.org/10.1074/jbc.M807869200; PMID: 19075016
- Chen WY, Wang DH, Yen RC, Luo J, Gu W, Baylin SB. Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell 2005; 123:437 - 48; http://dx.doi.org/10.1016/j.cell.2005.08.011; PMID: 16269335
- Kitagawa M, Aonuma M, Lee SH, Fukutake S, McCormick F. E2F-1 transcriptional activity is a critical determinant of Mdm2 antagonist-induced apoptosis in human tumor cell lines. Oncogene 2008; 27:5303 - 14; http://dx.doi.org/10.1038/onc.2008.164; PMID: 18521084
- Poyurovsky MV, Katz C, Laptenko O, Beckerman R, Lokshin M, Ahn J, et al. The C terminus of p53 binds the N-terminal domain of MDM2. Nat Struct Mol Biol 2010; 17:982 - 9; http://dx.doi.org/10.1038/nsmb.1872; PMID: 20639885
- Dai JM, Wang ZY, Sun DC, Lin RX, Wang SQ. SIRT1 interacts with p73 and suppresses p73-dependent transcriptional activity. J Cell Physiol 2007; 210:161 - 6; http://dx.doi.org/10.1002/jcp.20831; PMID: 16998810
- Verma R, Rigatti MJ, Belinsky GS, Godman CA, Giardina C. DNA damage response to the Mdm2 inhibitor nutlin-3. Biochem Pharmacol 2010; 79:565 - 74; http://dx.doi.org/10.1016/j.bcp.2009.09.020; PMID: 19788889
- Valentine JM, Kumar S, Moumen A. A p53-independent role for the MDM2 antagonist Nutlin-3 in DNA damage response initiation. BMC Cancer 2011; 11:79; http://dx.doi.org/10.1186/1471-2407-11-79; PMID: 21338495
- Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 1984; 22:27 - 55; http://dx.doi.org/10.1016/0065-2571(84)90007-4; PMID: 6382953