Abstract
mTORC1 inhibitors, including rapamycin and its analogs, have been actively studied both pre-clinically and clinically. However, the single treatment of mTORC1 inhibitors has been modest in most cancer types. We have previously demonstrated that the activation of PI3K/Akt and MEK/ERK signaling pathways attenuates the anticancer efficacy of mTORC1 inhibitors. In this study, we report that mTORC1 inhibition also phosphorylates and inactivates GSK3β, which is a tumor suppressor in lung cancer. Moreover, we show that perifosine, as an Akt inhibitor, decreases rapamycin-induced phosphorylation of GSK3β and elevated p-GSK3β levels in rapamycin-resistant cell lines. Combination of perifosine with mTORC1 inhibitors showed enhanced anticancer efficacy both in cell cultures and in a xenograft mouse model. In addition, perifosine inhibits the growth of both rapamycin sensitive and resistant A549 cells. However, inhibition of GSK3β by a selective inhibitor- LiCl, or downregulation of GSK3β expression by siRNA, reverses the growth inhibitory effects of perifosine on rapamycin resistant cells, suggesting the important role of GSK3β activation in enhancing mTORC1 inhibitors efficacy by perifosine. Thus, our results provide a potential therapeutic strategy to enhance mTORC1-targeted cancer therapy by using perifosine or targeting GSK3β.
Keywords: :
Introduction
Mammalian target of rapamycin (mTOR) is a serine/threonine kinase, and functions as a catalytic subunit in at least two complexes: mTORC1 (mTOR complex 1) and mTORC2. mTORC1 responds to growth factors, nutrients, and energetic status, and regulates cell growth, autophagy, and metabolism, whereas mTORC2 mainly responds to growth factors and regulates cell survival and the actin cytoskeleton. mTORC1 predominantly regulates protein translation through ribosomal p70 S6 Kinase (p70S6K) and eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1). mTORC2 functions mainly through directly activates Akt and other kinases, such as protein kinase C (PKC) and serum- and glucocorticoid-induced protein kinase 1 (SGK1). Recently, two sets of phosphoproteomics experiments uncovered a large fraction of new substrates, such as growth factor receptor-bound protein 10 (Grb10), or downstream targets of mTOR.Citation1-Citation3 Thus, mTOR locates in the node of multiple signaling pathways and has been an attractive therapeutic target in cancer and diabetes.
The first generation of mTORC1 inhibitors, including rapamycin and its derivatives-everolimus (RAD001) and temsirolimus (CCI-779), has shown limited efficacy as a single reagent in most cancer types, except for metastatic renal cell carcinoma and mantle cell lymphoma.Citation4-Citation6 PI3K/Akt or MEK/ERK signaling pathway activation, or mTORC2 dysregulation has been demonstrated to partly facilitate development of drug resistance, but the mechanisms of mTORC1 inhibitors resistance have not been fully elucidated.Citation3 Therefore, clarifying the mechanisms of resistance and developing new combination rationales are critically needed in preclinical studies and clinical trials.
Glycogen synthase kinase 3β (GSK3β) is an serine/threonine kinase that plays an important role in many cellular progresses, such as cell proliferation, apoptosis, and differentiation.Citation7 Dysregulated GSK3β has been implicated in diabetes, inflammation, neurodegenerative disorders, and malignancies.Citation8 It is a well-known substrate of Akt and p90 ribosomal S6 Kinase (p90RSK), and is also regulated by ERK, p70S6K, and PKC through phosphorylation.Citation7,Citation9 Phosphorylation at serine 9 inhibits GSK3β activity, whereas tyrosine phosphorylation positively regulates its activity.Citation10 Identified downstream targets of GSK3β include β-catenin, NFκB, c-Myc, Cyclin D1, AP-1 and so on. GSK3β acts as both a tumor promoter and a tumor suppressor, depending on cancer types and environments.Citation7 Studies in lung cancer showed increased levels of inactive form of GSK3β, suggesting activating GSK3β for cancer therapy in lung cancer.Citation11
Perifosine is a novel, oral alkylphospholipid and shows anticancer activity in many preclinical studies. It has been in phase I/II clinical trials, but it shows very limited anticancer efficacy when used as a single reagent in certain types of cancers.Citation12 Therefore, current studies are focusing on developing new strategies to combine it with other reagents. In recent preclinical studies, it showed synergistic effects when combined with histone deacetylase inhibitor, 3-Phosphoinositide-dependent protein kinase-1 (PDK1) inhibitor, epithelial growth factor receptor—tyrosine kinase inhibitors (EGFR-TKI), and some chemotherapeutic reagents.Citation13-Citation16 As an alkylphospholipid, it perturbs cell membrane and induces apoptosis, cell cycle arrest and autophagy through mechanisms that are still unclear.Citation17 It has been shown that inhibition of Akt or MAPK, induction of c-Jun NH2-terminal kinase (JNK), and upregulation of death receptor 5 (DR5) account partly for perifosine’s antitumor efficacy.Citation18-Citation21
We have found increased p-Akt and p-p44/42 levels in parallel with elevated p-GSK3β (Ser9) in our established rapamycin resistant cell lines.Citation22,Citation23 But for the role of GSK3β in rapamycin resistance is still unknown. Given that perifosine inhibits Akt and ERK and may activate GSK3β, we hypothesize that perifosine may augment mTORC1 inhibitor’s anticancer efficacy through regulation of GSK3β 17. In this study, we first confirmed regulation of GSK3β by rapamycin short- and long-term treatments. We then determined whether perifosine could decrease rapamycin-induced p-GSK3β (Ser9) and enhance mTORC1 inhibitors effects on the growth of culture cells and xenografts in nude mice. Moreover, we identified the role of GSK3β in rapamycin resistance and in the growth inhibitory effects of perifosine on our previously established rapamycin-resistant A549 cell lines. These findings provide a new rationale for combination perifosine with mTORC1 inhibitors in lung cancer.
