Abstract
The adverse side effects of doxorubicin, including cardiotoxicity and cancer treatment-related fatigue, have been associated with inflammatory cytokines, many of which are regulated by mitogen-activated protein kinases (MAPKs). ZAK is an upstream kinase of the MAPK cascade. Using mouse primary macrophages cultured from ZAK-deficient mice, we demonstrated that ZAK is required for the activation of JNK and p38 MAPK by doxorubicin. Nilotinib, ponatinib and sorafenib strongly suppressed doxorubicin-mediated phosphorylation of JNK and p38 MAPK. In addition, these small molecule kinase inhibitors blocked the expression of IL-1β, IL-6 and CXCL1 RNA and the production of these proteins. Co-administration of nilotinib and doxorubicin to mice decreased the expression of IL-1β RNA in the liver and suppressed the level of IL-6 protein in the serum compared with mice that were injected with doxorubicin alone. Therefore, by reducing the production of inflammatory mediators, the inhibitors identified in the current study may be useful in minimizing the side effects of doxorubicin and potentially other chemotherapeutic drugs.
Introduction
First discovered in the 1960s, the anthracycline, doxorubicin, is one of the most widely employed anticancer agents. In combination with other cytotoxic agents, doxorubicin is administered in the treatment of a variety of malignancies including breast, liver, bladder and ovarian cancer, childhood solid tumors, soft tissue sarcomas and aggressive lymphomas. Although doxorubicin is an effective anticancer agent, exposure to doxorubicin is associated with severe adverse side effects. Doxorubicin induces myocardial stress, cardiomyopathy and heart failure that is progressive and irreversible.Citation1 Although the underlying mechanisms of doxorubicin-mediated cardiotoxicity are still unclear, several mechanisms have been proposed including the induction of inflammatory cytokines.Citation2-Citation5 Administration of IL-1 receptor antagonist (IL-1RA), which blocks IL-1 signaling, protects mice from doxorubicin-mediated cardiotoxicity.Citation7 In addition, doxorubicin-mediated induction of inflammatory cytokines has been implicated in cancer treatment-related fatigue and associated symptoms.Citation8 Taken together, it is conceivable that suppressing the production of inflammatory cytokines may reduce the adverse side effects of doxorubicin and perhaps other cytotoxic chemotherapeutic agents. Elucidating the upstream transducers of doxorubicin-mediated inflammatory responses is essential to development of anti-inflammatory therapeutics.
At the cellular level, inflammatory cytokines such as IL-1β are regulated by multiple molecular pathways including the mitogen-activated protein kinase (MAPK) pathway.Citation9-Citation11 Doxorubicin activates MAPKs, including the Jun N-terminal kinases (JNK) and p38 MAPK, which are involved in various biochemical processes and regulate apoptosis and the production of many inflammatory mediators. When the activation of MAPKs is blocked in vitro using specific inhibitors such as SB 203580 and ML3403, the inflammatory and pro-apoptotic effects of doxorubicin are also suppressed.Citation6,Citation12 Due to the important role of MAPKs in many inflammatory diseases, a variety of small molecules have been developed as inhibitors that target JNK or p38 MAPK.Citation13,Citation14 Because it is known that inhibition of p38 MAPK may result in increased activity of JNK,Citation15,Citation16 it may be essential to block both JNK and p38 MAPK to achieve maximal suppression of the downstream effects of MAPK signaling, including toxic side effects.
MAPKs are phosphorylated by MAPK kinases (MAP2Ks), which, in turn, are phosphorylated by MAPK kinases (MAP3Ks). Using siRNA, we have previously shown that doxorubicin-mediated activation of JNK and p38 MAPK in keratinocytes is dependent on ZAK,Citation17 a MAP3K that contains a zipper sterile-α-motif.Citation18-Citation21 ZAK is known to have strong binding affinities to two small molecule kinase inhibitorsCitation22-Citation24: nilotinib, originally used to target BCR-ABL,Citation25 and sorafenib,Citation26 which targets B-Raf and several tyrosine protein kinases, such as vascular endothelial growth factor receptor (VEGFR) and platelet-derived growth factor receptor (PDGFR).Citation27 Ponatinib is a next-generation BCR-ABL inhibitor, currently in clinical trials, that also blocks VEGFR and PDGFR but has unknown binding affinity to ZAK.Citation28
The purpose of the current study has been to examine further the requirement of ZAK for the induction of doxorubicin-mediated inflammatory signaling in primary mouse macrophages. Our data demonstrated that the doxorubicin-mediated activation of MAPKs was strongly suppressed in primary macrophages isolated from ZAK-deficient mice or in macrophages from wild-type mice exposed to nilotinib, ponatinib and sorafenib. These inhibitors were also able to suppress the expression of IL-1β, IL-6 and CXCL1 in wild-type macrophages. We further demonstrated that co-administration of nilotinib and doxorubicin to wild-type mice was effective in decreasing the expression of IL-1β RNA in the liver and in suppressing the level of IL-6 protein in the serum compared with mice that were injected with doxorubicin alone. Our studies suggest that small molecule kinase inhibitors may be useful in minimizing the proinflammatory effects of doxorubicin.
