Abstract
In the search for new strategies to efficiently fight colorectal cancer, efforts are being increasingly focused on targeting regulatory signaling pathways involved in cancer-specific features. As a result, several studies have recently addressed the therapeutic potential of molecularly-targeted drugs capable of inhibiting the activity of protein kinases involved in relevant signaling cascades. Here we show that simultaneous inhibition of the DFG-in and DFG-out conformations of p38α by means of type-I and type-II inhibitors is beneficial to impair more efficiently its kinase activity. Moreover, we found that SB202190 (type-I) and sorafenib (type-II) synergize at the molecular and biological level, as co-treatment with these compounds enhances tumor growth inhibition and induction of apoptosis both in colorectal cancer cell lines and animal models. These results support the need to reconsider sorafenib as a therapeutic agent against colorectal cancer and provide new insights that underline the importance to elucidate the activity of protein kinase inhibitors for the treatment of colorectal carcinoma.
Introduction
Colorectal cancer (CRC) is the second cause of death from cancer in the western world. 35% of affected patients have metastatic (stage IV) CRC at presentation and 20–50% of patients with lower stages will progress to stage IV, whose current five-year survival rate remains approximately 10%.Citation1 Various molecularly-targeted drugs moved from bench to bedside in CRC treatment, and classical chemotherapy (FOLFIRI or FOLFOX) is currently combined with monoclonal antibodies against VEGF (bevacizumab) or EGFR (cetuximab and panitumumab).Citation2 Because of the limited efficacy of these combination regimens and in the search for new strategies to efficiently fight CRC, great interest has been recently aroused in the possibility to specifically target regulatory signaling pathways involved in cancer-specific features, such as uncontrolled proliferation, survival under nutrient and oxygen shortage conditions, aerobic glycolysis, neoangiogenesis, invasion and metastasis. As a result, several studies have focused on the modulation of the activity of kinases involved in relevant cascades.Citation3 However, difficulties in achieving appropriate selectivity profiles increased the interest in the design of new inhibitors capable of targeting specific conformations and/or allosteric sites. In particular, one of the emerging strategies in the last years has been to conceive molecules that can target the so-called “DFG-out” kinase conformation (i.e., type II inhibitors), which refers to an inactive state of the protein in which the Asp-Phe-Gly (DFG) motif is deviated from its active conformation.
We recently showed that the p38α kinase, despite having a tumor suppressive role in some cell types, is overactive in a large number of CRC cell lines, where it is the main p38 isoform.Citation4 Moreover, we found high levels of phospho-activated p38 in a subset of high-grade human CRC specimens.Citation5 In CRC cells, p38α sustains the expression of HIF1α target genes encoding for glycolytic rate-limiting enzymes, and its blockade by type-I inhibitors (i.e., compounds that target the DFG-in conformation) causes a drastic decrease in ATP intracellular levels. Pharmacologic or genetic inactivation of p38α triggers AMPK-mediated activation of FoxO-dependent transcription, leading to autophagy, cell cycle arrest and non-apoptotic cell death in vitro, in xenografted nude mice and in azoxymetane (AOM)-treated APCMin mice.Citation4,Citation6-Citation8 Furthermore, p38α inhibition by type-I or type-II compounds induces increased expression of the RTK receptor HER3 in a FoxO3A- and Sirt1-dependent manner, thus leading to MEK/ERK overactivation in CRC cells and tissues.Citation5 Of note, inhibition of MEK1 triggers the phospho-activation of p38, indicating the existence of a p38α/ERK crosstalk in CRC cells, which is independent from the mutational status of KRAS and BRAF. Indeed, these MEK/ERK upstream kinases have been found overactive in 50% of CRC patients,Citation9 and BRAF and MEK1 inhibitors exert anticancer effects in cell lines and xenografted tumors.Citation10,Citation11 However, the MEK1 inhibitor CI-1040 showed low antitumor activity against CRC in phase II clinical trials.Citation12 Combined inhibition of p38α and MEK by type-I ligands specifically fosters cell death by inducing apoptosis to a higher extent than either single treatment in CRC cell lines, in CRC xenografted nude mice and in the AOM/DSS colitis-associated carcinoma mouse preclinical model.Citation5 At the molecular level, inhibition of p38α causes transcriptional upregulation of TRAIL and activation of caspase-8, while concomitant MEK inhibition triggers Bax-dependent apoptosis by enabling signaling propagation through t-Bid and caspase-3.Citation5 These data, which have been validated in preclinical models, emphasize the therapeutic potential of combined inhibition of p38α and MEK and suggest that the cross-talk between p38α and ERK may play an important, reciprocal and compensatory role in CRC progression and cell survival. Thus, a single agent targeting both pathways could represent a useful tool in CRC therapy.
Sorafenib is reported to potently inhibit nine protein kinases, including BRAF. It has also been shown to target the DFG-out conformational state of p38α and to affect p38α kinase activity in vitro.Citation13,Citation14 Sorafenib induces apoptosis by downregulating Mcl-1 in several cell lines and xenografted tumors, including HT-29 and HCT-116 CRC cells.Citation13,Citation15,Citation16 It has been approved for clinical use by the FDA in hepatocellular carcinoma (HCC) and renal cell carcinoma (RCC), but failed to prove efficacy in CRC clinical trials,Citation13,Citation17 most probably because of its inability to completely impair p38α activity.
Based on these data, we tested the association of sorafenib, a type-II kinase inhibitor, with SB202190, a type-I inhibitor, in CRC cell lines and animal models in order to investigate whether combination of these compounds may entail enhanced effects allowing to control more efficiently cancer cell growth. Our results indicate that simultaneous inhibition of p38α DFG-in and -out conformations and BRAF leads to a synergistic increase of the apoptotic response in CRC cells.