Results
Rapamycin induces GSK3β phosphorylation at serine 9 in NSCLC cells
We and others have previously reported that PI3K/Akt, Ras/MAPK, and eukaryotic translation initiation factor (eIF4E) signaling pathways activation play important roles in rapamycin resistance, but the mechanisms of mTORC1 inhibitor’s resistance have not been fully elucidated. Given that GSK3β is regulated by many kinases correlated with mTOR signaling pathway, such as Akt, ERK, PKC, p70S6K, we first determined how it was regulated by rapamycin. Even though p-p70S6K (Thr389) and its downstream signal p-S6 (S235/S236) were downregulated, p-Akt (Ser473), p-p44/42 (Thr202/Tyr204), and p-PKCα (Ser657) were upregulated after rapamycin 48 h treatment, p-GSK3β (Ser9) was increased in three of four NSCLC cells, including H157, A549, and H460 cells (). Total Akt, p44/42, and GSK3β levels were not altered in all these cells, whereas total p70S6K decreased in H157 and A549 cells. It suggests that GSK3β phosphorylation may not be a simple consequence of Akt activation, and it may be regulated by complicated signaling pathways after rapamycin treatment.
Figure 1. Rapamycin increases GSK3β phosphorylation at serine 9 in NSCLC cell lines. (A) the indicated cell lines were treated with 10 nmol/L rapamycin for 48 h. (B) A549-P and A549-RR cells were treated with rapamycin 1 nmol/L or 1 μmol/L as indicated for 24 h. After the aforementioned treatments, the cells were subjected to preparation of whole-cell protein lysates and subsequent western blot analysis for detection of the proteins as presented. D, DMSO; ra, rapamycin; ra1, rapamycin 1 nmol/L; ra2, rapamycin 1 μmol/L; L.E., longer exposure.
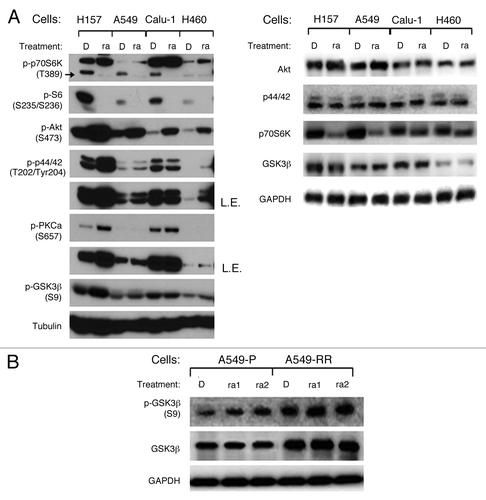
We have previously reported that p-Akt and p-p44/42 increased without alteration of total protein levels in our established rapamycin resistant cells (A549-RR) compared with its parental rapamycin sensitive cells (A549-P), and p-GSK3β (Ser9), as an indicator of Akt activity, is also increased in A549-RR cells.Citation22,Citation23 In this study, we found that both phosphorylated and total GSK3β were elevated in A549-RR compared with A549-P. 1nmol/L and 1µmol/L rapamycin treatments for 24 h, could increase p-GSK3β in A549-P cells, but not in A549-RR cells, whereas total GSK3β levels were not regulated in both cell lines (). Since GSK3β phosphorylation at serine 9 indicates its inactivation, these results suggest that rapamycin-induced inhibition of GSK3β may correlate with its resistance.
Perifosine inhibits rapamycin-induced p-GSK3β (Ser9)
Perifosine is an Akt inhibitor, which inhibits Akt activity by interfering with its recruitment to the cell membrane and phosphorylation by PI3K. It has also been reported to modulate many other kinases, including inhibition of p44/42 and p70S6K, and activation of GSK3β. First, we showed that perifosine decreased p-Akt, p-p44/42, p-p70S6K, p-GSK3β and c-Myc in a dose-dependent manner in A549 cells (). We also found that total Akt and p70S6K protein levels were decreased, but total p44/42 and GSK3β levels were not markedly changed. These results suggest that perifosine downregulates Akt, ERK and p70S6K signaling pathways, whereas activates the GSK3β signaling pathway.
Figure 2. Perifosine decreased GSK3β phosphorylation at serine 9 in NSCLC cells. (A) A549 cells were treated with different concentrations of perifosine as indicated for 24h. (B) A549 cells were treated with rapamycin 1nmol/L alone, 2.5 or 5 μmol/L perifosine alone, or their respective combinations as indicated for 48 h. (C) A549-P and A549-RR cells were treated with perifosine in different concentrations as indicated for 24 h. After the aforementioned treatments, cells were subjected for whole-cell protein lysates and subsequent western blot analysis for detection of the proteins as presented.
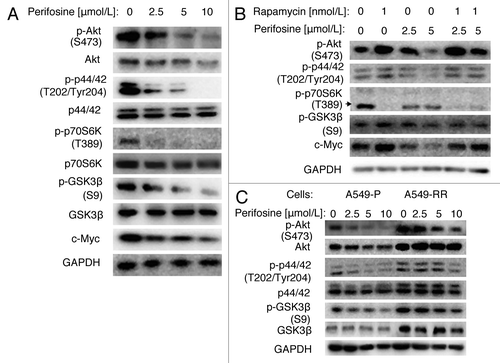
Then, we found that even though rapamycin differently regulated upstream signals of GSK3β, with significant decrease of p-p70S6K and increase of p-Akt, and a slight increase of p-p44/42, the p-GSK3β and c-Myc were increased, which suggests activation of this survival pathway. Moreover, perifosine co-treatment reversed rapamycin-induced p-Akt, p-GSK3β and c-Myc, but only minimally regulated p-p44/42 (). These findings suggest that perifosine has the potency of overcoming rapamycin-activated survival pathways, especially Akt/GSK3β pathway.