Materials and Methods
Chemicals
For in vitro experiments, doxorubicin (Sigma) was dissolved in water and stored at -20°C; nilotinib and sorafenib (LC Laboratories) and ponatinib (Tocris Bioscience) were dissolved in DMSO and stored at -80°C. For in vivo experiments, doxorubicin was purchased as a lyophilized 10 mg tablet from Bedford Labs; nilotinib was purchased as lyophilized 150 mg tablet from Novartis Pharma AG and dissolved in vehicle (7% N-methyl-2-pyrrolidone, 93% polyethylene glycol 300; both from Sigma). Trichloroacetic acid (TCA) was purchased from Fisher Scientific. Antibody against IL-1β (ab9722) was purchased from Abcam, phospho-JNK (9251) and phospho-p38 MAPK (9211) from Cell Signaling Technology and p38 MAPK (sc-535) from Santa Cruz Biotechnology.
Mice
All animal procedures were approved by the Institutional Animal Care and Use Committee at Oregon Health and Science University (OHSU). C57BL/6J mice (10–12 wk of age) were purchased from the Jackson Laboratory. Generation of ZAK+/− and ZAK−/− mice is described in a separate manuscript under preparation. For in vivo experiments, mice were housed five to a cage in pathogen-free rooms (12 h light-dark cycle) with ad libitum access to drinking water. Mice were treated with 0.1 mL of vehicle or nilotinib (75 mg/kg) by oral gavage daily for 6 d to obtain a sustained inhibition of MAPKs. On the sixth day, mice were also injected intraperitoneally with 1 mL of saline or doxorubicin (25 mg/kg). After injection, mice were returned to their home cages, at which time mouse chow was removed to eliminate any potential effects of food intake. Sixteen hours after injection with saline or doxorubicin, mice were terminally sedated using isofluorane according to protocols established at OHSU Department of Comparative Medicine. Peripheral blood was collected by cardiac puncture, and liver samples were removed, snap-frozen in liquid nitrogen and stored at -80°C.
Isolation and treatment of bone marrow-derived macrophages (BMDM)
Mice, 8–10 wk of age, were used throughout the experiments. Marrow was flushed from the femurs and tibias of wild-type C57BL/6 (WT), ZAK+/− and ZAK−/− mice with PBS and cultured in α-Minimum Essential Medium (Cellgro), supplied with 10% fetal bovine serum (Cellgro), 50 μg/ml gentamicin and 100 ng/mL recombinant mouse colony-stimulating factor 1 (R&D Systems) for 72 h on non-tissue culture-treated 10-cm Petri dishes. BMDM were passaged and cultured for an additional 72 h. Each confluent 10-cm dish was transferred into one 6-well or one 12-well tissue culture plate and cultured for 24 h before initiating experimental treatment. BMDM were treated with 5 µM doxorubicin (which corresponds to the peak plasma concentration in adults), either continuously for 12 or 24 h (used in many in vitro studies) or for 2 h followed by incubation in medium for an additional 10 or 22 h (which closely replicates the clinical situation in which levels of doxorubicin in the serum or tissues rapidly decrease after a distribution phase of 2 h).Citation29 Nilotinib, ponatinib and sorafenib, all at 1 µM, were added half an hour before the addition of doxorubicin.
Immunoblotting
BMDM were lysed in 2X electrophoresis sample buffer. Proteins in the cell lysates were separated on a denaturing polyacrylamide gel in the presence of sodium dodecyl sulfate and transferred onto polyvinylidene difluoride membranes according to standard laboratory procedures. Proteins from BMDM media supernatants were precipitated using TCA plus 200 μg insulin carrier protein and separated on 13% gels. Membranes were incubated with the indicated antibodies and the corresponding horseradish peroxidase-conjugated secondary antibodies. Signals were detected by using enhanced chemiluminescence.
Real-time RT-PCR
Total RNA from frozen tissues was isolated using TRIzol (Invitrogen) following the manufacturer's instructions. RNA was treated with DNase I (Invitrogen) and reverse-transcribed with SuperScript II and oligo dT primer (Invitrogen). Real-time PCR was performed using SYBR Green reagents on a ViiA 7 Real-Time PCR System (Applied Biosystems); fold induction was calculated with the absolute quantification method using levels of glyceraldehyde phosphate dehydrogenase (GAPDH) for normalization. The nucleotide sequences of the primers used in this study have been previously published.Citation30 Experiments were repeated at least three times, and representative data are shown.
Measurement of inflammatory cytokines from serum or culture supernatant
Peripheral blood obtained by cardiac puncture was allowed to clot at room temperature for 60 min and subjected to centrifugation in a microcentrifuge tube at 10,000 rpm for 2 min. Serum was removed and immediately frozen at -80°C prior to cytokine analysis. Serum levels of IL-1β, IL-6 and CXCL1/Gro-α in the serum or from culture supernatant were measured in duplicate in two separate experiments, using a bead-based multiplex immunofluorescence assay. Cytokine analysis kits were obtained from EMD Millipore, and assays were performed according to the protocol supplied by the manufacturer. Data were collected and analyzed using the Luminex-100 system Version IS (Luminex). A four- or five-parameter regression formula was used to calculate the sample concentrations from the standard curves.