Results
Sorafenib synergizes with the p38α DFG-in inhibitor SB202190 to abrogate phosphorylation of MK2 in CRC
We previously reported that p38α is overactive in a large number of CRC and ovarian cancer cell lines, where it is the main p38 isoformCitation4,Citation18 and that SB202190 is able to inhibit its kinase activity in these cells, as shown by the analysis of p38α endogenous targets with phospho-specific antibodies.Citation6 Conversely, the biological activity of sorafenib on p38α has not been studied in detail. In particular, it is still unclear whether a combination of type-I and type-II inhibitors may entail synergistic effects by acting on both the DFG-in and DFG-out conformations and, additionally, whether the pre-existence of DFG-in/DFG-out conformations is a prerequisite for inhibitor binding to p38α ().
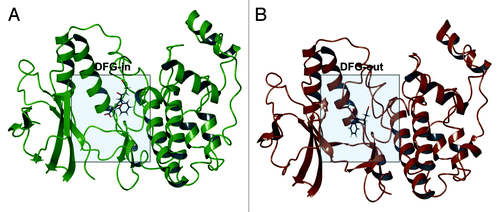
Figure 1. p38α DFG-in co-crystal structure with SB203580 (PDB ID: 1A9U) (A); p38α with sorafenib (PDB ID: 3HEG) with missing activation loop (in proximity of the DFG motif) and the glycine rich loop reconstructed by homology modeling starting from PDB ID: 1W83 (B).
To clarify these issues, the activity of p38α has been monitored by immunoblot with phospho-specific antibodies directed against p38α target genes (p-MK2, p-HSP27) in logarithmically growing HT-29 and HCT-116 cells treated with SB202190 (type-I inhibitor), sorafenib (type-II inhibitor) or a combination of both (). These CRC cell lines were selected based on the different mutations they carry in the RAS/RAF/ERK and p53 pathways. Indeed HT-29 cells contain a G273A mutation in the TP53 gene causing overproduction of a pathogenic form of p53; KRAS is wild type, while the BRAF gene carries the oncogenic V600E activating mutation. Conversely, in HCT-116 cells TP53 and BRAF genes are wild type, while KRAS is mutated (KRASD13) and over-active. MK2 (MAPKAPK-2) is directly phosphorylated by p38α on threonine 334. This promotes MK2 activation and the consequent phosphorylation of downstream targets such as HSP27, which is phosphorylated on serine 82.
Figure 2. Logarithmically growing HT-29 (A) and HCT-116 (B) cells were cultured for 24h (A) or 48h (B) in the presence of Dmso, SB202190 (10 μM), increasing concentrations of sorafenib (5–40 μM) or a combination of sorafenib (10 μM) and SB202190 (10 μM) (A), or Dmso or a combination of sorafenib (10 μM) and SB202190 (10 μM) (B). Immunoblots were performed with the indicated antibodies.
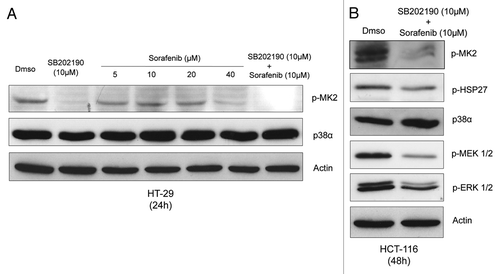
Our results show that sorafenib inhibits phosphorylation of MK2 in a dose-dependent manner () and that a concentration of 40 μM is capable of inhibiting p38α activity to a degree comparable to the DFG-in inhibitor SB202190 at 10 μM. Moreover, combination of both inhibitors completely abrogates phosphorylation of MK2 in both HT-29 and HCT-116 CRC cells and a concentration of 10 μM sorafenib is sufficient to synergize with SB202190 10 μM (). Additional analyses also showed that the concomitant treatment significantly reduces phosphorylation of HSP27 (). Moreover, while SB202190 triggers MEK/ERK overactivation, sorafenib is able to downregulate this pathway independently of KRAS and BRAF status ().
Taken together, these results show that sorafenib targets p38α in CRC cell lines and suggest that simultaneous inhibition of the DFG-in/DFG-out conformations might be beneficial to impair more efficiently p38α kinase activity.
Co-treatment with SB202190 and sorafenib enhances cell growth inhibition and activation of caspase 3
To evaluate the effects of these two inhibitors on cell growth, we performed a colony formation assay on CRC cells lines cultured in the presence of increasing concentrations of sorafenib and SB202190 for 48 h. Separated administration of SB202190 and sorafenib was able to inhibit cancer cell growth in both cell lines, as previously described (). Of note, combination of both compounds indicated that they could synergize at the biological level () and that their effects on cancer cell growth are largely independent of p53 status.
Figure 3. Logarithmically growing HCT-116 (A) and HT-29 (B) cells were cultured for 48h in the presence of Dmso, SB202190 (5–10 μM), sorafenib (5–20 μM) or a combination of SB202190 (5–10 μM) and sorafenib (5–20 μM). After 48 h, living cells were stained with Coomassie blue. Relative colony density is indicated for each time-point analyzed. Living cells were subjected to FACS analysis and the sub-G1 DNA content was measured by flow cytometry (C). Means from triplicates ± SD are shown.