Given that p-Akt, p-p44/42, and p-GSK3β levels were higher in A549-RR cells than in A549-P cells, we further examined whether these signals were regulated by perifosine. showed that 2.5–10 µmol/L perifosine inhibited p-Akt and p-p44/42 dose-dependently in both cell lines. Total Akt levels were decreased in A549-P cells but not in A549-RR cells, whereas total p44/42 levels were not markedly altered in both cell lines, suggesting downregulation of these two important survival pathways by perifosine. In addition, perifosine decreased p-GSK3β levels in both A549-P and A549-RR cells, but minimally regulated total GSK3β levels, suggesting perifosine activated GSK3β. Collectively, we conclude that perifosine can resume the activity of GSK3β after short- or long-term rapamycin treatment.
Perifosine enhanced mTORC1 inhibitor’s anticancer efficacy in vitro and in vivo
Since perifosine released the inhibitory effects of rapamycin on GSK3β, we suspected that perifosine might enhance rapamycin’s effects on cell growth. As shown in , rapamycin inhibited A549 cells growth dose-dependently. The combination of 0.05, 0.1, 0.5, 1 nmol/L rapamcyin and 1.25 µmol/L perifosine respectively exhibited more growth inhibitory effects on A549 cells by cell viability assay. Moreover, a greater inhibitory effect was observed when H460 cells were treated with 1 nmol/L rapamycin in combination with 2.5 or 5 µmol/L perifosine compared with each single reagent treatment by a 14-d colony formation assay (). In addition, A549-RR cells were as sensitive as A549-P cells to different concentrations of perifosine treatment (). These results suggest that perifosine increases the inhibitory potency of rapamycin.
Figure 3. Perifosine enhances mTORC1 inhibitors’ anticancer efficacy in cell culture (A, B, C) and in nude mice (D, E). (A) A549 cells were treated with different concentrations of rapamycin alone, 1.25 μmol/L perifosine alone, and perifosine plus rapamycin as indicated for 3 d and subjected for SRB assay. (B) H460 cells were treated with 1 nmol/L rapamycin alone, 2.5 and 5.0 μmol/L perifosine alone, and perifosine plus rapamycin as indicated for 14 d and subjected for colony formation assay. (C) A549-P and A549-RR cells were treated with different concentrations of perifosine as indicated for 3 d and subjected for SRB assay. Columns and points, means of four replicate determinations; bars, SD. (D) four groups of mice with A549 xenografts were treated with vehicle control, RAD001 (3 mg/kg/day o.g.) alone, perifosine 7.5 mg/kg/day o.g.) alone, and RAD001 plus perifosine on the same day after grouping. After 14 d, the mice were sacrificed. During the treatments, tumor sizes were measured once every two days. E, on day 14, the tumors were removed and weighed. Each group included 6 mice. Points and columns, means; bars, SD; *, p < 0.05 vs control; #p < 0.05 vs RAD001; $p < 0.05 vs perifosine.
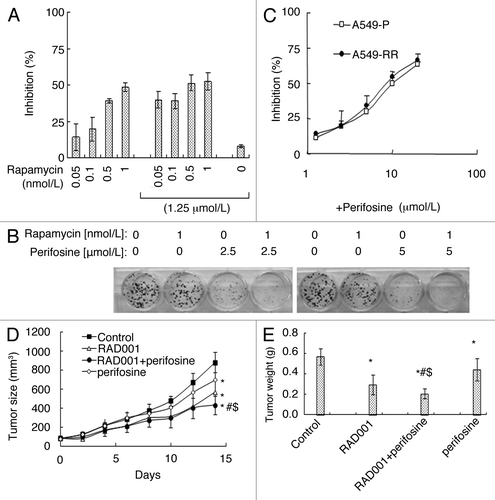
We further examined the combination effects of perifosine and RAD001, a derivative of rapamycin and an oral mTORC1 inhibitor, on the growth of lung cancer xenografts in nude mice. As presented in , both RAD001 and perifosine alone significantly inhibited the growth of A549 lung cancer xenografts. However, their combination exhibited the best effects, which was superior to either reagent alone (p < 0.05). Measuring the tumor weights at the end of the experiments also confirmed this finding (p < 0.05) (). These results suggest that combining perifosine and rapamycin has better anticancer potency.
Inactivation of GSK3β partially abrogated perifosine’s effects on the growth of rapamycin resistant cells
Even though short-term rapamycin treatment increases p-GSK3β, and long-term rapamycin treatment induces an increase of both phosphorylated and total GSK3β expression in A549-RR cells, the role of GSK3β in rapamycin resistance is still unknown. To clarify this, we first determined whether activation of GSK3β could sensitize rapamycin’s inhibitory effects on the growth of A549-RR cells. A549-RR cells were transfected with the HA GSK3 β S9A pcDNA plasmid which encoded the constitutively active form of GSK3β (S9A mutant) or the control pcDNA plasmid, and then treated with rapamcyin. As shown in , p-GSK3β was decreased and total GSK3β was increased significantly, suggesting successfully overexpression of active GSK3β proteins. Further SRB assay showed that rapamycin’s inhibitory effects increased significantly (p < 0.05) after S9A mutant expression compared with control (). These results suggest that recovery of GSK3β activity sensitizes rapamycin’s effects.