Statistical analysis
Individual groups were compared using unpaired t-test analysis. To determine p values, all statistical analyses were interpreted in a two-tailed manner, and p values < 0.05 were considered to be statistically significant.
Results
Doxorubicin activates MAPKs and increases expression of inflammatory genes in macrophages
We first examined whether a clinically relevant dose of doxorubicin would activate MAPKs in primary mouse macrophages. Macrophages were incubated in medium containing 5 μM doxorubicin, a dose within the range of clinical relevance.Citation31,Citation32 After 12 h of continuous exposure to doxorubicin, JNK and p38 MAPK became phosphorylated (). Levels of total p38 MAPK were invariant and used as a loading control. After 24 h of continuous exposure to doxorubicin, phosphorylation of JNK and p38 MAPK was observed, but at lower levels compared with 12 h. Total p38 MAPK levels, which are commonly used as a loading control, were also diminished by 24 h, at which time cells were apoptotic (not shown). When BMDM were treated for doxorubicin for 2 h, washed and incubated with doxorubicin-free medium, phosphorylation of JNK and p38 MAPK was not observed, suggesting that phosphorylation of these proteins may have returned to basal level at the end of the subsequent 10 or 22 h of incubation in medium after removal of doxorubicin.
Figure 1. MAPK activation and gene expression in doxorubicin-treated BMDM. BMDM were plated onto 12-well plates. After serum deprivation for 0.5 h, BMDM were untreated, treated with 5 µM of doxorubicin for 2 h, then washed away and replaced by medium, or treated continuously with 5 µM of doxorubicin for 12 or 24 h. (A) Western blot of cell lysates using antibodies against phospho-JNK, phospho-p38 MAPK or total p38 MAPK. (B) Measurement of gene expression in cells after identical treatments using real-time RT-PCR. Mean values ± SD are shown. ** p < 0.01; *** p < 0.001 compared with values corresponding to wild-type.
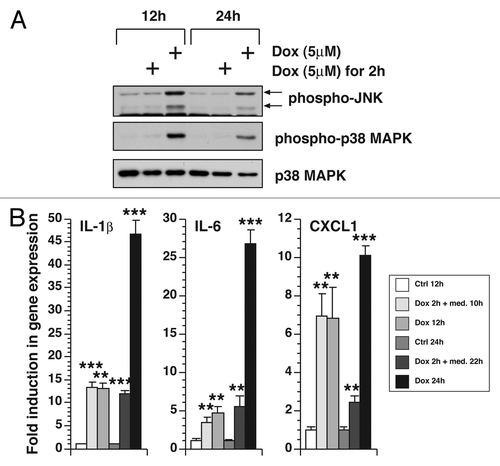
We also measured the effects of doxorubicin on the expression of RNA-encoding inflammatory gene products by RT-PCR (). Compared with untreated cells, BMDM treated with doxorubicin expressed increased levels of IL-1β and IL-6. The levels of expression were similar whether the cells were treated continuously with doxorubicin for 12 h or acutely for 2 h followed by 10 h post-incubation in doxorubicin-free medium. Expression of IL-1β and IL-6 was significantly higher when treatment was continued for 24 h compared with other treatments. Expression of CXCL1 in response to doxorubicin was similar whether doxorubicin was present only for the initial 2 h followed by 10 h of post-incubation or for the entire 12 h. However, the doxorubicin-mediated increase in CXCL1 expression was reduced after 22 h post-incubation, but proceeded to higher levels if doxorubicin was present for the entire 24 h. There was no increase in expression in TNF-α and CCL2 by any treatments (data not shown).
Taken together, these data demonstrated that doxorubicin is able to induce the phosphorylation of JNK and p38 MAPK as well as the expression of IL-1β, IL-6 and CXCL1 in BMDM. If doxorubicin was removed following a 2-h pulse, MAPK phosophorylation was not sustained, but expression of RNA encoding inflammatory mediators remained at elevated levels.
ZAK is essential for the activation of MAPKs by doxorubicin
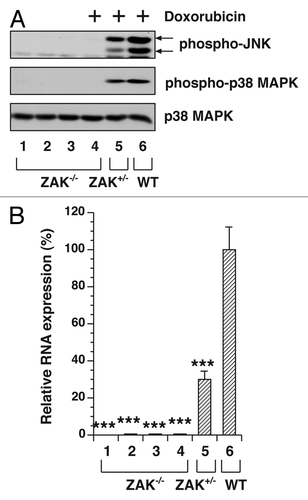
We previously demonstrated that suppression of ZAK levels by siRNA strongly reduced doxorubicin-mediated MAPK activation in a human keratinocyte cell line,Citation17 suggesting that ZAK signaling is essential for activation of JNK and p38 MAPK in these cells. To determine whether ZAK is similarly required in doxorubicin-mediated MAPK activation and inflammatory gene expression in macrophages, we performed experiments using BMDM isolated from WT, ZAK+/− and ZAK−/− mice (). To determine whether doxorubicin would induce the phosphorylation of JNK and p38 MAPK in macrophages deficient in ZAK, we employed a supramaximal dose of doxorubicin (500 μM) in order to determine the extent to which maximal stimulation of JNK and p38 MAPK would depend on the presence of ZAK. Compared with doxorubicin-induced phosphorylation of JNK and p38 MAPK in WT cells (lane 6), the phosphorylation of these kinases in ZAK−/− cells (lane 4) was undetectable after treatment with doxorubicin at even as high as 500 μM, whereas ZAK+/− (lane 5) showed intermediate levels of phospho-JNK and phospho-p38 MAPK (). These experiments demonstrated a requirement for ZAK in stimulating doxorubicin-mediated phosphorylation of JNK and p38 MAPK. Because a useful antibody against ZAK protein is not commercially available, we employed real-time RT-PCR to confirm the absence of expression of ZAK RNA from ZAK−/− mice and the reduction of ZAK RNA from ZAK+/− mice ().
