Abstract
DNA damaging agents such as radiotherapy and gemcitabine are frequently used for the treatment of pancreatic cancer. However, these treatments typically provide only modest benefit. Improving the low survival rate for pancreatic cancer patients therefore remains a major challenge in oncology. Inhibition of the key DNA damage response kinase ATR has been suggested as an attractive approach for sensitization of tumor cells to DNA damaging agents, but specific ATR inhibitors have remained elusive. Here we investigated the sensitization potential of the first highly selective and potent ATR inhibitor, VE-821, in vitro. VE-821 inhibited radiation- and gemcitabine-induced phosphorylation of Chk1, confirming inhibition of ATR signaling. Consistently, VE-821 significantly enhanced the sensitivity of PSN-1, MiaPaCa-2 and primary PancM pancreatic cancer cells to radiation and gemcitabine under both normoxic and hypoxic conditions. ATR inhibition by VE-821 led to inhibition of radiation-induced G2/M arrest in cancer cells. Reduced cancer cell radiosurvival following treatment with VE-821 was also accompanied by increased DNA damage and inhibition of homologous recombination repair, as evidenced by persistence of γH2AX and 53BP1 foci and inhibition of Rad51 foci, respectively. These findings support ATR inhibition as a novel approach to improve the efficacy and therapeutic index of standard cancer treatments across a large proportion of pancreatic cancer patients.
Keywords: :
Introduction
Pancreatic cancer is the tenth most common site of new cancers, being responsible for 6% of all cancer-related deaths.Citation1 Approximately 55% of all patients have distant disease and 25% have regional spread at the time of diagnosis, with a 5-year overall survival (OS) of < 5%.Citation1 Surgery is the only curative treatment but even if the tumor is completely resected, patient outcome in early stage disease remains poor.Citation2 Additionally, neoadjuvant chemoradiation is an emerging concept in the management of patients that is expected to decrease the rate of local failures and prolong survival by enhancing resectability.Citation3
Several targeted therapeutics have been tested in phase II and III trials though results to date have been disappointing. Tipifarnib, an oral farnesyltransferase inhibitor that blocks RAS signaling, did not show significant improvement in OS when combined with gemcitabine despite the presence of KRAS mutations in 90% of pancreatic cancers.Citation4 Similarly, treatment with angiogenesis or matrix metalloproteinase inhibitors failed to prolong survival.Citation5-Citation8 Moreover, no clinical benefit was observed upon addition of the epidermal growth factor receptor (EGFR) inhibitor cetuximab to gemcitabine in patients with advanced pancreatic cancer.Citation9 Only a small benefit in median OS (6.24 vs. 5.91 mo) was found with the combination of gemcitabine with the EGFR inhibitor, erlotinib.Citation10 A possible reason of the limited success of these targeted therapies in pancreatic cancer could be that at the time of diagnosis, the tumor has become less dependent on oncogenic signaling than it was during the initial stages of carcinogenesis.Citation11 This lack of success of conventional and recent targeted therapies clearly shows the need for new strategies to improve pancreatic cancer treatment.
Radiotherapy and most forms of chemotherapy exert their cytotoxic effect by causing DNA damage.Citation12,Citation13 This damage leads to activation of the DNA-damage response, involving activation of cell cycle checkpoint and DNA repair. Two key kinases involved in DNA signaling are Ataxia-telangiectasia mutated (ATM) and ATM and Rad3-related (ATR), generally thought to be involved in recognizing double strand DNA breaks and single stranded DNA, respectively.
In tumor cells, oncogenic mutations inducing senescence and replication stress can give selective pressure for developing mutations in genes involved in DNA damage signaling or repair. As a result, tumor cells differ significantly from normal cells in their DNA damage response (DDR), lacking certain DNA repair pathways or having a deregulated cell cycle checkpoint signaling. In fact, defective DNA damage signaling through loss of ATM, or p53 mutation occurs in 70% of cases of pancreatic cancer.Citation2,Citation14-Citation16 Single nucleotide polymorphisms (SNPs) of ATM can occur in up to 95% of patients and have also been shown to correlate with adverse overall survival.Citation16
These differences in DNA repair signaling between normal and tumor cells can potentially be exploited to selectively increase the sensitivity of cancer cells to DNA damaging agents without harming normal cells.Citation17 It has been hypothesized that cells with disrupted ATM signaling may become more reliant on ATR.Citation18-Citation20 As p53 mutations abrogate efficient G1 checkpoint signaling, these cells depend on the ATR-activated G2/M checkpoint for cell cycle arrest in response to DNA damage.Citation21 For this reason, inhibition of ATR is expected to sensitize tumor cells to DNA damage but should not sensitize normal cell with wild type p53.Citation18 Furthermore, ATR inhibition is thought to be toxic to cells with high levels of replication stress, a frequent feature of tumor cells.Citation22 Despite the attractiveness of ATR as a target for cancer therapy, potent and selective ATR inhibitors have remained elusive. Recently we reported the characterization of the novel ATR inhibitor, VE-821, and showed that it sensitizes cancer cells but not normal cells to chemotoxic treatment.Citation19 In this paper, we show VE-821 can act as a sensitizer of radiotherapy and chemotherapy treatment of pancreatic cancer cells in both normoxic and hypoxic conditions.