Figure 4. Inactivation of GSK3β by LiCl (A, B) or knockdown of GSK3β expression by siRNA (C, D) in A549-RR cells reversed perifosine’s growth inhibitory effects. (A) A549-RR cells were transfected with the HA GSK3 β S9A pcDNA plasmid or control pcDNA plasmid in 6-well plates for 24h and then reseeded to 96-well plates or 6-well plates. Cells in 6-well plates were cultured for another 24h and subjected for western blot analysis. (B) cells in 96-well plates were added with different concentrations of rapamycin as indicated on the same day for 3 d and then subjected for SRB assay. Points, means of four replicate determinations; bars, SD *p < 0.05 vs control pcDNA. (C) A549-RR cells were treated with 10 µmol/L perifosine alone, 10 mmol/L LiCl alone, and perifosine plus LiCl for 24 h, and then subjected for western blot analysis. (D) A549-RR cells were treated with different concentrations of perifosine without or with 5, 10, or 15 mmol/L LiCl for 3 d and subjected for SRB assay. Points, means of four replicate determinations; bars, SD; *p < 0.05 vs perifosine alone. E, A549-RR cells were transfected with GSK3β siRNA or control siRNA in 6-well plates for 24 h and then reseeded to 6-well plates or 96-well plates. Cells in 6-well plates were treated with or without perifosine for another 24h, and then subjected for qRT-PCR assay and western blot analysis. Columns, means of three replicate determinations; bars, SD *p < 0.05 vs control siNRA. F, cells in 96-well plates were added with different concentrations of perifosine on the second day as indicated for 3 d and then subjected for SRB assay. Points, means of four replicate determinations; bars, SD *p < 0.05 vs control siNRA.
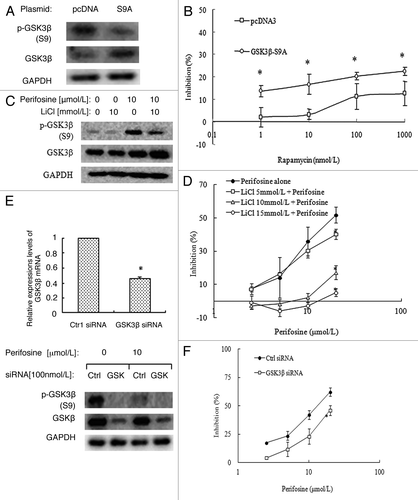
We then explored whether perifosine-induced activation of GSK3β played an important role to impact rapamycin’s effects. We first observed the effects of a selective GSK3β inhibitor-LiCl on the growth of A549-RR cells with perifosine treatment. Western blot analysis showed that p-GSK3β increased significantly after 10 mmol/L LiCl treatment as a single reagent, and perifosine-induced decrease of p-GSK3β was greatly resumed with LiCl cotreatment, suggesting LiCl inhibited perifosine-induced activation of GSK3β (). SRB assays showed that LiCl ranging from 5, 10, or 15 mmol/L reversed perifosine’s inhibitory effects on cell growth dose-dependently ().
Since LiCl inhibits both GSK3α and GSK3β, we then evaluated the role of GSK3β by silencing its expression using a specific siRNA. showed a 0.54-fold decrease of GSK3β mRNA levels after 100 nmol/L GSK3β siRNA transfection for 48 h compared with control siRNA transfection in A549-RR cells by quantitative RT-PCR assay. A significant decrease of both p-GSK3β and total GSK3β protein levels was also observed by western blot analysis, indicating a successful GSK3β knockdown (). Then, we found that perifosine inhibits the growth of A549-RR cells with control siRNA transfection in a dose-dependent manner, whereas its inhibitory effects decreased significantly after GSK3β siRNA transfection (). These results suggest that activation of GSK3β plays an important role in mediating the growth inhibitory effects of perifosine on A549-RR cells.
Discussion
In this study, we show that rapamycin short-term or long-term treatment increases the levels of p-GSK3β (Ser9) in some human lung cancer cell line. Moreover, exogenous expression of the constitutively active form of GSK3β sensitizes rapamycin’s growth inhibitory effects on A549-RR cells, suggesting that inactivation of GSK3β plays a role in mTORC1 inhibitors resistance. In addition, among the upstream regulators of GSK3β, p-Akt, p-p44/42 and p-PKCα were increased, while p-p70S6K was decreased, suggesting that rapamycin-induced p-GSK3β may not be a simple consequence of Akt activation, but an output of the very complicated signaling transduction network. We and others have previously reported that mTORC1 inhibition activates multiple survival pathways, including PI3K/Akt, MEK/ERK, and eIF4E, and co-targeting these pathways can enhance mTORC1-targeted cancer therapy.Citation22,Citation24-Citation26 In this study, we further suggest the importance of GSK3β inactivation in the acquired resistance of mTORC1 inhibitors and suggest a rationale of co-targeting GSK3β and mTORC1 in lung cancer therapy.
Perifosine, as an alkylphospholipid, exerts its antitumor effects mainly through targeting cell membrane, such as membrane structure, metabolism, and permeability.Citation12 Currently, the mechanism of its tumor-inhibitory activity has not been fully elucidated. Even though many preclinical studies have convinced it as an Akt inhibitor, clinical studies to correlate Akt inhibition with perifosine’s efficacy are still lacking. Kondapaka et al.Citation27 has shown that perifosine decreases p-Akt and inhibits Akt activity by interfering Akt translocation to cell membrane through its pleckstrin homology (PH) domain. Other studies also showed its regulation on MAPK, JNK and DR5 et al.Citation18-Citation21 GSK3β, as a well-known downstream target of Akt and ERK, has been shown to be activated by perifosine, whereas the mechanism is quite unclear. It is possible that perifosine directly attenuates the translocation of GSK3β to the cell membrane and the consequent phosphorylation by kinases in addition to Akt and ERK. In this study, we found that perifosine not only inhibited rapamycin short-time treatment-induced p-GSK3β, but also significantly decreased the elevated p-GSK3β and GSK3β levels in A549-RR cells in a dose-dependent manner. Taken together, these findings suggest that perifosine can counteract multiple survival pathways that activated by mTORC1 inhibition including GSK3β. Therefore, we suspect that combination of perifosine and rapamycin may be more effective than each single reagent alone in inhibiting cell growth partly through regulation the activity of GSK3β.