Figure 2. MAPK activation in WT, ZAK+/− and ZAK−/− BMDM. Serum-deprived BMDM were untreated or treated with 500 μM doxorubicin for 3 h. (A) Western blot of cell lysates from ZAK−/− (lanes 1 and 4), ZAK+/− (lanes 2 and 5) and WT (lanes 3 and 6) mice. (B) Measurement of gene expression in cells from four individual ZAK−/− mice (lanes 1–4), one ZAK+/− mouse (lane 5) and one WT mouse (lane 6) using real-time RT-PCR. Mean values ± SD are shown. *** p < 0.001 compared with values of the corresponding control treatments.
Small molecule kinase inhibitors block MAPK phosphorylation and inflammatory gene expression in a dose-dependent manner following exposure to doxorubicin
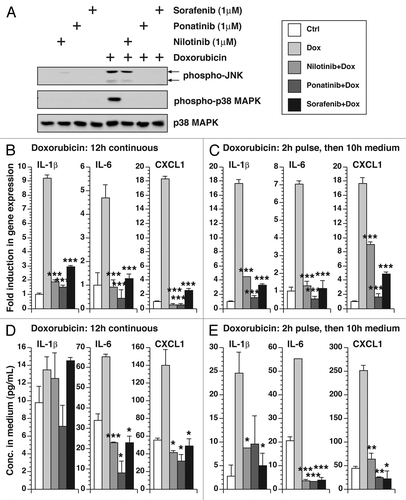
We tested the ability of small molecule kinase inhibitors to suppress the activation of MAPKs and the expression of inflammatory cytokines in BMDM by doxorubicin. Nilotinib, ponatinib and sorafenib all reduced the doxorubicin-mediated phosphorylation of JNK and p38 MAPK, although the inhibition of JNK by nilotinib was partial (). These inhibitors were also able to suppress the expression of IL-1β, IL-6 and CXCL1 RNAs, whether doxorubicin was administered continuously () or only for 2 h (). The inhibition of CXCL1 RNA was partial if doxorubicin was administered in a 2-h pulse. The inhibitors were also effective in reducing secreted IL-1β, IL-6 and CXCL1 proteins to basal levels (), demonstrating the ability of these inhibitors to reduce the expression of the inflammatory RNAs and their encoded proteins.
Figure 3. Effect of small molecule kinase inhibitors on doxorubicin-mediated MAPK phosphorylation, gene expression and cytokine secretion. (A) Western blot of cell lysates from BMDM treated with 500 μM doxorubicin for 3 h in the presence or absence of 1 µM nilotinib, ponatinib or sorafenib. B, measurement of gene expression in cells untreated or treated continuously with 5 µM doxorubicin for 12 h in the presence or absence of 1 µM nilotinib, ponatinib or sorafenib using real-time RT-PCR. (C) Measurement of gene expression in cells untreated or treated with 5 µM of doxorubicin for 2 h in the presence or absence of 1 µM nilotinib, ponatinib or sorafenib then washed away and replaced by medium in the presence or absence of 1 µM nilotinib, ponatinib or sorafenib for 10 h. (D) Cytokine measurement of the culture supernatant from (B) using bead-based multiplex assay. (E) Cytokine measurement of the culture supernatant from (C). Mean values ± SD are shown. * p < 0.005; ** p < 0.01; *** p < 0.001 compared with values corresponding to treatment with doxorubicin alone.
Nilotinib inhibits doxorubicin-mediated inflammatory responses in vivo
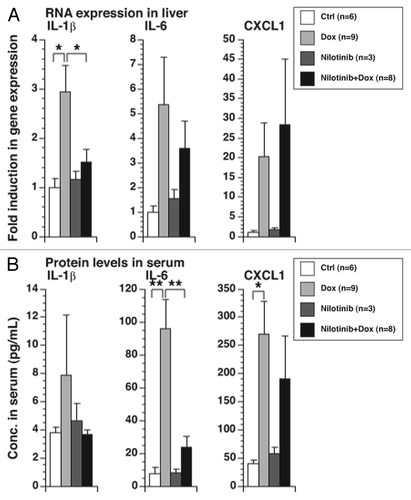
Doxorubicin administration in mice results in an increase in IL-1β and several other pro-inflammatory cytokines and chemokines.Citation17 To determine whether nilotinib could block the inflammatory effects of doxorubicin in vivo, mice were pretreated with nilotinib prior to injection with doxorubicin. Levels of IL-1β RNA in the liver were increased after doxorubicin treatment, decreasing to basal levels if the mice were pre-treated with nilotinib. In contrast, differences in levels of IL-6 and CXCL1 RNA were not statistically significant between treatment groups (). Serum levels of IL-6 protein increased after doxorubicin treatment and decreased to basal levels if the mice were pretreated with nilotinib (). In contrast, levels of IL-1β and CXCL1 proteins did not show statistically significant differences between treatment groups ().