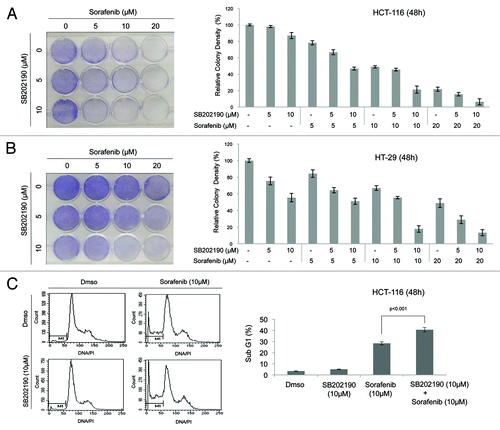
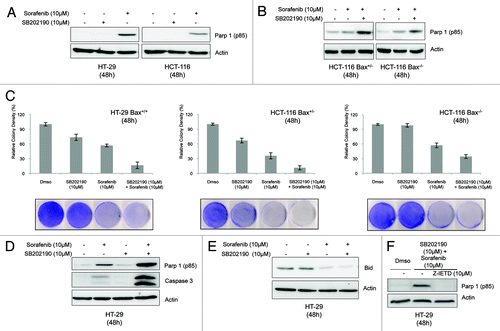
In CRC cells, SB202190 has a time-dependent biological effect. Indeed, in the first 48 h it mainly causes cell cycle arrest, while afterwards it induces non-apoptotic cell death with autophagic features.Citation4,Citation6,Citation7 Thus, we chose this time-point to evaluate the effect of SB202190 and sorafenib on CRC cell fate. FACS analysis revealed that sorafenib-dependent cell death was significantly increased by addition of SB202190 (from 28,35% to 40.6%) (). Sorafenib has been shown to affect tumor growth by inducing apoptosis in several cancer cell lines, including HT-29 and HCT-116. To get insight into the type of cell death promoted by the combined treatment, we analyzed PARPp85 cleavage, a proteolytic modification dependent on caspase 3 activation. We found that activation of caspase 3 was enhanced by the co-treatment when compared with administration of either inhibitor alone (), and was unaffected by Bax knock-down in HCT-116 cells (). We also compared the colony-forming ability of HT-29 Bax+/+ cells, HCT-116 Bax+/− cells and an isogenic HCT-116 cell line knocked down for the Bax gene (HCT-116 Bax−/−). Our results indicate that the effect of sorafenib and SB202190 + sorafenib on cancer cell growth is largely independent of the copy number of Bax (). These experiments confirmed published data showing that the activity of sorafenib in cancer cells is independent of Bax status. Indeed, sorafenib acts through downregulation of Mcl-1, a negative regulator of Bak.Citation19 Intriguingly, the effect of SB202190 on HCT-116 cell growth is abrogated by Bax genetic inactivation, suggesting a role for this gene in SB202190-dependent non apoptotic cell death. Upon co-treatment with the two inhibitors, HT-29 cells exhibited enhanced cleavage of PARP and Bid (with consequent generation of tBid, data not shown), indicating activation of caspase 3 and 8, respectively (). SB202190 + sorafenib-dependent caspase 3 activation was abrogated by the inhibition of caspase 8 ().
Figure 4. The indicated CRC cells were cultured for 48h in the presence of Dmso, SB202190 (10 μM), sorafenib (20 μM) or a combination of SB202190 (10 μM) and sorafenib (20 μM). Then, cells were harvested and subjected to immunoblot with the indicated antibodies (A, B, D, E, F), or living cells were stained with Coomassie blue (C). To inhibit caspase 8 activity, Z-IEDT FMK (10 μM) was added to the culture for the last 12h (F).
We previously reported that p38α inhibition in CRC induces autophagy, which represents a survival response in the first 48 h of treatment.Citation4,Citation6,Citation7 Evaluation of the autophagic marker Map1lc3-II (the lipidated and autophagosome-bound form of the protein)Citation20 indicated that sorafenib is also able to induce autophagy in CRC cells, and that the synergistic effect of SB202190 and sorafenib is more evident in HCT-116 cells (Fig. S1A and B). However, further experiments will be needed to elucidate whether induction of autophagy accompanies apoptosis or instead buffers cell death.
Combined use of SB202190 and sorafenib in vivo reduces CRC growth by inducing apoptosis
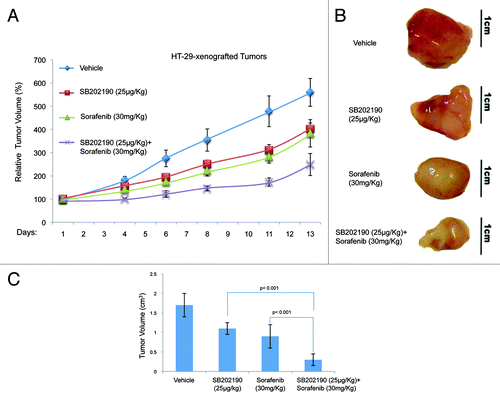
To test whether the effects described above were confined to cell culture conditions, we injected HCT-116 or HT-29 cells into each flank of athymic nude mice (n = 80). Once tumors (n = 91) had reached a measurable volume, mice were divided in four groups and treated with vehicle (control; mice: n = 20, tumors: n = 20), or sorafenib (30 mg/kg/d; mice: n = 20, tumors: n = 22), or SB202190 (25 μg/kg/d; mice: n = 20, tumors: n = 23), or a combination of sorafenib (30 mg/kg/d) and SB202190 (25 μg/kg/d) (mice: n = 20, tumors: n = 26). Drug treatment was given daily by gavage for sorafenib and intraperitoneally for SB202190. Mice were treated for 12–13 d and tumor volume and body weight were recorded every 2–3 d. At the end of the treatment, mice were sacrificed and tumors explanted for histologic and immunohistochemical analysis. In HCT-116-derived colorectal tumors, administration of SB202190, sorafenib or a combination of both gave similar results in terms of measurement of external tumor size (around 58% growth reduction compared with control tumors) (). However, the gross appearance of the explanted tumors indicated that while the effects of the treatments with the single inhibitors were similar, combination of SB202190 and sorafenib significantly reduced the tumor mass (). In HT-29-derived colorectal tumors, the enhanced effect of the co-treatment was evident also in terms of external measurements. Indeed, SB202190 induced a 28% reduction of tumor growth, compared with a 31% reduction promoted by sorafenib, while combination of both drugs reduced tumor growth by 55% (). These data suggest that, similarly to what we documented in vitro, combined use of SB202190 and sorafenib results in a synergistic effect also in xenograft models.