We have previously demonstrated that the RAD001 combined with the PI3K inhibitor LY294002, or EGFR inhibitor erlotinib, which inhibited both PI3K/Akt and MEK/ERK signaling pathways, enhanced anticancer efficacy in vivo.Citation22,Citation26 Given our findings aforementioned, we examined the effects of the combination of perifosine with rapamycin or RAD001 both in cultured cells and in a mouse xenograft model. The combination showed enhanced growth-inhibitory effects both in vitro and in vivo, as reported in other types of cancers, such as hematologic malignancies and multiple myeloma.Citation28,Citation29 In addition, A549-RR cells were as sensitive as A549-P cells to perifosine treatment, suggesting perifosine was still effective for cancer cells already developed resistance to mTORC1 inhibitors. Therefore, perifosine can enhance mTORC1 inhibitors anticancer efficacy and can be a substitute for patients with acquired mTORC1 inhibitors resistance.
As we further evaluated the role of GSK3β in perifosine’s effects on rapamycin resistance, we take advantage of our previously established A549-RR cells. Even though LiCl, a selective inhibitor of GSK3β, completely blocks perifosine’s effects on A549-RR cells, we should note that it has many off-target effects except for GSK3α and GSK3β inhibition.Citation30 Further study using siRNA to specifically knockdown of GSK3β expression was performed. Results showed a partial reversed effect of siRNA on perifosine’s growth inhibitory effects in A549-RR cells. It may be because GSK3β only partly accounts for perifosine’s effects. It is also possible that the inhibition of other signals, such as Akt, ERK, and their downstream targets, contributes to perifosine’s effects. By the way, we observed the same partial reverse effect of GSK3β siRNA in A549-P cells (data were not showed here). Therefore, GSK3β only partly accounts for perifosine’s effects, and further investigation is needed to clarify the mechanisms of perifosine.
The activity of GSK3β is regulated through phophorylation at multiple sites by several kinases, including Akt, MAPK, PKC, p70S6K, p90RSK, and cyclic AMP-dependeng protein kinase.Citation31 In this study, we found that both Akt and ERK were activated by rapamycin and inhibited by perifosine, but the combination treatment showed that only Akt activation induced by rapamycin was reversed by perifosine, while ERK activation was minimally affected. Moreover, GSK3β’s regulation by the combination treatment showed the same pattern as Akt. These findings suggest that Akt is more important than ERK in perifosine’s effects on rapamycin-induced p-GSK3β. It is well-known that p90RSK locates downstream of ERK and directly phosphorylates GSK3β at serine 9 and inhibits its activity.Citation9,Citation32 We have shown that ERK/p90RSK signaling pathway activation is more significant in H157 cells than in A549 cells.Citation23 Therefore, p90RSK activation may be involved in the inhibition of GSK3β by rapamycin and in the development of rapamycin resistance. The role of p90RSK-regulated GSK3β in rapamycin resistance needs further investigation.
GSK3β regulates stability of oncogenic transcription factors (TFs), such as cyclin D1, c-Myc, NFκB, β-catenin, Snail, AP-1, to regulate cell cycle progression, epithelial-mesenchymal transition (EMT), and apoptosis.Citation7 GSK3β has been shown to be both a tumor suppressor (such as skin cancer, oral cancer, and melanoma cancer) and a tumor promoter (such as prostate cancer and gastro-intestinal cancer) depending on different substrates that it regulated in different cancer types.Citation31 In lung cancer, high levels of inactive form of GSK3β are observed, suggesting that activation of GSK3β may be therapeutic.Citation11 Therefore, elucidating how rapamycin modulates GSK3β signaling pathway to develop resistance is interesting and deserves further study.
In summary, the currently study demonstrates that rapamycin inhibits GSK3β to counteract its antitumor effects in addition to activation of other survival pathways, such as PI3K/Akt and MEK/ERK signaling pathways. Perifosine can release the inhibitory effects of rapamycin on GSK3β, and downregulation of GSK3β partly reverses perifosine’s effects on cell growth. Combination of perifosine and mTORC1 inhibitors generates enhanced anticancer efficacy both in cell cultures and in a xenograft mouse model. These findings indicate new rationales for enhancing mTORC1-targeted cancer therapy by using perifosine or targeting GSK3β.
Materials and Methods
Reagents
Rapamycin was purchased from LC Laboratories (R-5000). Perifosine was purchased from Selleck Chemicals LLC (S1037), dissolved in PBS at a concentration of 50 mmol/L, and stored at -20°C. RAD001 powder, formulated RAD001, and matched placebo control were provided by Novartis Pharmaceuticals Corporation. The powders of these reagents were dissolved in DMSO at a concentration of 10 mmol/L, and stored at -80°C. LiCl was purchased from Sigma-Aldrich (31048), dissolved in distilled water at a concentration of 1 mol/L. Lipofectamine 2000 transfection reagent was purchased from Life Technologies Co. Invitrogen (11668–019). Rabbit polyclonal antibodies against p-Akt (Ser473)(9271), p-p44/42 (Thr202/Tyr204)(9101), p-GSK3β (Ser9)(9336), p-p70S6K (Thr389)(9205), p-S6 (Ser235/236)(4858), Akt (9272), p70S6K (9202) and rabbit monoclonal antibody against GSK3β (9315) were purchased from Cell Signaling Technology, Inc. Mouse monoclonal antibodies against c-Myc (sc-40), actin (sc-130300), β-tubulin (sc-5274) and rabbit polyclonal antibodies against p-PKCα (Ser657)(sc-12356) were purchased from Santa Cruz Biotechnology Inc. Rabbit polyclonal anti-GAPDH (AP0063) and anti-ERK1/2 (BS3627) antibodies were purchased from Bioworld Technology Inc.