Figure 4. Effect of nilotinib in doxorubicin-mediated RNA expression and cytokine production in mice. Mice were treated with 0.1 mL of vehicle or nilotinib (75 mg/kg) daily for 6 d by oral gavage. On the sixth day, mice were also injected with saline or doxorubicin (25 mg/kg). Sixteen hours post-injection, blood was collected by cardiac puncture and livers were snap-frozen for RNA isolation. (A) Measurement of gene expression in liver samples using real-time PCR. (B) Cytokine measurement of serum samples using bead-based multiplex assay. Mean values ± SEM are shown. * p < 0.005; ** p < 0.01.
Discussion
In clinical cancer therapy, peak plasma concentration of doxorubicin ranges between 5–15 μM and has an average half-life of 25 h.Citation31,Citation32 In experiments reported herein, we found that exposure of macrophages to a clinically relevant dose of doxorubicin (5 μM) resulted in phosphorylation of JNK and p38 MAPK () and in the increased expression of RNA-encoding inflammatory mediators, when examined 24 h post-addition ( and ). A similar rationale was employed to determine the ability of sorafenib, nilotinib and ponatinib to suppress MAPK activation ().
Despite a difference in the phosphorylation levels of MAPKs (), levels of IL-1β, IL-6 and CXCL1 RNA were comparable whether doxorubicin was present continuously for 12 h or only for a 2-h pulse (, and ). Therefore, sustained MAPK activation does not appear to be necessary for the expression of these inflammatory mediators. Interestingly, an increase in secreted IL-1β protein was only detected when cells were given an acute 2-h dose of doxorubicin (). The absence of IL-1β protein after extended exposure to doxorubicin may be explained by a gradual decrease in translation caused by doxorubicin.Citation17
The expression of many inflammatory mediators is regulated by MAPKs.Citation9-Citation11 Levels of phosphorylation of both JNK and p38 MAPK in doxorubicin-treated BMDM from ZAK−/− mice were at nearly basal levels compared with those from WT mice even at an extremely high doses of doxorubicin (), confirming that the presence of ZAK was required for doxorubicin-mediated activation of JNK and p38 MAPK. Using siRNA in different cell types, previous studies have shown that agents that cause ribotoxic stress,Citation38 including ricin, Shiga toxin, anisomycin, UV radiation, daunorubicin and doxorubicin, all require ZAK to transduce signals between ribosomes and MAPKs,Citation6,Citation39,Citation40 thereby supporting the selection of ZAK as a unique target to block the downstream events associated with ribotoxic stress.
We previously reported that nilotinib and sorafenib block doxorubicin-mediated phosphorylation of JNK and p38 MAPK in human keratinocytes.Citation17 Because nilotinib targets BCR-ABLCitation25 and sorafenib targets VEGFR and PDGFR,Citation27 we suspected that ponatinib, which targets BCR-ABL, VEGFR and PDGFR,Citation28 may bind with high affinity to other kinases that are targets of nilotinib. In the present study, we confirmed that sorafenib and nilotinib inhibited doxorubicin-mediated inflammatory responses in mouse macrophages and further found that ponatinib was also a potent inhibitor (). Unlike inhibitors directed against either p38 MAPK or JNK,Citation13,Citation14 the small molecule kinase inhibitors employed in the current study blocked the phosphorylation of both JNK and p38 MAPK, suggesting that they inhibit one or more kinases that are situated further upstream. Nilotinib was also able to suppress doxorubicin-induced activation of MAPKs and expression of inflammatory mediators in vivo, although the effects that we detected were limited to suppression of IL-1β RNA in the liver and IL-6 protein levels in the serum (). Additional experiments are necessary to determine the anti-inflammatory property of ponatinib and sorafenib in vivo. Nilotinib and sorafenib are currently used in human patients to treat chronic myeloid leukemia and hepatocellular carcinoma, respectively. Ponatinib is currently in clinical trials as a treatment of chronic myeloid leukemia.Citation28 Because these inhibitors are well-tolerated and have relatively few known side effects, they may be attractive candidates for suppressing the proinflammatory side effects of doxorubicin that may be responsible for cancer treatment-related fatigue and associated symptoms. Because doxorubicin-induced cardiotoxicity has been associated with increased levels of IL-1β,Citation5,Citation7 these agents may also prove to be effective in reducing cardiotoxicity, although our studies have not sought to determine the association of ZAK or inflammatory cytokines with the cytotoxic actions of doxorubicin in relevant cell types.
Nilotinib and sorafenib have strong binding affinities to ZAK.Citation22-Citation24 Direct measurement of the ability of these inhibitors to suppress ZAK kinase activity is needed. It is also currently unclear whether the anti-inflammatory effects of these small molecule kinase inhibitors result exclusively from inhibition of ZAK or from blocking the activity of other kinases that activate the inflammatory cascade in response to doxorubicin.