Figure 5. Growth kinetics of HCT-116-xenografted tumors (A). HCT-116 cells were injected subcutaneously into each flank of CD1 athimic nude mice. Mice were treated daily with SB202190 or sorafenib or a combination of both or the vehicle alone starting when tumors had reached a mean volume of 100 mm3. Animals were sacrificed when control tumors reached a mean volume of around 700 mm3. Representative tumors explanted from mice after the indicated treatments (B). Graph showing the volume of explanted tumors (mean ± SD) (C).
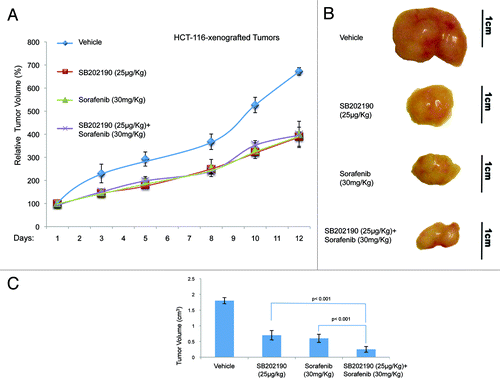
Figure 6. Growth kinetics of HT-29-xenografted tumors (A). HT-29 cells were injected subcutaneously into each flank CD1 athimic nude mice. Mice were treated daily with SB202190 or sorafenib or a combination of both or the vehicle alone starting when tumors had reached a mean volume of 100 mm3. Animals were sacrificed when control tumors reached a mean volume of around 550 mm3. Representative tumors explanted from mice after the indicated treatments (B). Graph showing the volume of explanted tumors (mean ± SD) (C).
Histopathological analysis of HCT-116- and HT-29-derived colorectal tumor specimens revealed central areas of necrosis with neutrophilic and lymphoid infiltrates that were significantly detectable in all tumors treated with SB202190 or sorafenib ( and ; ). Necro-inflammatory areas were sometimes delimited by fibroblastic and vessel proliferation representing reparative tissue replacing neoplastic cells. No considerable neoplastic regression was present in untreated CRC-derived tumors, while co-treatment with both compounds significantly increased cancer cell death in both HCT-116- and HT-29-xenografted tumors ( and ; ). These data further strengthen the efficacy of the combined use of both drugs against colorectal neoplasms. Moreover, evaluation of apoptotic markers revealed a significant increase of TUNEL-positive (apoptotic) cells in tumor sections from SB202190 + sorafenib-treated compared with single drug-treated animals ( and ; ). These data validate in vivo the combined use of SB202190 and sorafenib as a promising approach to reduce CRC growth by inducing apoptosis.
Figure 7. Hematoxylin and eosin (H&E) staining and immunohistochemical analysis of HCT-116-xenografted tumors with antibodies recognizing phosphorylated ERK (p-ERK) and MK2 (p-MK2). Apoptosis was scored by TUNEL assay.
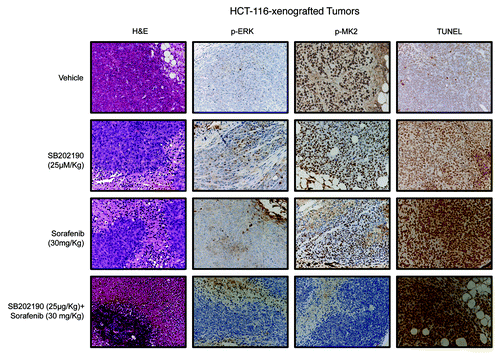
Figure 8. Hematoxylin and eosin (H&E) staining and immunohistochemical analysis of HT-29-xenografted tumors with antibodies recognizing phosphorylated ERK (p-ERK) and MK2 (p-MK2). Apoptosis was scored by TUNEL assay.
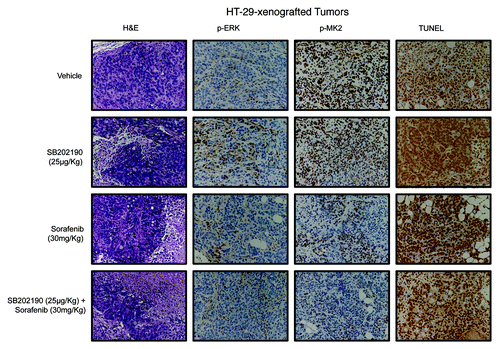
Table 1. Histological and immunohistochemical evaluation of HCT-116- and HT-29-driven colorectal tumors based on the data represented in , , and
Immunohistochemical analyses confirmed the crosstalk between the p38α and the ERK pathways,Citation5 since SB202190-treated tumors showed increased levels of phosphorylated ERK (p-ERK) ( and ; ). Evaluation of MK2 revealed the ability of both SB202190 and sorafenib to inhibit p38α activity also in vivo and highlighted the synergistic effect of these drugs when used in combination ( and ; ).