Cell lines and cell treatment
Human lung cancer cell lines were purchased from the American Type Culture Collection (ATCC). Rapamycin sensitive and resistant cells were established as we previously described.Citation22 These cell lines were grown in monolayer cultured in RPMI 1640 medium supplemented with 5% fetal bovine serum at 37°C in a humidified atmosphere consisting of 5% CO2.
Sulforhodamine B assay
Cells were seeded in 96-well plates by 3,000 cells/well. On the second day cells were treated or co-treated with reagents diluted in media. For co-treatment, different reagents were added simultaneously. After 3 d, monolayer cells were fixed and stained with sulforhodamine B (SRB). Finally, the dye was dissolved in 10 mmol/L Tris base and subjected for quantification as we previously described.Citation33 Absorbance was measured at 500 nm using μQuant Universal Microplate Spectrophotometer (BioTek Instruments, Inc.).
Colony formation assay
Cells were seeded in 12-well plates by 200 cells/well. On the second day cells were treated with rapamycin, perifosine, or rapamycin plus perifosine diluted in growth media. Every 3 d, the media were replaced with fresh media containing these reagents. After 14 d, cell colonies were stained with crystal violet. Pictures were taken by using a digital camera.
Western blot analysis
Whole-cell protein lysis buffer contains 1% Triton, 40 mmol/L HEPES, 120 mmol/L NaCl, 1 mmol/L EDTA, 10 mmol/L pyrophosphate, 10 mmol/L glycerophosphate, 50 mmol/L NaF, 0.5 mmol/L Na3VO4, and was stored at 4°C. Protease inhibitors were added just before use. Cells were harvested at the end of treatment. Protein concentration was confirmed by the Bradford assay. Protein was separated by 10% SDS-PAGE and then transferred to PVDF membrane using standard procedures. The membrane was incubated with the primary antibody overnight at 4°C and washed. After incubation with secondary antibody and wash, membrane was subjected to Supersignal west Pico chemiluminescent substrate from Thermo Scientific Pierce (34080) and exposed.
Quantitative real-time polymerase chain reaction
Total RNA from cells was extracted using Trizol reagent (1596–026) and reverse transcription was conducted according to standard procedure using M-MLV reverse transcriptase from Promega (M1701). PCR reactions were conducted according to manufacturer's instructions using Power SYBR Green PCR Master Mix from Applied Biosystems Inc. (4369016). Forward (F) and reverse (R) primers were used as follows: GSK3β, F: 5′-CTAAGGATTCGTCAGGAACAG-3′ and R: 5′- TTGAGTGGTGAAGTTGAAGAG -3′; GAPDH, F: 5′-TGTTCGACAGTCAGCCGC-3′, and R: 5′-GGTGTCTGAGCGATGTGGC-3′, and synthesized by Invitrogen.Citation34,Citation35 All real-time amplifications were measured in triplicates and performed with the ABI Prism 7300 sequence detection system (Applied Biosystems). The fold-change of GSK3β mRNA was calculated using the 2-ΔΔCT method.
Gene knockdown by small interfering RNA
Control (non-target) small interfering RNA (siRNA) was purchased from Invitrogen. GSK3β siRNA that targets 5′- AAGAATCGAGAGCTCCAGATC -3′ was described previously and synthesized by Invitrogen.Citation36 Cells were seeded to 6-well plate at 5 × 105 cells/well and transfected with 100 nmol/L control or GSK3β siRNA respectively for 24 h using lipofectamine 2000 following its instruction (11668–019). Then cells were reseeded to 96-well plates, treated with perfosine for additional 3 d, and subjected for Sulforhodamine B assay. Left cells were reseeded to 6-well plates and subjected to total-mRNA extraction and whole-cell protein lysates preparation and subsequent qRT-PCR assay and western blot analysis respectively after another 24 h to confirm GSK3β knockdown efficacy.
Gene overexpression by plasmids
The HA GSK3 β S9A pcDNA plasmid (14754) that encodes the constitutively active form of GSK3β (GSK3β-S9A mutant) was described previouslyCitation32 and were purchased from Addgene, Inc. pcDNA plasmid was used as control. Transfection was conducted by using lipofectamine 2000 following its instruction. The transfected A549-RR cells were subjected for SRB assay and western blot analysis as described above.
Lung cancer xenografts and treatments
Animal experiments were approved by the Institutional Animal Care and Use Committee of Emory University. Four-to 6 weeks old female anthymic (nu/nu) mice were ordered from Taconic and housed under pathogen-free conditions in microisolator cages with laboratory chow and water ad libitum. A549 cells at 5 × 106 cells in serum-free medium were injected s.c. into the flank region of nude mice. When tumors reached certain size ranges (± 100 mm3), the mice were randomized into four groups (n = 6/group) according to tumor volumes and body weights for the following treatments: vehicle control, formulated RAD001 (3 mg/kg/day; og), perifosine (7.5 mg/kg/day; og), and the combination of RAD001 and perifosine. Tumor volumes were measured using caliper measurements once every two days and calculated with the formula V = π (length × widthCitation2)/6. After a 14-d treatment, the mice were sacrificed with CO2. The tumors were then removed and weighed.