Although nilotinib, ponatinib and sorafenib may be able to decrease the side effects of doxorubicin, there is an undesired possibility that they may also lower the ability of doxorubicin to kill cancer cells. However, nilotinib has been found to synergize with doxorubicin in the induction of apoptosis in human synovial sarcoma cells and leiomyosarcoma cells.Citation41 In addition, sorafenib and doxorubicin have been used together in a randomized human trial in the treatment of advanced hepatocellular carcinoma.Citation42 A reduction in adverse effects is not reported, but the median time to progression, overall survival and progression-free survival are all extended for patients when doxorubicin is used in combination with sorafenib compared with monotherapy with doxorubicin. Different treatment regimens need to be tested to achieve the best outcome. For example, it may be desirable to consider pretreatment with an inhibitor prior to the administration of doxorubicin as performed in our mouse model or to use a lower dose of the inhibitor so that the side effects of the inhibitor are minimized while maintaining the ability to reduce the side effects of doxorubicin at the same time.
| Abbreviations: | ||
| BMDM | = | bone marrow-derived macrophages |
| IL-1RA | = | IL-1 receptor antagonist |
| JNK | = | Jun N-terminal kinases |
| MAP2K | = | MAPK kinase |
| MAP3K | = | MAPK kinase kinase |
| MAPK | = | mitogen-activated protein kinase |
| PDGFR | = | platelet-derived growth factor receptor |
| TCA | = | trichloroacetic acid |
| VEGFR | = | vascular endothelial growth factor receptor |
Acknowledgments
These studies were supported by grants AI1059335 (to B.E.M.) and R01NR012479 (to L.J.W.) from the National Institutes of Health and grant RSGPB-05–212–01-CPPB (to L.J.W.) from the American Cancer Society.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Lipshultz SE, Miller TL, Scully RE, Lipsitz SR, Rifai N, Silverman LB, et al. Changes in cardiac biomarkers during doxorubicin treatment of pediatric patients with high-risk acute lymphoblastic leukemia: associations with long-term echocardiographic outcomes. J Clin Oncol 2012; 30:1042 - 9; http://dx.doi.org/10.1200/JCO.2010.30.3404; PMID: 22370326
- Bruynzeel AM, Abou El Hassan MA, Schalkwijk C, Berkhof J, Bast A, Niessen HW, et al. Anti-inflammatory agents and monoHER protect against DOX-induced cardiotoxicity and accumulation of CML in mice. Br J Cancer 2007; 96:937 - 43; http://dx.doi.org/10.1038/sj.bjc.6603640; PMID: 17325706
- Mann DL, Young JB. Basic mechanisms in congestive heart failure. Recognizing the role of proinflammatory cytokines. Chest 1994; 105:897 - 904; http://dx.doi.org/10.1378/chest.105.3.897; PMID: 8131560
- Matsumori A, Yamada T, Suzuki H, Matoba Y, Sasayama S. Increased circulating cytokines in patients with myocarditis and cardiomyopathy. Br Heart J 1994; 72:561 - 6; http://dx.doi.org/10.1136/hrt.72.6.561; PMID: 7857740
- Zhu J, Zhang J, Zhang L, Du R, Xiang D, Wu M, et al. Interleukin-1 signaling mediates acute doxorubicin-induced cardiotoxicity. Biomed Pharmacother 2011; 65:481 - 5; http://dx.doi.org/10.1016/j.biopha.2011.06.005; PMID: 22000485
- Sauter KA, Wood LJ, Wong J, Iordanov M, Magun BE. Doxorubicin and daunorubicin induce processing and release of interleukin-1β through activation of the NLRP3 inflammasome. Cancer Biol Ther 2011; 11:1008 - 16; http://dx.doi.org/10.4161/cbt.11.12.15540; PMID: 21464611
- Zhu J, Zhang J, Xiang D, Zhang Z, Zhang L, Wu M, et al. Recombinant human interleukin-1 receptor antagonist protects mice against acute doxorubicin-induced cardiotoxicity. Eur J Pharmacol 2010; 643:247 - 53; http://dx.doi.org/10.1016/j.ejphar.2010.06.024; PMID: 20599921
- Schubert C, Hong S, Natarajan L, Mills PJ, Dimsdale JE. The association between fatigue and inflammatory marker levels in cancer patients: a quantitative review. Brain Behav Immun 2007; 21:413 - 27; http://dx.doi.org/10.1016/j.bbi.2006.11.004; PMID: 17178209
- Baldassare JJ, Bi Y, Bellone CJ. The role of p38 mitogen-activated protein kinase in IL-1 beta transcription. J Immunol 1999; 162:5367 - 73; PMID: 10228013