Results
The ATR inhibitor VE-821 radiosensitizes pancreatic tumor cells
First we wanted to confirm that VE-821 inhibits ATR signaling in pancreatic cancer cells lines treated with radiation and/or gemcitabine, both of which are commonly used in pancreatic cancer treatment. We assessed phosphorylation of Chk1, a downstream target of ATR, by western blotting of PSN-1 and MiaPaCa-2 cells. In both cell lines, 1 µM VE-821 inhibited phosphorylation of Chk1 (Ser 345) after treatment with gemcitabine (100 nM), radiation (6 Gy) or both, at 2 h post-irradiation (). Importantly, we confirmed that VE-821 did not inhibit phosphorylation of ATM (Ser1981) or Chk2 (Thr68) under these conditions (Fig. S1A).
Figure 1. VE-821 radiosensitizes pancreatic tumor cells. (A) Western blot analysis of Chk1 inhibition. Cells were treated with 100 nM gemcitabine and 1 µM VE-821 1 h prior to irradiation at 6 Gy as indicated in the graphical representation. Drugs were left for the duration of the experiment and cells were lysed at 2 h post-irradiation and subjected to western blot analysis. (B) Effect of VE-821 on cell viability of pancreatic cancer cells with and without radiation treatment. PSN-1, PANC-1, and MiaPaCa-2 cells were treated with increasing concentrations of VE-821 for 72 h. Cells were irradiated at 4Gy 1h after VE-821 addition. Cell viability was measured after 10 d and shown as normalized to DMSO-treated cells. (C) Scheduling of VE-821 affects radiosensitivity. PSN-1 cells were plated as single cells, treated with 1 µM VE-821 at different time points in relation to 4 Gy irradiation and assessed for colony formation after 10 d as indicated in the graphical representation. The survival fraction at 4 Gy for each of the treatment schedules was determined by taking into account the relevant plating efficiency of unirradiated cells (see Fig. S1B). (D) Clonogenic survival of pancreatic cancer cells, MiaPaCa-2, PSN-1, PANC-1 and primary cancer cells PancM in response to irradiation and VE-821 treatment. Cells were treated according to the 72h VE-821 treatment regime described in (C) with 1 µM VE-821 added at 1 h prior to irradiation and removed 72 h post-irradiation and colony-forming ability being assessed after 10 to 21 d. N = 3; *p < 0.05; **p < 0.01 over DMSO-treated control.
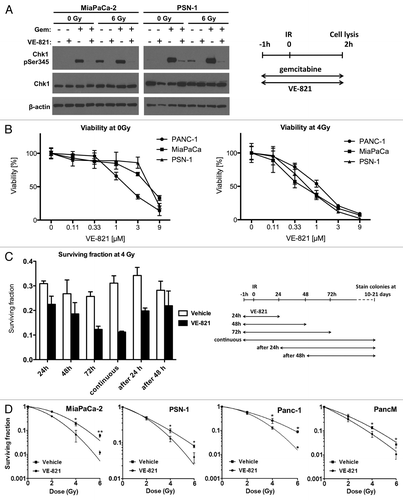
Next, we sought to establish suitable doses of VE-821 to use in radiosensitization experiments in pancreatic cancer cell lines. To this end, we performed a viability assay across a dose range of the VE-821 drug with and without radiation in a panel of pancreatic cancer cell lines, PSN-1, MiaPaCa-2 and PANC-1, all of which are mutant for p53. As VE-821 is effective in inhibiting cellular ATR activity within 1 h after drug addition,Citation19 a pre-incubation of 1 h prior to irradiation was chosen for these and subsequent experiments. Based on earlier data,Citation19 a dose range between 0.1 and 9 µM was chosen. As shown in , incubation with drug alone was associated with single agent toxicity above doses of 3 µM in all cell lines tested, whereas a radiosensitizing effect was observed from 1 µM. Based on these results, a concentration of 1 µM VE-821 was chosen for subsequent experiments.
Next, we assessed the impact of dose scheduling on the radiosensitizing effect in clonogenic assays. PSN-1 cells were treated with 1 µM VE-821 for various time periods in relation to irradiation. In the absence of irradiation, an incubation time of up to 72 h with drug alone did not decrease plating efficiency significantly, whereas continuous exposure to the drug was associated with increased single agent toxicity (Fig. S1B). When cells were irradiated in the presence of VE-821, a decrease in surviving fraction was observed and this radiosensitizing effect increased as the drug incubation time after irradiation was extended (). Interestingly, radiosensitization was still observed when cells were irradiated in the absence of VE-821 followed by drug addition at 24 h after radiation, suggesting that VE-821 also acts in the later stages of repair process (). However, the radiosensitizing effect was lost when drug was added 48 h after radiation. As a pre-incubation of 1 h and a total incubation period of 72 h showed maximum radiosensitization combined with limited single agent toxicity, this incubation schedule was used in subsequent clonogenic experiments. We confirmed that VE-821 caused radiosensitization in the pancreatic lines PSN-1, MiaPaCa-2 and PANC-1 (). Importantly, this effect was also seen in primary pancreatic tumor cells (PancM; ).