Statistical analysis
The statistical significance of differences in tumor size, weights, or cell viability between different treatments was analyzed with two-sided unpaired Student's t tests. Results were considered to be statistically significant at p < 0.05.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (No. 30873099, 81102458, 81172004) and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD). We thank Dr. Shi-Yong Sun in Winship Cancer Institute at Emory University for providing us with RAD001 and for helping us to conduct animal experiments.
Notes
† These authors contributed equally to this work.
References
- Yu Y, Yoon SO, Poulogiannis G, Yang Q, Ma XM, Villén J, et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science 2011; 332:1322 - 6; http://dx.doi.org/10.1126/science.1199484; PMID: 21659605
- Hsu PP, Kang SA, Rameseder J, Zhang Y, Ottina KA, Lim D, et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 2011; 332:1317 - 22; http://dx.doi.org/10.1126/science.1199498; PMID: 21659604
- Yea SS, Fruman DA. Cell signaling. New mTOR targets Grb attention. Science 2011; 332:1270 - 1; http://dx.doi.org/10.1126/science.1208071; PMID: 21659593
- Fasolo A, Sessa C. Current and future directions in mammalian target of rapamycin inhibitors development. Expert Opin Investig Drugs 2011; 20:381 - 94; http://dx.doi.org/10.1517/13543784.2011.541154; PMID: 21299441
- Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, et al, RECORD-1 Study Group. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet 2008; 372:449 - 56; http://dx.doi.org/10.1016/S0140-6736(08)61039-9; PMID: 18653228
- Hess G, Herbrecht R, Romaguera J, Verhoef G, Crump M, Gisselbrecht C, et al. Phase III study to evaluate temsirolimus compared with investigator’s choice therapy for the treatment of relapsed or refractory mantle cell lymphoma. J Clin Oncol 2009; 27:3822 - 9; http://dx.doi.org/10.1200/JCO.2008.20.7977; PMID: 19581539
- Mishra R. Glycogen synthase kinase 3 beta: can it be a target for oral cancer. Mol Cancer 2010; 9:144; http://dx.doi.org/10.1186/1476-4598-9-144; PMID: 20537194
- Wang Z, Iwasaki M, Ficara F, Lin C, Matheny C, Wong SH, et al. GSK-3 promotes conditional association of CREB and its coactivators with MEIS1 to facilitate HOX-mediated transcription and oncogenesis. Cancer Cell 2010; 17:597 - 608; http://dx.doi.org/10.1016/j.ccr.2010.04.024; PMID: 20541704
- Sutherland C, Leighton IA, Cohen P. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: new kinase connections in insulin and growth-factor signalling. Biochem J 1993; 296:15 - 9; PMID: 8250835
- Hur EM, Zhou FQ. GSK3 signalling in neural development. Nat Rev Neurosci 2010; 11:539 - 51; http://dx.doi.org/10.1038/nrn2870; PMID: 20648061
- Zheng H, Saito H, Masuda S, Yang X, Takano Y. Phosphorylated GSK3beta-ser9 and EGFR are good prognostic factors for lung carcinomas. Anticancer Res 2007; 27:5B 3561 - 9; PMID: 17972518
- Gills JJ, Dennis PA. Perifosine: update on a novel Akt inhibitor. Curr Oncol Rep 2009; 11:102 - 10; http://dx.doi.org/10.1007/s11912-009-0016-4; PMID: 19216841
- Rahmani M, Reese E, Dai Y, Bauer C, Payne SG, Dent P, et al. Coadministration of histone deacetylase inhibitors and perifosine synergistically induces apoptosis in human leukemia cells through Akt and ERK1/2 inactivation and the generation of ceramide and reactive oxygen species. Cancer Res 2005; 65:2422 - 32; http://dx.doi.org/10.1158/0008-5472.CAN-04-2440; PMID: 15781658
- Dasmahapatra GP, Didolkar P, Alley MC, Ghosh S, Sausville EA, Roy KK. In vitro combination treatment with perifosine and UCN-01 demonstrates synergism against prostate (PC-3) and lung (A549) epithelial adenocarcinoma cell lines. Clin Cancer Res 2004; 10:5242 - 52; http://dx.doi.org/10.1158/1078-0432.CCR-03-0534; PMID: 15297428
- Festuccia C, Gravina GL, Muzi P, Millimaggi D, Dolo V, Vicentini C, et al. Akt down-modulation induces apoptosis of human prostate cancer cells and synergizes with EGFR tyrosine kinase inhibitors. Prostate 2008; 68:965 - 74; http://dx.doi.org/10.1002/pros.20757; PMID: 18361408
- Nyåkern M, Cappellini A, Mantovani I, Martelli AM. Synergistic induction of apoptosis in human leukemia T cells by the Akt inhibitor perifosine and etoposide through activation of intrinsic and Fas-mediated extrinsic cell death pathways. Mol Cancer Ther 2006; 5:1559 - 70; http://dx.doi.org/10.1158/1535-7163.MCT-06-0076; PMID: 16818515
- Fu L, Kim YA, Wang X, Wu X, Yue P, Lonial S, et al. Perifosine inhibits mammalian target of rapamycin signaling through facilitating degradation of major components in the mTOR axis and induces autophagy. Cancer Res 2009; 69:8967 - 76; http://dx.doi.org/10.1158/0008-5472.CAN-09-2190; PMID: 19920197