Binding of the type-II kinase inhibitor sorafenib occurs through a pseudo-DFG-out conformation
Type-II kinase inhibitors have been largely studied as they are expected to provide a greater selectivity compared with conventional ATP-competitive inhibitors, since only few kinases have been demonstrated to undergo DFG-out conformationCitation21,Citation14. Sorafenib, a type-II inhibitor, has been shown to target p38α, and their recently established co-crystal structure also highlighted the mode of binding (). The arrangement of sorafenib is typical of other DFG-out kinases in which Phe169, in particular, is projected toward a solvent-exposed conformation in the ATP-binding site with concomitant opening of a buried hydrophobic pocket that is targeted by the complementary hydrophobic moiety of type-II ligands. Conversely, SB202190 binds to p38α in a DFG-in conformation () in which the DFG motif is arranged to occupy the hydrophobic pocket, thus interfering with the normal binding of type-II inhibitors. It is still a matter of debate whether protein kinases naturally adopt DFG-in and DFG-out conformations by simply cycling among low-energetic states or, vice versa, if this is a consequence of a fitting process induced upon ligand binding.
Figure 9. Binding mode of SB202190 to p38α in the DFG-in conformation (PDB ID: 1A9U) (A). Lowest energy binding mode of sorafenib to p38α in the DFG-in conformation after Induced Fit Docking (IFD) (B). Binding mode of sorafenib to p38α in the DFG-out conformation (PDB ID: 3HEG).
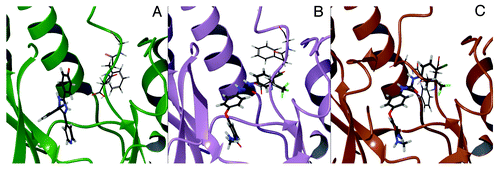
In order to elucidate these mechanisms at the molecular level for the binding of sorafenib to p38α, we performed induced fit docking (IFD) calculations in which we docked sorafenib by allowing conformational adaptation of the three DFG residues and taking the crystallographic structure of a DFG-in conformation as a template (). The lowest energy binding pose of sorafenib obtained with IFD is in good agreement with the one observed in the co-crystal structure of the DFG-out conformation (). In addition, and interestingly, the induced fit of sorafenib occurs by simple flipping out of Phe169 from the buried hydrophobic pocket to assume a conformation that is intermediate between a DFG-in and a DFG-out structure (). The existence of this pseudo-DFG-out conformation is consistent with other findings.Citation21 Thus, our computational results support the idea that the mechanism by which sorafenib binds to p38α is initially mediated by a small rearrangement of Phe169, which is subsequently followed by the major transition toward the DFG-out conformation (). This mechanism may also explain the synergistic effect that we observed for the co-treatment with SB202190 and sorafenib in CRC. In fact, since sorafenib is able to bind p38α, we hypothesize that a residual kinase activity, exerted by the remaining pool of p38α in a DFG-in conformation, may constitute the basis of clinical failure of sorafenib in CRC. Therefore, the concomitant inhibition with a type-I inhibitor such as SB202190 is expected to abrogate the residual activity of p38α with consequent synergistic effects. It is worth noting that the growing availability of three-dimensional structures of p38α is likely to facilitate the deployment of in silico techniques in search of novel compounds capable of simultaneously targeting its DFG-in and DFG-out conformations.Citation22
Discussion
The ability of chemotherapy to kill cancer cells relies on the induction of apoptosis. However, as cancer cells are exposed to a selective pressure, they evolve strategies to circumvent pro-apoptotic signaling pathways and favor pro-survival and proliferative cascades. For this reason, a promising therapeutic approach consists in targeting critical pathways/proteins that are essential for tumor growth and progression. This concept is exploited through the use of compounds that are able to target relevant signaling pathways, such as MEK/ERK and PI3K/AKT, with some of these molecules currently being tested in clinical trials.Citation23,Citation24 In CRC, the MEK/ERK pathway has been frequently found overactive due to mutations in the upstream kinases RAS and RAF. As the RAS/RAF pathway promotes cell proliferation and survival and is thus crucial for tumor initiation and progression, it has been widely used as a pharmacological target for small-molecule inhibitors.Citation23 All these kinases exert their catalytic activity by phosphorylating target proteins using an ATP molecule that binds a well-defined pocket, the ATP-binding site. The ATP-binding site has been targeted for a long time now with small-molecule inhibitors that compete with ATP. However, this strategy has two main disadvantages: i) inhibitors display a relatively low specificity and, ii) inhibitors have to compete with high concentrations of ATP in the cell. In fact, although initial results demonstrated that inhibitors of RAF or MEK1 exerts cytostatic and cytotoxic effects in vitro and in xenograft models, the MEK1 inhibitor CI-1040 showed poor antitumor activity in phase II clinical trials. In addition, inhibition of RAF in tumor cells harboring mutant BRAF entailed inhibition of ERK signaling but, unexpectedly, enhanced ERK signaling in cells with wild-type BRAF or mutant KRAS. During tumorigenesis, signaling pathways that have an anti-tumorigenic activity in healthy cells might become involved in the preservation of tumor cells and in the promotion of cell migration. The p38α MAP kinase drives one of these pathways and is required for cell cycle arrest and apoptosis in some cell types, but acquires a tumor-promoting activity in cancer cells.