VE-821 radiosensitizes tumor cells under hypoxic conditions
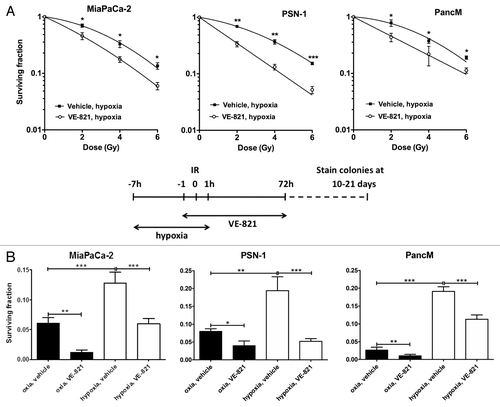
Hypoxia is a common feature of pancreatic cancerCitation23 and as hypoxic cells can be up to 3-fold more radioresistant, we sought to investigate whether the radiosensitizing effect of VE-821 can still be seen under hypoxic conditions. Using the same dosing schedule as was used under normoxia, we treated PSN-1, MiaPaCa-2 and PancM with 1 µM VE-821 and irradiation under hypoxic conditions (0.5% O2). To avoid any hypoxia-associated toxicity, cells were treated in hypoxic conditions for a minimum time period (from 6 h prior to 1 h post irradiation). Treatment with VE-821 under these conditions did not result in increased toxicity in the absence of irradiation (data not shown). All cell lines showed increased radioresistance under hypoxic conditions, confirming the hypoxic effect in our experimental settings. Addition of VE-821 reduced survival significantly for all three cell lines in hypoxia (). These results confirm that VE-821 can radiosensitize tumor cells in hypoxic as well as normoxic conditions.
Figure 2. VE-821 radiosensitizes pancreatic tumor cells under hypoxic conditions. (A) Clonogenic survival curves of cells treated with 1 µM VE-821 and irradiation under hypoxic conditions as indicated in the graphical representation. Plated cells were transferred to hypoxia (0.5% O2) and acclimatized for 6 h. VE-821 (1 µM) was then added at 1h prior to irradiation and left for 72 h upon which the medium was replaced. Cells were transferred to normoxia at 1 h post-irradiation. (B) Bar graph representation of clonogenic survival of cells after irradiation with 6 Gy and treatment with 1 µM VE-821 in oxic and hypoxic (0.5% O2) conditions, as shown above and in . N = 3; *p < 0.05; **p < 0.01; ***p < 0.001 over DMSO-treated control.
VE-821 sensitizes normoxic and hypoxic cells to gemcitabine
The nucleoside analog gemcitabine is the standard chemotherapeutic agent in pancreatic cancer. We tested whether VE-821 would increase the cytotoxic effects of gemcitabine. As shown in , this was indeed the case, with the strongest sensitization being observed in the PSN-1 cells.
Figure 3. VE-821 sensitizes pancreatic cancer cells to gemcitabine treatment. (A) Clonogenic survival of cells treated with gemcitabine and 1 µM VE-821. Cells were treated with increasing concentrations of gemcitabine for 24 h followed by 72 h treatment of 1 µM VE-821. Colony forming ability was assessed after 10 to 21 d (see graphical representation in panel E). (B) Clonogenic survival of cells treated with gemcitabine in hypoxia. Plated cells were transferred to hypoxia (0.5% O2) and acclimatized for 6 h. Cells were then treated with increasing concentrations of gemcitabine for 24 h followed by 72 h treatment of 1 µM VE-821. Hypoxic cells were transferred to normoxia 1 h after VE-821 addition. (C) Bar graph representation of clonogenic survival after treatment with 20 nM gemcitabine and VE-821 in oxic and hypoxic (0.5% O2) conditions, shown in (A and B). (D) Clonogenic survival of cells treated with gemcitabine and irradiation. PSN-1 and MiaPaCa-2 cells were treated with 5 nM or 10 nM gemcitabine, respectively, for 24 h, medium was then replaced and 1 µM VE-821 was added from 1 h prior to 72 h post 4 Gy irradiation. Colony forming ability was assessed after 10 to 21 d. (n = 3). *, p < 0.05; **, p < 0.01; ***, p < 0.001 over DMSO-treated control. (E) Graphical representation for the treatment regimes used in panels [A, B, (C) and D].
![Figure 3. VE-821 sensitizes pancreatic cancer cells to gemcitabine treatment. (A) Clonogenic survival of cells treated with gemcitabine and 1 µM VE-821. Cells were treated with increasing concentrations of gemcitabine for 24 h followed by 72 h treatment of 1 µM VE-821. Colony forming ability was assessed after 10 to 21 d (see graphical representation in panel E). (B) Clonogenic survival of cells treated with gemcitabine in hypoxia. Plated cells were transferred to hypoxia (0.5% O2) and acclimatized for 6 h. Cells were then treated with increasing concentrations of gemcitabine for 24 h followed by 72 h treatment of 1 µM VE-821. Hypoxic cells were transferred to normoxia 1 h after VE-821 addition. (C) Bar graph representation of clonogenic survival after treatment with 20 nM gemcitabine and VE-821 in oxic and hypoxic (0.5% O2) conditions, shown in (A and B). (D) Clonogenic survival of cells treated with gemcitabine and irradiation. PSN-1 and MiaPaCa-2 cells were treated with 5 nM or 10 nM gemcitabine, respectively, for 24 h, medium was then replaced and 1 µM VE-821 was added from 1 h prior to 72 h post 4 Gy irradiation. Colony forming ability was assessed after 10 to 21 d. (n = 3). *, p < 0.05; **, p < 0.01; ***, p < 0.001 over DMSO-treated control. (E) Graphical representation for the treatment regimes used in panels [A, B, (C) and D].](/cms/asset/3a353264-6b8c-4ca3-81a1-8ba14cb5474f/kcbt_a_10921093_f0003.gif)
Because hypoxia markedly reduces the efficacy of chemotherapy drugs in pancreatic cancer,Citation24,Citation25 we tested the effect of combining gemcitabine and VE-821 under hypoxic conditions (0.5% O2). In line with previous findings, we observed an increase in resistance to gemcitabine treatment in hypoxia, which was reduced when cells were co-treated with VE-821. We established that this sensitizing effect could be seen across a range of gemcitabine doses ().