- Kent LM, Smyth LJ, Plumb J, Clayton CL, Fox SM, Ray DW, et al. Inhibition of lipopolysaccharide-stimulated chronic obstructive pulmonary disease macrophage inflammatory gene expression by dexamethasone and the p38 mitogen-activated protein kinase inhibitor N-cyano-N’-(2-[8-(2,6-difluorophenyl)-4-(4-fluoro-2-methylphenyl)-7-oxo-7,8-dihydropyrido[2,3-d] pyrimidin-2-yl]aminoethyl)guanidine (SB706504). J Pharmacol Exp Ther 2009; 328:458 - 68; http://dx.doi.org/10.1124/jpet.108.142950; PMID: 19004925
- Obata T, Brown GE, Yaffe MB. MAP kinase pathways activated by stress: the p38 MAPK pathway. Crit Care Med 2000; 28:Suppl N67 - 77; http://dx.doi.org/10.1097/00003246-200004001-00008; PMID: 10807318
- Elsea CR, Roberts DA, Druker BJ, Wood LJ. Inhibition of p38 MAPK suppresses inflammatory cytokine induction by etoposide, 5-fluorouracil, and doxorubicin without affecting tumoricidal activity. PLoS One 2008; 3:e2355; http://dx.doi.org/10.1371/journal.pone.0002355; PMID: 18523641
- Kaminska B. MAPK signalling pathways as molecular targets for anti-inflammatory therapy--from molecular mechanisms to therapeutic benefits. Biochim Biophys Acta 2005; 1754:253 - 62; http://dx.doi.org/10.1016/j.bbapap.2005.08.017; PMID: 16198162
- Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer 2009; 9:537 - 49; http://dx.doi.org/10.1038/nrc2694; PMID: 19629069
- Avdi NJ, Malcolm KC, Nick JA, Worthen GS. A role for protein phosphatase-2A in p38 mitogen-activated protein kinase-mediated regulation of the c-Jun NH(2)-terminal kinase pathway in human neutrophils. J Biol Chem 2002; 277:40687 - 96; http://dx.doi.org/10.1074/jbc.M204455200; PMID: 12186863
- Lahti A, Sareila O, Kankaanranta H, Moilanen E. Inhibition of p38 mitogen-activated protein kinase enhances c-Jun N-terminal kinase activity: implication in inducible nitric oxide synthase expression. BMC Pharmacol 2006; 6:5; http://dx.doi.org/10.1186/1471-2210-6-5; PMID: 16504051
- Sauter KA, Magun EA, Iordanov MS, Magun BE. ZAK is required for doxorubicin, a novel ribotoxic stressor, to induce SAPK activation and apoptosis in HaCaT cells. Cancer Biol Ther 2010; 10:258 - 66; http://dx.doi.org/10.4161/cbt.10.3.12367; PMID: 20559024
- Bloem LJ, Pickard TR, Acton S, Donoghue M, Beavis RC, Knierman MD, et al. Tissue distribution and functional expression of a cDNA encoding a novel mixed lineage kinase. J Mol Cell Cardiol 2001; 33:1739 - 50; http://dx.doi.org/10.1006/jmcc.2001.1437; PMID: 11549352
- Gotoh I, Adachi M, Nishida E. Identification and characterization of a novel MAP kinase kinase kinase, MLTK. J Biol Chem 2001; 276:4276 - 86; http://dx.doi.org/10.1074/jbc.M008595200; PMID: 11042189
- Gross EA, Callow MG, Waldbaum L, Thomas S, Ruggieri R. MRK, a mixed lineage kinase-related molecule that plays a role in gamma-radiation-induced cell cycle arrest. J Biol Chem 2002; 277:13873 - 82; http://dx.doi.org/10.1074/jbc.M111994200; PMID: 11836244
- Liu TC, Huang CJ, Chu YC, Wei CC, Chou CC, Chou MY, et al. Cloning and expression of ZAK, a mixed lineage kinase-like protein containing a leucine-zipper and a sterile-alpha motif. Biochem Biophys Res Commun 2000; 274:811 - 6; http://dx.doi.org/10.1006/bbrc.2000.3236; PMID: 10924358
- Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol 2008; 26:127 - 32; http://dx.doi.org/10.1038/nbt1358; PMID: 18183025
- Manley PW, Drueckes P, Fendrich G, Furet P, Liebetanz J, Martiny-Baron G, et al. Extended kinase profile and properties of the protein kinase inhibitor nilotinib. Biochim Biophys Acta 2010; 1804:445 - 53; http://dx.doi.org/10.1016/j.bbapap.2009.11.008; PMID: 19922818
- Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, et al. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol 2011; 29:1046 - 51; http://dx.doi.org/10.1038/nbt.1990; PMID: 22037378
- Weisberg E, Manley P, Mestan J, Cowan-Jacob S, Ray A, Griffin JD. AMN107 (nilotinib): a novel and selective inhibitor of BCR-ABL. Br J Cancer 2006; 94:1765 - 9; http://dx.doi.org/10.1038/sj.bjc.6603170; PMID: 16721371
- Blay JY, von Mehren M. Nilotinib: a novel, selective tyrosine kinase inhibitor. Semin Oncol 2011; 38:Suppl 1 S3 - 9; http://dx.doi.org/10.1053/j.seminoncol.2011.01.016; PMID: 21419934