We recently showed that p38α activity is required for CRC cell growth in vitro and in preclinical mouse models of the human colon cancer syndrome Familial Adenomatous Polyposis. In addition, p38α inhibition induces autophagic cell death and reduces tumor growth in CRC cells and in AOM-treated APCmin/+ mice.Citation6,Citation7 The activity of p38α is also required to maintain a glycolytic metabolism, which is a typical hallmark of cancer cells, and pharmacological inhibition of p38α leads to an acute state of energy demand that is compensated through the activation of a FoxO3A-dependent autophagic response.Citation6,Citation7
Despite drug discovery efforts focusing on protein kinase inhibitors yielded high-profile approved agents, such as Gleevec (imatinib), Sprycel (dasatinib), Iressa (gefitinib), and sorafenib for p38α, relapse is still a major clinical problem as drug resistance is developed also against type-II inhibitors. The target-centric approach consisting in the design of a single small-molecule having optimal selectivity profiles against one kinase conformation has been very successful in some cases, but failed to provide the expected results in other circumstances. Several studies are now being performed to collect information on the polypharmacological profile of kinase inhibitors. These data are of particular importance, as they will allow favoring selective vs. promiscuous effects depending on the therapeutic application.Citation25
In this context, the scientific rationale behind this study comes from experiments performed in our lab showing that the combination of inhibitors targeting the p38α and the MEK/ERK pathways promote CRC cell death by inducing apoptosis in cell lines and animal models. Conversely, single inhibition of p38α or MEK1 results in autophagic cell death and cell cycle arrest, respectively.Citation5 Interestingly, activation of p38α has been observed in both HT-29 and HCT-116 CRC cells upon inhibition of MEK1, but not in human primary non-tumorigenic colon cells,Citation5 suggesting that a reciprocal compensatory role between these two pathways may exist in cancer cells. Therefore, a single agent or a combination of agents capable of efficiently targeting both pathways could prove a promising tool in CRC therapy. Sorafenib could represent the ideal candidate for this therapeutic approach since it has a multikinase inhibitor activity, being able to target the DFG-out conformation () of BRAF and p38α.Citation13,Citation14 Notwithstanding, clinical trials failed to demonstrate a beneficial effect for sorafenib in CRC patients.
Overall, the results we presented here suggest that simultaneous inhibition of the DFG-in and DFG-out conformations is be beneficial to impair more efficiently the biological functions of p38α. Indeed, the combined use of SB202190 and sorafenib results not only in significantly reduced phosphorylation of endogenous targets, but also in an enhanced anti-cancer activity mediated by the induction of apoptosis, the most suitable type of cell death in tumor treatment. These observations are consistent with structural data and other studies showing that p38α can be targeted by type-I and type-II kinase inhibitors in its DFG-in and DFG-out conformations, respectively.Citation14,Citation21,Citation22 In addition, in agreement with other findings,Citation21 our computational results support the idea that the mechanism by which the type-II inhibitor sorafenib binds to p38α is initially through a presudo-DFG-out conformation, that is likely to turn into a DFG-out structure after major structural rearrangement.
This work might constitute the basis for a novel drug design paradigm of kinase inhibition, in which both the DFG-in and DFG-out conformations are simultaneously targeted, either with a combination of inhibitors (type-I and type-II inhibitors) or with one single agent acting against both conformations.Citation24 To this purpose, specific molecular design techniques focused on DFG conformations, which are available as three-dimensional structures from public sources (Protein Data Bank), might be used to carry out time- and cost-effective computer-aided drug design campaignsCitation21,Citation22,Citation27,Citation28 in search of novel combination of p38α inhibitors. In addition, provided that similar additive effects are also demonstrated for other kinases, these molecular design efforts might involve not only a combination of DFG-in/DFG-out conformations, but also different kinases, such as BRAF.
Materials and Methods
Cell culture and reagents
HT-29, HCT-116 and HCT-116 BAX−/− cells were maintained in DMEM supplemented with 10% FBS, avoiding confluence at any time. SB202190 (5–10 μM) was purchased from Calbiochem or Sigma-Aldrich. Caspase 8 inhibitor Z-IEDT FMK (10μM) was purchased from PD PharMingen. Sorafenib tosylate was generously provided by Bayer Pharmaceuticals Corporation. For cell culture experiments, it was used at a final concentration of 5–40 μM. For in vivo experiments, sorafenib was dissolved in Chremophor EL/Ethanol (50:50). It was weighed out at the beginning of the study and stored away from light, then dissolved fresh each day prior to dosing. The dosing volume used was 0.1 ml/10 g body weight. The mixture was heated (up to 60°C) for 10 min and sonicated for 2 min to get the compound in solution. Once the compound was in solution, the aqueous component was added slowly while mixing to generate the 1X dosing solution (3 parts of water to one part of 4X stock solution). Sorafenib was given to mice by oral gavage.
Immunoblot
Immunoblots were performed according to Cell Signaling's instructions. Briefly, cells were homogenized in 1X lysis buffer (50 mMTRIS-HCl pH7.4; 5 mM EDTA; 250 mMNaCl; 0,1% Triton X-100) supplemented with protease and phosphatase inhibitors (1 mM PMSF; 1.5 μM pepstatin A; 2 μM leupeptin; 10 μg/ml aprotinin, 5 mMNaF; 1 mM Na3VO4). Fifteen to 20 μg of protein extracts from each sample were denatured in 5x Laemmli sample buffer and used for western blots analysis. Western blots were performed using anti-actin (Sigma), anti-p38α, anti-phosphoMAPKAPK2 (Thr334), anti-phosphoHSP27 (Ser82), BID, Caspase 3 (all from Cell Signaling), PARPp85 (Promega), MAP1LC3 (Novus Biologicals). Western blots were developed with the ECL-plus chemiluminescence reagent (GE Healthcare) as per manufacturer's instructions.