- Hideshima T, Catley L, Yasui H, Ishitsuka K, Raje N, Mitsiades C, et al. Perifosine, an oral bioactive novel alkylphospholipid, inhibits Akt and induces in vitro and in vivo cytotoxicity in human multiple myeloma cells. Blood 2006; 107:4053 - 62; http://dx.doi.org/10.1182/blood-2005-08-3434; PMID: 16418332
- Elrod HA, Lin YD, Yue P, Wang X, Lonial S, Khuri FR, et al. The alkylphospholipid perifosine induces apoptosis of human lung cancer cells requiring inhibition of Akt and activation of the extrinsic apoptotic pathway. Mol Cancer Ther 2007; 6:2029 - 38; http://dx.doi.org/10.1158/1535-7163.MCT-07-0004; PMID: 17604333
- Papa V, Tazzari PL, Chiarini F, Cappellini A, Ricci F, Billi AM, et al. Proapoptotic activity and chemosensitizing effect of the novel Akt inhibitor perifosine in acute myelogenous leukemia cells. Leukemia 2008; 22:147 - 60; http://dx.doi.org/10.1038/sj.leu.2404980; PMID: 17928881
- Tazzari PL, Tabellini G, Ricci F, Papa V, Bortul R, Chiarini F, et al. Synergistic proapoptotic activity of recombinant TRAIL plus the Akt inhibitor Perifosine in acute myelogenous leukemia cells. Cancer Res 2008; 68:9394 - 403; http://dx.doi.org/10.1158/0008-5472.CAN-08-2815; PMID: 19010914
- Wang X, Yue P, Kim YA, Fu H, Khuri FR, Sun SY. Enhancing mammalian target of rapamycin (mTOR)-targeted cancer therapy by preventing mTOR/raptor inhibition-initiated, mTOR/rictor-independent Akt activation. Cancer Res 2008; 68:7409 - 18; http://dx.doi.org/10.1158/0008-5472.CAN-08-1522; PMID: 18794129
- Wang X, Hawk N, Yue P, Kauh J, Ramalingam SS, Fu H, et al. Overcoming mTOR inhibition-induced paradoxical activation of survival signaling pathways enhances mTOR inhibitors’ anticancer efficacy. Cancer Biol Ther 2008; 7:1952 - 8; http://dx.doi.org/10.4161/cbt.7.12.6944; PMID: 18981735
- Wang X, Sun SY. Enhancing mTOR-targeted cancer therapy. Expert Opin Ther Targets 2009; 13:1193 - 203; http://dx.doi.org/10.1517/14728220903225008; PMID: 19694499
- Wang X, Yue P, Chan CB, Ye K, Ueda T, Watanabe-Fukunaga R, et al. Inhibition of mammalian target of rapamycin induces phosphatidylinositol 3-kinase-dependent and Mnk-mediated eukaryotic translation initiation factor 4E phosphorylation. Mol Cell Biol 2007; 27:7405 - 13; http://dx.doi.org/10.1128/MCB.00760-07; PMID: 17724079
- Wang X, Hawk N, Yue P, Kauh J, Ramalingam SS, Fu H, et al. Overcoming mTOR inhibition-induced paradoxical activation of survival signaling pathways enhances mTOR inhibitors’ anticancer efficacy. Cancer Biol Ther 2008; 7:1952 - 8; http://dx.doi.org/10.4161/cbt.7.12.6944; PMID: 18981735
- Kondapaka SB, Singh SS, Dasmahapatra GP, Sausville EA, Roy KK. Perifosine, a novel alkylphospholipid, inhibits protein kinase B activation. Mol Cancer Ther 2003; 2:1093 - 103; PMID: 14617782
- Witzig TE, Kaufmann SH. Inhibition of the phosphatidylinositol 3-kinase/mammalian target of rapamycin pathway in hematologic malignancies. Curr Treat Options Oncol 2006; 7:285 - 94; http://dx.doi.org/10.1007/s11864-006-0038-1; PMID: 16916489
- Cirstea D, Hideshima T, Rodig S, Santo L, Pozzi S, Vallet S, et al. Dual inhibition of akt/mammalian target of rapamycin pathway by nanoparticle albumin-bound-rapamycin and perifosine induces antitumor activity in multiple myeloma. Mol Cancer Ther 2010; 9:963 - 75; http://dx.doi.org/10.1158/1535-7163.MCT-09-0763; PMID: 20371718
- Martinez A. Preclinical efficacy on GSK-3 inhibitors: towards a future generation of powerful drugs. Med Res Rev 2008; 28:773 - 96; http://dx.doi.org/10.1002/med.20119; PMID: 18271054
- Patel S, Woodgett J. Glycogen synthase kinase-3 and cancer: good cop, bad cop?. Cancer Cell 2008; 14:351 - 3; http://dx.doi.org/10.1016/j.ccr.2008.10.013; PMID: 18977324
- Stambolic V, Woodgett JR. Mitogen inactivation of glycogen synthase kinase-3 beta in intact cells via serine 9 phosphorylation. Biochem J 1994; 303:701 - 4; PMID: 7980435
- Sun SY, Rosenberg LM, Wang X, Zhou Z, Yue P, Fu H, et al. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res 2005; 65:7052 - 8; http://dx.doi.org/10.1158/0008-5472.CAN-05-0917; PMID: 16103051
- Wang L, Lin HK, Hu YC, Xie S, Yang L, Chang C. Suppression of androgen receptor-mediated transactivation and cell growth by the glycogen synthase kinase 3 beta in prostate cells. J Biol Chem 2004; 279:32444 - 52; http://dx.doi.org/10.1074/jbc.M313963200; PMID: 15178691
- Dubrovska A, Kim S, Salamone RJ, Walker JR, Maira SM, García-Echeverría C, et al. The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem-like cell populations. Proc Natl Acad Sci U S A 2009; 106:268 - 73; http://dx.doi.org/10.1073/pnas.0810956106; PMID: 19116269
- Liao X, Zhang L, Thrasher JB, Du J, Li B. Glycogen synthase kinase-3beta suppression eliminates tumor necrosis factor-related apoptosis-inducing ligand resistance in prostate cancer. Mol Cancer Ther 2003; 2:1215 - 22; PMID: 14617795