Finally, as patients often receive gemcitabine combined with radiotherapy for treatment of pancreatic cancer,Citation3 we wanted to establish that VE-821 could still increase tumor kill in a chemoradiotherapeutic setting. As shown in , VE-821 potentiates the effect of chemoradiation in both PSN-1 and MiaPaCa-2 cells.
VE-821 disrupts damage-induced cell cycle checkpoints
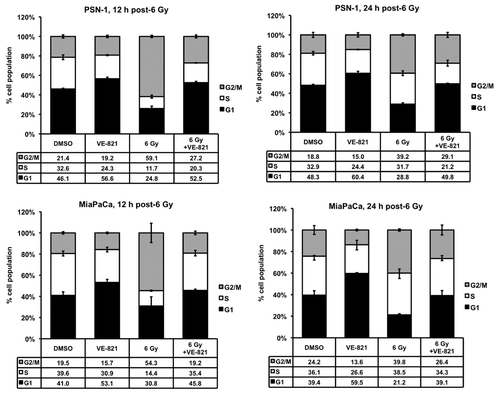
To establish whether VE-821 modifies the cell cycle checkpoint that is normally activated after DNA damage, PSN-1 and MiaPaCa-2 cells were pre-treated with VE-821 at 1 h prior to radiation and their cell cycle profile was subsequently analyzed at 12 h and 24 h after radiation. In the absence of irradiation, VE-821 caused a decrease in the proportion of cells in the S-phase ( and Fig. S2A), similarly to what has been shown before.Citation19 As expected, radiation caused a significant increase of the proportion of cells in the G2/M phase, indicative of the cell cycle checkpoint activation ( and Fig. S2A). However, in VE-821-treated cells, this increase in G2/M phase was much reduced and delayed, indicating inhibition of the DNA-damage induced checkpoint. Similarly to normoxia, addition of VE-821 in hypoxic conditions abrogated the G2/M checkpoint post-irradiation (Fig. S2B).
Figure 4. VE-821 perturbs the irradiation-induced cell cycle checkpoint in pancreatic cancer cells. VE-821 (1 µM) was added 1 h prior to 6 Gy irradiation and left for the duration of the experiment. Cells were lifted and fixed at 12 h or 24 h after irradiation, stained with propidium iodide and analyzed for cell cycle distribution by flow cytometry. Bar graphs represent mean +/− SEM (n = 6). Individual DNA histograms of representative samples are shown in Figure S2A.
VE-821 inhibits repair of DNA damage by homologous recombination
Diminished DNA repair efficiency can be demonstrated by delayed resolution of post-irradiation γH2AX and 53BP1 foci, which accumulate at sites of DNA breaks. To confirm that the ATR inhibitor caused increased DNA damage foci, MiaPaCa-2 and PSN-1 cells were irradiated at 6 Gy and fixed and stained for γH2AX and 53BP1 foci at 24 h after irradiation (). In accordance with the increased radiosensitivity shown earlier, cells pretreated with VE-821 had a higher number of γH2AX and 53BP1 foci. No significant increase in foci number was seen in cells treated with VE-821 in the absence of radiation (data not shown). Interestingly, addition of VE-821 several hours after the radiation dose still led to an increased number of residual DNA damage foci. These results mirror the data obtained with the clonogenic survival experiments shown in , which showed that the drug does not need to be present during irradiation but rather during subsequent stages of DNA repair.
Figure 5. VE-821 increases 53BP1 and γH2AX foci number and reduces Rad51 foci formation. Cells were treated with 1 µM VE-821 at various time points in relation to 6 Gy irradiation, as indicated in the legends and the graphical representation, and fixed at 24 h post-irradiation. Subsequently, cells were stained for (A) γH2AX and (B) 53BP1 foci and the percentage of cells with more than 7 and 5 foci per cell was quantitated, respectively. (C) For analyzing Rad51 foci formation, cells were fixed at 6 h post-irradiation as indicated in the graphical representation and the percentage of cells with more than 9 foci per cell was quantitated. Representative images are shown on the right. N = 4; *p < 0.05 over DMSO treated controls.
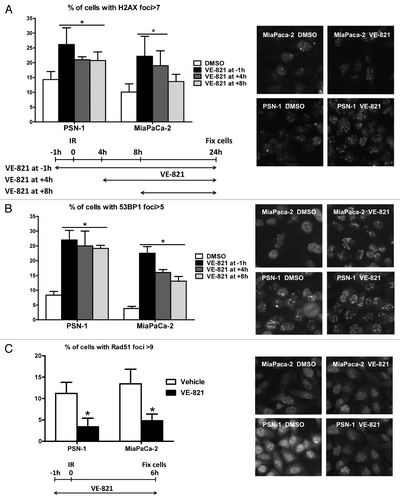
To further investigate the radiosensitizing mechanisms of the ATR inhibitor, we also looked at Rad51 foci, which accumulate at sites of DNA damage being repaired by homologous recombination (HRR).Citation17 Cells treated with VE-821 prior to irradiation showed a decrease in Rad51 (), suggesting that VE-821 inhibits HRR.