Colony formation assay
CRC cells were cultured in 60mm dishes in the presence or the absence of SB202190, sorafenib or a combination of both. After 48 h the medium was discarded and cells were washed twice with PBS 1X. Two ml of Coomassie brilliant blu (Bradford) were added in each dish for 5 min. Cells were then washed with ethanol 70% to remove the excess of Coomassie. Plates were dried at room temperature. The densitometric evaluation was performed by the ImageJ software.
FACS analysis
Cells were collected, ethanol-fixed and stained with propidium iodide (Sigma). Cell cycle distribution and sub-G1 DNA content were measured with a FACS Vantage flow cytometer and analyzed using the Cell Quest-PRO software (BD Bioscience).
In vivo studies
Female CD-1 athymic nude mice (6–8 week old) were purchased from the Charles River Laboratories. 10 × 106 HT-29 or 10x106 HCT-116 cells were injected subcutaneously into the flanks (0.2 ml per flank) of female athymic nude CD-1 mice. The volume of the tumors was measured every 2–3 d. The tumor volume was calculated using the following formula: volume (mm3) = (width)2 × length × 0.5. When the tumor volume reached 60 mm3, mice were randomized into four treatment groups (n ≥ 6 for each group): vehicle (control), sorafenib 30 mg/kg/d, SB202190 25 μg/kg/d, and sorafenib 30 mg/kg/d plus SB202190 25 μg/kg/d. Drug treatment was given daily by gavage for sorafenib and intraperitoneally for SB202190. Mice were treated for 12 d and tumor volume and body weight were recorded every 2–3 d. At the end of the treatment, mice were sacrificed and tumors explanted for histologic and immunohistochemical analysis.
Procedures involving animals and their care were conducted in conformity with the institutional guidelines that are in compliance with national and international laws and policies.
Histology, immunohistochemistry and apoptosis assays
HCT-116- and HT-29-derived mice colorectal tumor specimens were fixed overnight in 10% neutral-buffered formalin, embedded in paraffin, sectioned at a thickness of 4 μm and stained with hematoxylin-eosin. Additional sections, collected on poly-l-lysine coated slides, were used for immunohistochemical stains, that were performed with avidin-biotin-based detection systems. Sections were incubated with antibodies against Ki-67 (Dako), phospho-MK2 (Cell Signaling), phospho-ERK 1/2 (Cell Signaling), with overnight incubations at 4°C. Appropriate negative controls, obtained by replacing primary antibodies with pre-immune serum, and positive controls were included in the procedure. TUNEL assays (Roche) were performed on tissue sections following manufacturer's instructions.
In silico studies
The p38α co-crystal structure with SB203580 (PDB ID: 1A9U) was used as a template for the DFG-in conformation. The ligand was manually edited in order to reflect the structure of SB202190. The crystal structure of p38α with sorafenib (PDB ID: 3HEG) was taken as a representative DFG-out conformation. The missing activation loop (in proximity of the DFG motif) and the glycine rich loop were reconstructed by homology modeling with the Prime (Schrödinger) software starting the crystallographic structure PDB ID: 1W83. Induced fit docking (IFD) was performed with the Prime and Glide (Schrödinger) software by using the trim side chains option on Asp168, Phe169 and Gly170, refining residues within 8.0 Å from the ligand pose and with SP precision protocol.
Additional material
Download Zip (180.8 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This article is dedicated to the memory of Margherita D’Aprile and Gianni Iannuzzi. We thank Dr Francesco Paolo Jori for his helpful discussion during the preparation of the manuscript and editorial assistance, and Dr Giovanna Forte for contribution in preparing the illustrations. This work was partially supported by an Investigator Grant 2010(IG10177) (to C.S.) and by the Start-Up Grant (n° 6266) (to A.D.R) from the Italian Association for Cancer Research (AIRC). Bayer Italia is gratefully acknowledged for providing Sorafenib.
Supplemental Materials
Supplemental materials may be found here:
http://www.landesbioscience.com/journals/cbt/article/22254/
References
- Chiacchiera F, Simone C. Signal-dependent regulation of gene expression as a target for cancer treatment: inhibiting p38alpha in colorectal tumors. Cancer Lett 2008; 265:16 - 26; http://dx.doi.org/10.1016/j.canlet.2008.02.061; PMID: 18395970
- Segal NH, Saltz LB. Evolving treatment of advanced colon cancer. Annu Rev Med 2009; 60:207 - 19; http://dx.doi.org/10.1146/annurev.med.60.041807.132435; PMID: 19630571
- Blume-Jensen P, Hunter T. Oncogenic kinase signaling. Nature 2001; 411:335 - 6; http://dx.doi.org/10.1038/35077225
- Comes F, Matrone A, Lastella P, Nico B, Susca FC, Bagnulo R, et al. A novel cell type-specific role of p38α in the control of autophagy and cell death in colorectal cancer cells. Cell Death Differ 2007; 14:693 - 702; http://dx.doi.org/10.1038/sj.cdd.4402076; PMID: 17159917