- Ranieri G, Gadaleta-Caldarola G, Goffredo V, Patruno R, Mangia A, Rizzo A, et al. Sorafenib (BAY 43-9006) in hepatocellular carcinoma patients: from discovery to clinical development. Curr Med Chem 2012; 19:938 - 44; http://dx.doi.org/10.2174/092986712799320736; PMID: 22214462
- O’Hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM, Wang F, et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell 2009; 16:401 - 12; http://dx.doi.org/10.1016/j.ccr.2009.09.028; PMID: 19878872
- Johansen PB. Doxorubicin pharmacokinetics after intravenous and intraperitoneal administration in the nude mouse. Cancer Chemother Pharmacol 1981; 5:267 - 70; http://dx.doi.org/10.1007/BF00434396; PMID: 7261254
- Korcheva V, Wong J, Corless C, Iordanov M, Magun B. Administration of ricin induces a severe inflammatory response via nonredundant stimulation of ERK, JNK, and P38 MAPK and provides a mouse model of hemolytic uremic syndrome. Am J Pathol 2005; 166:323 - 39; http://dx.doi.org/10.1016/S0002-9440(10)62256-0; PMID: 15632024
- Vrignaud P, Eghbali H, Hoerni B, Iliadis A, Robert J. Pharmacokinetics and metabolism of epirubicin during repetitive courses of administration in Hodgkin’s patients. Eur J Cancer Clin Oncol 1985; 21:1307 - 13; http://dx.doi.org/10.1016/0277-5379(85)90309-8; PMID: 3865777
- Mross K, Maessen P, van der Vijgh WJ, Gall H, Boven E, Pinedo HM. Pharmacokinetics and metabolism of epidoxorubicin and doxorubicin in humans. J Clin Oncol 1988; 6:517 - 26; PMID: 3162516
- Kalyanaraman B, Joseph J, Kalivendi S, Wang S, Konorev E, Kotamraju S. Doxorubicin-induced apoptosis: implications in cardiotoxicity. Mol Cell Biochem 2002; 234-235:119 - 24; http://dx.doi.org/10.1023/A:1015976430790; PMID: 12162424
- Hassan F, Islam S, Mu MM, Ito H, Koide N, Mori I, et al. Lipopolysaccharide prevents doxorubicin-induced apoptosis in RAW 264.7 macrophage cells by inhibiting p53 activation. Mol Cancer Res 2005; 3:373 - 9; http://dx.doi.org/10.1158/1541-7786.MCR-05-0046; PMID: 16046548
- Riad A, Bien S, Westermann D, Becher PM, Loya K, Landmesser U, et al. Pretreatment with statin attenuates the cardiotoxicity of Doxorubicin in mice. Cancer Res 2009; 69:695 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-08-3076; PMID: 19147586
- Riad A, Bien S, Gratz M, Escher F, Westermann D, Heimesaat MM, et al. Toll-like receptor-4 deficiency attenuates doxorubicin-induced cardiomyopathy in mice. Eur J Heart Fail 2008; 10:233 - 43; http://dx.doi.org/10.1016/j.ejheart.2008.01.004; PMID: 18321777
- Korcheva V, Wong J, Lindauer M, Jacoby DB, Iordanov MS, Magun B. Role of apoptotic signaling pathways in regulation of inflammatory responses to ricin in primary murine macrophages. Mol Immunol 2007; 44:2761 - 71; http://dx.doi.org/10.1016/j.molimm.2006.10.025; PMID: 17257680
- Iordanov MS, Pribnow D, Magun JL, Dinh TH, Pearson JA, Chen SL, et al. Ribotoxic stress response: activation of the stress-activated protein kinase JNK1 by inhibitors of the peptidyl transferase reaction and by sequence-specific RNA damage to the alpha-sarcin/ricin loop in the 28S rRNA. Mol Cell Biol 1997; 17:3373 - 81; PMID: 9154836
- Jandhyala DM, Ahluwalia A, Obrig T, Thorpe CM. ZAK: a MAP3Kinase that transduces Shiga toxin- and ricin-induced proinflammatory cytokine expression. Cell Microbiol 2008; 10:1468 - 77; http://dx.doi.org/10.1111/j.1462-5822.2008.01139.x; PMID: 18331592
- Wang X, Mader MM, Toth JE, Yu X, Jin N, Campbell RM, et al. Complete inhibition of anisomycin and UV radiation but not cytokine induced JNK and p38 activation by an aryl-substituted dihydropyrrolopyrazole quinoline and mixed lineage kinase 7 small interfering RNA. J Biol Chem 2005; 280:19298 - 305; http://dx.doi.org/10.1074/jbc.M413059200; PMID: 15737997
- Villar VH, Vögler O, Martínez-Serra J, Ramos R, Calabuig-Fariñas S, Gutiérrez A, et al. Nilotinib counteracts P-glycoprotein-mediated multidrug resistance and synergizes the antitumoral effect of doxorubicin in soft tissue sarcomas. PLoS One 2012; 7:e37735; http://dx.doi.org/10.1371/journal.pone.0037735; PMID: 22662203
- Abou-Alfa GK, Saltz LB. Doxorubicin and sorafenib for treatment of advanced hepatocellular cancer. Gastroenterology 2011; 141:e19 - 20, author reply e20-1; http://dx.doi.org/10.1053/j.gastro.2011.04.063; PMID: 21794834