Discussion
Pancreatic cancer is the most lethal solid tumor with a five-year survival rate of less than 5%.Citation1 Therapeutic options for this disease are highly restricted, particularly for non-resectable diseases. Chemoradiotherapy achieves higher local control rates than chemotherapy and leads to better progression-free survival.Citation26 Thus chemoradiotherapy is under intensive investigation in randomized clinical trials (LAP07, SCALOP). However, chemoradiotherapy is currently reliant on cytotoxic drugs like gemcitabine that have considerable effects on normal tissue (reviewed in ref. 3). This is why more tumor selective radiosensitizing strategies are urgently needed and combinations of radiotherapy with molecular targeted agents are investigated.
One mechanism of resistance to therapy in pancreatic cancer is proficient DNA repair. However it is clear that cancer cells carry multiple mutations in their DNA repair machinery, which likely places increased burden on the remaining pathways. Importantly, 50 to 75% of pancreatic cancers harbor mutations in p53,Citation2,Citation14 which is expected to produce an increased sensitivity to ATR-Chk1 pathway inhibition. In addition it appears that a number of common oncogenes act to increase replicative stress which could exert a further burden on remaining DDR pathways. Most notably, pancreatic cancer is KRAS mutated in more than 90% of the cases.Citation2,Citation27 Previous work has shown that complete ATR inhibition in combination with oncogenic RAS expression synergistically killed tumor cells and was synthetically lethal.Citation28 Furthermore, pancreatic tumors have been also been shown to overexpress Rad51, leading to the prediction that they might rely more heavily on HRR and thus be more sensitive to ATR-Chk1 inhibition than normal cells.Citation29 Consequently, it is possible that both p53 defective and p53 wild-type tumors may be sensitized through ATR inhibition. Last but not least, pancreatic cancer has been shown to be highly hypoxic,Citation23 with emerging evidence suggesting a critical role for an ATR-driven DDR in survival from hypoxia and hypoxia-associated radioresistance.Citation12 Taken together, these findings underscore the potential of ATR inhibition in pancreatic cancer therapy.
In this report we describe the effects of using the recently described ATR inhibitor, VE-821, in conjunction with radiotherapy in pancreatic cancer cells. We showed that ATR inhibition by VE-821 sensitizes pancreatic tumor cells to radiotherapy. We also showed that VE-821 synergizes with gemcitabine, the major chemotherapeutic agent in pancreatic cancer with and without radiotherapy. Both effects were not only observed in normal oxygen levels but also under hypoxic conditions that are frequently encountered in pancreatic cancer. Radiosensitization by VE-821 was associated with disruption of damage-induced cell cycle checkpoints. This resulted in increased persistence of DNA damage, evidenced by persistent γH2AX and 53BP1 foci.
The ATR/Chk1 pathway is responsible for reducing replication stress, as generated by oncogenic signaling.Citation30 Recent studies have shown that inhibition of ATR results in increased killing of oncogene-driven (K-ras, H-ras and c-Myc) tumors, through exacerbation of replication stress.Citation31-Citation33 The growth of malignancies harboring oncogenic mutations could be inhibited by genetic suppression of ATR with minimal impact on normal tissue homeostasis.Citation32
In line with our observations, clonogenic survival of pancreatic cancer cells is reduced with the Chk1/2 inhibitor AZD7762 alone and more dramatically in conjunction with gemcitabine.Citation34 Furthermore, another ATR inhibitor, NU6027, was shown to cause chemo- and radiosensitization in breast and ovarian cancer cell lines, confirming our results with ATR inhibition by VE-821.Citation35 As the sensitization of cancer cells to chemotherapy and radiotherapy by ATR inhibition is very potent and in some cases much greater than that seen with Chk1 inhibition (reviewed in refs. Citation22 and Citation36), the discovery of a selective ATR inhibitor represents a very exciting development. Taken together, our data strongly support development and evaluation of ATR inhibitors that can be used in vivo in the preclinical and clinical context in combination with gemcitabine-radiation in patients with locally advanced pancreatic cancer.
Conclusion
In the present study we demonstrate the potential for VE-821 to sensitize normoxic and hypoxic pancreatic malignant cells to radiation and chemotherapy. These findings support the testing of highly specific ATR inhibitors in pancreatic cancer models in vivo as a conduit to Phase I clinical trials.
Methods
Drugs, cell lines and antibodies
The ATR inhibitor VE-821 was obtained from Vertex Pharmaceuticals under a material transfer agreement. The drug was dissolved in DMSO in 10 mM aliquots and stored at -80°C. Gemcitabine (Eli Lilly) was dissolved in DMSO and stored in 100 mM aliquots. Pancreatic tumor cell lines, PSN-1, MiaPaCa-2 and PANC-1 were obtained from ATCC. Cells were cultured in DMEM containing 4.5 g/L glucose (Lonza) supplemented with 10% fetal bovine serum (HyClone). The primary pancreatic cancer cell line PancM (passage 17) and the culture conditions are described in detail elsewhere.Citation37 HEPES (1 mM; Sigma) was added to cells exposed to hypoxia to account for pH and glucose alterations. Mouse anti-human γH2AX (Ser 139) was from Millipore (clone JBW301), Rabbit anti-human Rad51 from Santa Cruz (sc-8349), Rabbit anti-human 53BP1, Mouse anti-human Chk1 and rabbit anti-human Chk1 phoshoSer345 antibodies and rabbit anti-human Chk2 phosphoThr68 were from New England Biolabs, Mouse anti-human β-actin (clone AC-15) rabbit anti-human ATM were from Sigma. Phospho ATM (Ser1981) antibody was from Epitomics, and total Chk2 from Millipore. HRP conjugated secondary antibodies were from ThermoFisher. Alexafluor 488 and Alexafluor 568 conjugated secondary antibodies were from Invitrogen.