- Chiacchiera F, Grossi V, Cappellari M, Peserico A, Simonatto M, Germani A, et al. Blocking p38/ERK crosstalk affects colorectal cancer growth by inducing apoptosis in vitro and in preclinical mouse models. Cancer Lett 2012; 324:98 - 108; http://dx.doi.org/10.1016/j.canlet.2012.05.006; PMID: 22579651
- Chiacchiera F, Matrone A, Ferrari E, Ingravallo G, Lo Sasso G, Murzilli S, et al. p38alpha blockade inhibits colorectal cancer growth in vivo by inducing a switch from HIF1alpha- to FoxO-dependent transcription. Cell Death Differ 2009; 16:1203 - 14; http://dx.doi.org/10.1038/cdd.2009.36; PMID: 19343039
- Chiacchiera F, Simone C. Inhibition of p38alpha unveils an AMPK-FoxO3A axis linking autophagy to cancer-specific metabolism. Autophagy 2009; 5:1030 - 3; http://dx.doi.org/10.4161/auto.5.7.9252; PMID: 19587525
- Chiacchiera F, Simone C. The AMPK-FoxO3A axis as a target for cancer treatment. Cell Cycle 2010; 9:1091 - 6; http://dx.doi.org/10.4161/cc.9.6.11035; PMID: 20190568
- Fang JY, Richardson BC. The MAPK signalling pathways and colorectal cancer. Lancet Oncol 2005; 6:322 - 7; http://dx.doi.org/10.1016/S1470-2045(05)70168-6; PMID: 15863380
- Sebolt-Leopold JS, Dudley DT, Herrera R, Van Becelaere K, Wiland A, Gowan RC, et al. Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nat Med 1999; 5:810 - 6; http://dx.doi.org/10.1038/10533; PMID: 10395327
- Wickenden JA, Jin H, Johnson M, Gillings AS, Newson C, Austin M, et al. Colorectal cancer cells with the BRAF(V600E) mutation are addicted to the ERK1/2 pathway for growth factor-independent survival and repression of BIM. Oncogene 2008; 27:7150 - 61; http://dx.doi.org/10.1038/onc.2008.335; PMID: 18806830
- Rinehart J, Adjei AA, Lorusso PM, Waterhouse D, Hecht JR, Natale RB, et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol 2004; 22:4456 - 62; http://dx.doi.org/10.1200/JCO.2004.01.185; PMID: 15483017
- Wilhelm S, Carter C, Lynch M, Lowinger T, Dumas J, Smith RA, et al. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov 2006; 5:835 - 44; http://dx.doi.org/10.1038/nrd2130; PMID: 17016424
- Namboodiri HV, Bukhtiyarova M, Ramcharan J, Karpusas M, Lee Y, Springman EB. Analysis of imatinib and sorafenib binding to p38alpha compared with c-Abl and b-Raf provides structural insights for understanding the selectivity of inhibitors targeting the DFG-out form of protein kinases. Biochemistry 2010; 49:3611 - 8; http://dx.doi.org/10.1021/bi100070r; PMID: 20337484
- Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res 2004; 64:7099 - 109; http://dx.doi.org/10.1158/0008-5472.CAN-04-1443; PMID: 15466206
- Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther 2008; 7:3129 - 40; http://dx.doi.org/10.1158/1535-7163.MCT-08-0013; PMID: 18852116
- Dal Lago L, D’Hondt V, Awada A. Selected combination therapy with sorafenib: a review of clinical data and perspectives in advanced solid tumors. Oncologist 2008; 13:845 - 58; http://dx.doi.org/10.1634/theoncologist.2007-0233; PMID: 18695262
- Matrone A, Grossi V, Chiacchiera F, Fina E, Cappellari M, Caringella AM, et al. p38alpha is required for ovarian cancer cell metabolism and survival. Int J Gynecol Cancer 2010; 20:203 - 11; http://dx.doi.org/10.1111/IGC.0b013e3181c8ca12; PMID: 20169663
- Gillissen B, Wendt J, Richter A, Richter A, Müer A, Overkamp T, et al. Endogenous Bak inhibitors Mcl-1 and Bcl-xL: differential impact on TRAIL resistance in Bax-deficient carcinoma. J Cell Biol 2010; 188:851 - 62; http://dx.doi.org/10.1083/jcb.200912070; PMID: 20308427
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008; 4:151 - 75; PMID: 18188003
- Filomia F, De Rienzo F, Menziani MC. Insights into MAPK p38alpha DFG flip mechanism by accelerated molecular dynamics. Bioorg Med Chem 2010; 18:6805 - 12; http://dx.doi.org/10.1016/j.bmc.2010.07.047; PMID: 20724167
- Kufareva I, Abagyan R. Type-II kinase inhibitor docking, screening, and profiling using modified structures of active kinase states. J Med Chem 2008; 51:7921 - 32; http://dx.doi.org/10.1021/jm8010299; PMID: 19053777
- Carnero A. The PKB/AKT pathway in cancer. Curr Pharm Des 2010; 16:34 - 44; http://dx.doi.org/10.2174/138161210789941865; PMID: 20214616
- Montagut C, Settleman J. Targeting the RAF-MEK-ERK pathway in cancer therapy. Cancer Lett 2009; 283:125 - 34; http://dx.doi.org/10.1016/j.canlet.2009.01.022; PMID: 19217204
- Dar AC, Das TK, Shokat KM, Cagan RL. Chemical genetic discovery of targets and anti-targets for cancer polypharmacology. Nature 2012; 486:80 - 4; http://dx.doi.org/10.1038/nature11127; PMID: 22678283
- Knight ZA, Lin H, Shokat KM. Targeting the cancer kinome through polypharmacology. Nat Rev Cancer 2010; 10:130 - 7; http://dx.doi.org/10.1038/nrc2787; PMID: 20094047
- Pierotti MA, Tamborini E, Negri T, Pricl S, Pilotti S. Targeted therapy in GIST: in silico modeling for prediction of resistance. Nat Rev Clin Oncol 2011; 8:161 - 70; http://dx.doi.org/10.1038/nrclinonc.2011.3; PMID: 21364689
- Liu Y, Gray NS. Rational design of inhibitors that bind to inactive kinase conformations. Nat Chem Biol 2006; 2:358 - 64; http://dx.doi.org/10.1038/nchembio799; PMID: 16783341