Irradiation
All in vitro irradiation experiments were performed using an IBL634 cesium irradiator at a dose rate of 0.89 Gy/min at room temperature.
Cell viability assays
MiaPaCa-2, PSN-1 and Panc1 (5 x 104) were plated in 96-well plates and after 4 h treated with increasing concentrations of VE-821 at 1 h before irradiation with a single dose of 4 Gy. Medium was replaced 72 h post-irradiation at which point viability was measured using the using the Alamar Blue assay (Resazurin substrate, SIGMA). Cells were allowed to proliferate and cell viability was again analyzed at day 10 for the different treatment conditions. Cell viability and surviving fraction were normalized to the untreated (control) group.
Clonogenic assay
Clonogenic survival assays were performed as described before.Citation38 Briefly, logarithmically growing cells were plated in triplicate in 6-well tissue culture dishes under oxic (21% O2) or hypoxic conditions (0.5% O2) using an InVivo2 300 chamber (Ruskinn Technology, UK). Cells were incubated for 6 h before irradiation under oxia or hypoxia using tightly sealed chambers. The target O2 level was achieved within 6 h of gassing and maintained during irradiation, as confirmed by an OxyLite oxygen probe (Oxford Optronix). Cells irradiated under hypoxia were exposed to normoxia at 1 h post-irradiation. As standard, VE-821 (1 µM) was added 1 h prior to irradiation (6 Gy) and was washed away 72 h after irradiation. For the chemotherapy experiments, cells were initially exposed to increasing concentrations of gemcitabine (5, 10 and 20 nM) for 24 h before addition of the VE-821 (1 µM) for another 72 h. The effect of triple combination of irradiation with VE-821 and gemcitabine was examined as well. Cells were incubated for 10–21 d until colonies were stained with 0.5% crystal violet and counted in a CellCount automated colony counter (Oxford Optronix). Clonogenic survival was calculated and data were fitted in GraphPad Prism 4.0 (GraphPad Software, CA).
Western blot
MiaPaCa-2 and PSN-1 cells were exposed to 100 nM gemcitabine and/or 1 µM VE-821 drug 1 h prior to irradiation with a single dose of 6 Gy. Cells were lysed in RIPA buffer 2 h post-irradiation and subjected to SDS-PAGE electrophoresis and immunoblotting. Chemoluminescence (SuperSignal, Millipore) and film exposure was used to detect antibody binding. Exposed film was digitized and figures were assembled using Microsoft PowerPoint.
Nuclear foci analysis
Cells growing in 96-well plates were treated with 1 µM VE-821 drug 1 h prior to 6 Gy irradiation and fixed in 3% formaldehyde at multiple time points. Cells were subsequently permeabilized and blocked in PBS with 0.1% Triton 1% BSA (w/v). Cells were incubated with primary antibody overnight at 4°C and after a PBS wash incubated with fluorescently labeled secondary antibody and nuclear staining with DAPI followed by a PBS wash. Images were acquired and foci quantitated using the In Cell Analyzer 1000 automated epifluorescence microscope and analysis software (GE Healthcare).
Cell cycle analysis
Cells growing in 6-well dishes were treated with 1 µM VE-821 drug 1 h prior to 6 Gy irradiation. Cells were incubated for 6 h before irradiation under oxia (21% O2) or hypoxia (0.5% O2) using tightly sealed chambers. At multiple time points, cells were lifted in trypsin and fixed in 70% ethanol and stored at 4°C. Cells were incubated with propidium iodide (50 μg/ml in PBS containing 200 μg/ml RNase) for 1 h at room temperature and analyzed by flow cytometry (FACSort, Becton Dickinson). Cell cycle phase was quantitated using ModFit Cell Cycle Analysis software.
Statistical analyses
The values were expressed as means ± SD. The significance of differences between the means was measured by two-tailed t-test using the GraphPad Prism program version 4.0 (GraphPad Software). A value p < 0.05 was considered statistically significant
Additional material
Download Zip (1.7 MB)Acknowledgments
This work was supported by grants from Cancer Research UK, the Medical Research Council and by Vertex Pharmaceuticals.
Conflict of Interest
This study was partly funded by a grant from Vertex Pharmaceuticals. The authors based at Vertex Pharmaceuticals are full time employees of Vertex Pharmaceuticals (Europe), Ltd. and hold equity in Vertex Pharmaceuticals, Inc.
Notes
† Co-first author
Related Research Data
References
- Raimondi S, Maisonneuve P, Lowenfels AB. Epidemiology of pancreatic cancer: an overview. Nat Rev Gastroenterol Hepatol 2009; 6:699 - 708; http://dx.doi.org/10.1038/nrgastro.2009.177; PMID: 19806144
- Hidalgo M. Pancreatic cancer. N Engl J Med 2010; 362:1605 - 17; http://dx.doi.org/10.1056/NEJMra0901557; PMID: 20427809
- Brunner TB, Scott-Brown M. The role of radiotherapy in multimodal treatment of pancreatic carcinoma. Radiat Oncol 2010; 5:64; http://dx.doi.org/10.1186/1748-717X-5-64; PMID: 20615227
- Van Cutsem E, van de Velde H, Karasek P, Oettle H, Vervenne WL, Szawlowski A, et al. Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J Clin Oncol 2004; 22:1430 - 8; http://dx.doi.org/10.1200/JCO.2004.10.112; PMID: 15084616
- Bramhall SR, Rosemurgy A, Brown PD, Bowry C, Buckels JA, Marimastat Pancreatic Cancer Study Group. Marimastat as first-line therapy for patients with unresectable pancreatic cancer: a randomized trial. J Clin Oncol 2001; 19:3447 - 55; PMID: 11481349
- Kindler HL, Friberg G, Singh DA, Locker G, Nattam S, Kozloff M, et al. Phase II trial of bevacizumab plus gemcitabine in patients with advanced pancreatic cancer. J Clin Oncol 2005; 23:8033 - 40; http://dx.doi.org/10.1200/JCO.2005.01.9661; PMID: 16258101
- Kindler HL, Ioka T, Richel DJ, Bennouna J, Létourneau R, Okusaka T, et al. Axitinib plus gemcitabine versus placebo plus gemcitabine in patients with advanced pancreatic adenocarcinoma: a double-blind randomised phase 3 study. Lancet Oncol 2011; 12:256 - 62; http://dx.doi.org/10.1016/S1470-2045(11)70004-3; PMID: 21306953
- Moore MJ, Hamm J, Dancey J, Eisenberg PD, Dagenais M, Fields A, et al, National Cancer Institute of Canada Clinical Trials Group. Comparison of gemcitabine versus the matrix metalloproteinase inhibitor BAY 12-9566 in patients with advanced or metastatic adenocarcinoma of the pancreas: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 2003; 21:3296 - 302; http://dx.doi.org/10.1200/JCO.2003.02.098; PMID: 12947065
- Philip PA, Benedetti J, Corless CL, Wong R, O’Reilly EM, Flynn PJ, et al. Phase III study comparing gemcitabine plus cetuximab versus gemcitabine in patients with advanced pancreatic adenocarcinoma: Southwest Oncology Group-directed intergroup trial S0205. J Clin Oncol 2010; 28:3605 - 10; http://dx.doi.org/10.1200/JCO.2009.25.7550; PMID: 20606093
- Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, et al, National Cancer Institute of Canada Clinical Trials Group. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 2007; 25:1960 - 6; http://dx.doi.org/10.1200/JCO.2006.07.9525; PMID: 17452677
- Stathis A, Moore MJ. Advanced pancreatic carcinoma: current treatment and future challenges. Nat Rev Clin Oncol 2010; 7:163 - 72; http://dx.doi.org/10.1038/nrclinonc.2009.236; PMID: 20101258
- Begg AC, Stewart FA, Vens C. Strategies to improve radiotherapy with targeted drugs. Nat Rev Cancer 2011; 11:239 - 53; http://dx.doi.org/10.1038/nrc3007; PMID: 21430696
- Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer 2008; 8:193 - 204; http://dx.doi.org/10.1038/nrc2342; PMID: 18256616
- Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008; 321:1801 - 6; http://dx.doi.org/10.1126/science.1164368; PMID: 18772397
- Li D, Frazier M, Evans DB, Hess KR, Crane CH, Jiao L, et al. Single nucleotide polymorphisms of RecQ1, RAD54L, and ATM genes are associated with reduced survival of pancreatic cancer. J Clin Oncol 2006; 24:1720 - 8; http://dx.doi.org/10.1200/JCO.2005.04.4206; PMID: 16520463
- Okazaki T, Jiao L, Chang P, Evans DB, Abbruzzese JL, Li D. Single-nucleotide polymorphisms of DNA damage response genes are associated with overall survival in patients with pancreatic cancer. Clin Cancer Res 2008; 14:2042 - 8; http://dx.doi.org/10.1158/1078-0432.CCR-07-1520; PMID: 18381943
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature 2009; 461:1071 - 8; http://dx.doi.org/10.1038/nature08467; PMID: 19847258
- López-Contreras AJ, Fernandez-Capetillo O. The ATR barrier to replication-born DNA damage. DNA Repair (Amst) 2010; 9:1249 - 55; http://dx.doi.org/10.1016/j.dnarep.2010.09.012; PMID: 21036674
- Reaper PM, Griffiths MR, Long JM, Charrier JD, Maccormick S, Charlton PA, et al. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat Chem Biol 2011; 7:428 - 30; http://dx.doi.org/10.1038/nchembio.573; PMID: 21490603
- Ruzankina Y, Schoppy DW, Asare A, Clark CE, Vonderheide RH, Brown EJ. Tissue regenerative delays and synthetic lethality in adult mice after combined deletion of Atr and Trp53. Nat Genet 2009; 41:1144 - 9; http://dx.doi.org/10.1038/ng.441; PMID: 19718024
- Tao Y, Leteur C, Yang C, Zhang P, Castedo M, Pierré A, et al. Radiosensitization by Chir-124, a selective CHK1 inhibitor: effects of p53 and cell cycle checkpoints. Cell Cycle 2009; 8:1196 - 205; http://dx.doi.org/10.4161/cc.8.8.8203; PMID: 19305158
- Toledo LI, Murga M, Fernandez-Capetillo O. Targeting ATR and Chk1 kinases for cancer treatment: a new model for new (and old) drugs. Mol Oncol 2011; 5:368 - 73; http://dx.doi.org/10.1016/j.molonc.2011.07.002; PMID: 21820372
- Koong AC, Mehta VK, Le QT, Fisher GA, Terris DJ, Brown JM, et al. Pancreatic tumors show high levels of hypoxia. Int J Radiat Oncol Biol Phys 2000; 48:919 - 22; http://dx.doi.org/10.1016/S0360-3016(00)00803-8; PMID: 11072146
- Onozuka H, Tsuchihara K, Esumi H. Hypoglycemic/hypoxic condition in vitro mimicking the tumor microenvironment markedly reduced the efficacy of anticancer drugs. Cancer Sci 2011; 102:975 - 82; http://dx.doi.org/10.1111/j.1349-7006.2011.01880.x; PMID: 21255190
- Yokoi K, Fidler IJ. Hypoxia increases resistance of human pancreatic cancer cells to apoptosis induced by gemcitabine. Clin Cancer Res 2004; 10:2299 - 306; http://dx.doi.org/10.1158/1078-0432.CCR-03-0488; PMID: 15073105
- Huguet F, André T, Hammel P, Artru P, Balosso J, Selle F, et al. Impact of chemoradiotherapy after disease control with chemotherapy in locally advanced pancreatic adenocarcinoma in GERCOR phase II and III studies. J Clin Oncol 2007; 25:326 - 31; http://dx.doi.org/10.1200/JCO.2006.07.5663; PMID: 17235048
- Jimeno A, Hidalgo M. Molecular biomarkers: their increasing role in the diagnosis, characterization, and therapy guidance in pancreatic cancer. Mol Cancer Ther 2006; 5:787 - 96; http://dx.doi.org/10.1158/1535-7163.MCT-06-0005; PMID: 16648548
- Gilad O, Nabet BY, Ragland RL, Schoppy DW, Smith KD, Durham AC, et al. Combining ATR suppression with oncogenic Ras synergistically increases genomic instability, causing synthetic lethality or tumorigenesis in a dosage-dependent manner. Cancer Res 2010; 70:9693 - 702; http://dx.doi.org/10.1158/0008-5472.CAN-10-2286; PMID: 21098704
- Han H, Bearss DJ, Browne LW, Calaluce R, Nagle RB, Von Hoff DD. Identification of differentially expressed genes in pancreatic cancer cells using cDNA microarray. Cancer Res 2002; 62:2890 - 6; PMID: 12019169
- López-Contreras AJ, Fernandez-Capetillo O. The ATR barrier to replication-born DNA damage. DNA Repair (Amst) 2010; 9:1249 - 55; http://dx.doi.org/10.1016/j.dnarep.2010.09.012; PMID: 21036674
- Murga M, Campaner S, Lopez-Contreras AJ, Toledo LI, Soria R, Montaña MF, et al. Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat Struct Mol Biol 2011; 18:1331 - 5; http://dx.doi.org/10.1038/nsmb.2189; PMID: 22120667
- Schoppy DW, Ragland RL, Gilad O, Shastri N, Peters AA, Murga M, et al. Oncogenic stress sensitizes murine cancers to hypomorphic suppression of ATR. J Clin Invest 2012; 122:241 - 52; http://dx.doi.org/10.1172/JCI58928; PMID: 22133876
- Toledo LI, Murga M, Zur R, Soria R, Rodriguez A, Martinez S, et al. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat Struct Mol Biol 2011; 18:721 - 7; http://dx.doi.org/10.1038/nsmb.2076; PMID: 21552262
- Morgan MA, Parsels LA, Zhao L, Parsels JD, Davis MA, Hassan MC, et al. Mechanism of radiosensitization by the Chk1/2 inhibitor AZD7762 involves abrogation of the G2 checkpoint and inhibition of homologous recombinational DNA repair. Cancer Res 2010; 70:4972 - 81; http://dx.doi.org/10.1158/0008-5472.CAN-09-3573; PMID: 20501833
- Peasland A, Wang LZ, Rowling E, Kyle S, Chen T, Hopkins A, et al. Identification and evaluation of a potent novel ATR inhibitor, NU6027, in breast and ovarian cancer cell lines. Br J Cancer 2011; 105:372 - 81; http://dx.doi.org/10.1038/bjc.2011.243; PMID: 21730979
- Wagner JM, Kaufmann SH. Prospects for the Use of ATR Inhibitors to Treat Cancer. Pharmaceuticals (Ott) 2010; 3:1311 - 34; http://dx.doi.org/10.3390/ph3051311
- Martin NE, Brunner TB, Kiel KD, DeLaney TF, Regine WF, Mohiuddin M, et al. A phase I trial of the dual farnesyltransferase and geranylgeranyltransferase inhibitor L-778,123 and radiotherapy for locally advanced pancreatic cancer. Clin Cancer Res 2004; 10:5447 - 54; http://dx.doi.org/10.1158/1078-0432.CCR-04-0248; PMID: 15328183
- Prevo R, Deutsch E, Sampson O, Diplexcito J, Cengel K, Harper J, et al. Class I PI3 kinase inhibition by the pyridinylfuranopyrimidine inhibitor PI-103 enhances tumor radiosensitivity. Cancer Res 2008; 68:5915 - 23; http://dx.doi.org/10.1158/0008-5472.CAN-08-0757; PMID: 18632646