Abstract
EGFR is a validated therapeutic target in many human cancers. EGFR targeted therapies are in widespread clinical use in patients with non-small cell lung cancer and other tumor types. Despite the clinical success of EGFR targeted therapy, resistance to treatment is a significant barrier to the optimized use of EGFR inhibitors to cure patients with lung and other cancers. Here, we review established and emerging mechanisms of resistance to EGFR targeted therapy and highlight strategies that could overcome treatment resistance and therefore enhance clinical outcomes.
Introduction
Lung cancer accounted for approximately one third of all cancer deaths in the United States in 2011, making it the foremost cause of cancer mortality in the United States and worldwide. Unfortunately, less than 15% of patients with lung cancer survive five years.Citation1-Citation3 Thus, conventional approaches to the diagnosis and management of lung cancer patients have achieved limited success. Until recently, lung cancer was primarily diagnostically classified on a histological basis as either non-small cell (NSCLC, ~85% of cases) or small cell (SCLC, ~15% of cases). NSCLC encompasses adenocarcinoma, squamous cell carcinoma and large-cell carcinoma.Citation4 Patients with either advanced stage NSCLC or SCLC traditionally have been treated with systemic cytotoxic chemotherapy. This approach has been largely unsuccessful and resulted in heterogeneous, incomplete and transient clinical responses (with few notable exceptions) and little progress in reducing lung cancer-related mortality. Over the past decade, the identification of specific genetic alterations (oncogene drivers) in lung (and other) cancers has paved the way for a new classification of lung cancer wherein distinct clinical subsets are defined by molecular criteria. These discoveries have simultaneously led to the development of more effective therapies that capitalize on the key tumor-specific genetic changes upon which tumor growth is dependent (oncogene dependence).Citation5 Thus, clinical practice has moved into an era in which individualized, precision approaches to the diagnosis and treatment of many lung cancer patients are leading to improved clinical outcomes, although resistance to these personalized approaches also develops. In this review, we will focus on mechanisms of drug resistance in EGFR-driven lung cancers and briefly address the role of EGFR signaling and EGFR-directed therapies in other cancers.
Somatic activating mutations in the epidermal growth factor receptor (EGFR) gene occur in approximately 15–30% of NSCLCs.Citation6 EGFR belongs to the ErbB/HER family of ligand-activated receptor tyrosine kinases (RTKs), whose members play important roles in the regulation of cellular processes including cell proliferation, survival and migration. Regulation of EGFR kinase activity can be disrupted by several mechanisms, including increased production of ligands (i.e., EGF, TGFα and Heregulin), upregulation of EGFR expression, impaired downregulation of EGFR, cross-talk with other receptors (i.e., other ErbB family members, RTKs, cell adhesion molecules, cytokine receptors, ion channels and G-protein coupled receptors) and activating mutations.Citation7 Since 2004, several activating lesions in EGFR have been identified in exons 18 to 21 and are localized around the ATP-binding site of the kinase domain.Citation8,Citation9 The most common mutations are an in-frame deletion within exon 19 that removes residues amino acids 746–750 (del746–750) and an exon 21 missense mutation replacing leucine 858 with arginine (L858R).Citation10 The crystal structures of the L858R and G719S EGFR mutants provide evidence that these mutations increase kinase activity by 50- and 10-fold, respectively, through abrogation of autoinhibitory interactions proximal to the ATP-binding cleft of the EGFR protein.Citation11 Additionally, the EGFR juxtamembrane domain functions differently from other receptor tyrosine kinases in that it activates, rather than inhibits, the kinase domain through stabilization of an asymmetric kinase dimer.Citation12,Citation13 Indeed, a lung cancer mutation (V665M) identified within the activating region of the EGFR juxtamembrane domain has been shown to activate EGFR constitutively through increased receptor dimerization.Citation14 Thus far, the crystal structure of EGFR (del746–750) has not been determined.
Targeting EGFR Mutations
EGFR-mutant NSCLC cells are a characteristic example of oncogene dependence because these cells are addicted to the aberrant signaling of the mutant kinase for their growth and survival. This characteristic provides a tumor-specific vulnerability and therapeutic opportunity to target mutant EGFR specifically in the tumor cells while sparing normal cells. The ATP-competitive EGFR tyrosine kinase inhibitors (TKIs), erlotinib (and gefitinib in Europe and Asia), are currently approved for first-line use in lung cancer patients whose tumors harbor an EGFR activating mutation. These small molecule inhibitors of EGFR achieve clinical efficacy by suppressing the activity of the mutant kinase and downregulating downstream signaling.Citation15-Citation17 Importantly, activating EGFR mutations show significantly greater sensitivity to treatment with EGFR TKIs than wild type EGFR. For example, the EGFR L858R mutant is approximately 100 times more sensitive to EGFR TKI inhibition than wild type EGFR.Citation18-Citation20 This exquisite response is attributed to the preferential binding of TKI-sensitive mutants to erlotinib or gefitinib vs. ATP.Citation4 Since the presence of activating EGFR mutations is associated with enhanced clinical responsiveness to EGFR TKI therapy,Citation15-Citation17,Citation21 initial molecular analysis of NSCLC tumors for activating EGFR mutations is now standard of care for patients with advanced NSCLC.Citation22
Another approach to targeted EGFR inhibition is the use of monoclonal antibodies that bind to the extracellular domain of EGFR. Antibody binding prevents ligand-induced receptor dimerization and activation or induces receptor endocytosis and/or degradation. Cetuximab is a monoclonal human-murine chimeric antibody approved for the treatment of colorectal cancer and head and neck squamous cell carcinoma (HNSCC), but its effectiveness in EGFR-mutant NSCLC has been limited to date.Citation23-Citation25 Cetuximab interferes with EGFR ligand-binding and activation of downstream signaling cascades and may also activate the complement pathway to induce antibody-dependent cytotoxicity. Panitumumab is a similar, but fully humanized, monoclonal antibody directed against EGFR that has been approved for the treatment of colorectal cancers that harbor wild type KRAS. Panitumumab also acts by blocking the binding of EGFR ligands to EGFR, but in contrast with cetuximab, does not lead to activation of the complement pathway.Citation26,Citation27 Unlike the positive correlation between EGFR mutations and response to EGFR TKIs, the role of EGFR mutations as predictive markers for sensitivity to anti-EGFR antibodies has not been demonstrated. While antibodies are capable of blocking ligand-induced receptor activation and promoting antibody-mediated receptor downregulation, mutations that confer ligand independence, such as those found in EGFR in NSCLCs, may abrogate the efficacy of anti-EGFR antibody therapy.Citation28
Mechanisms of Resistance to EGFR-Targeted Therapies
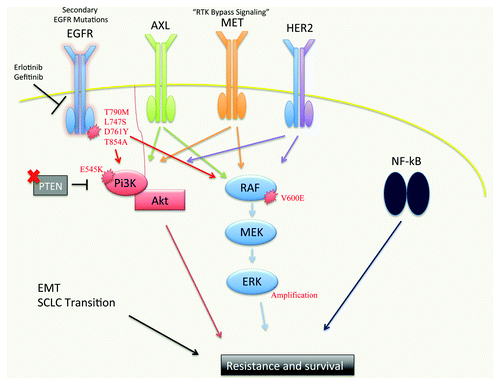
Despite the effectiveness of EGFR TKIs in NSCLC patients with EGFR activating mutations, the success of EGFR-targeted therapy is limited by the challenge of drug resistance (). Some patients exhibit de novo resistance to EGFR inhibition and are refractory to therapy that is expected to be effective based on the biology or genetics of the cancer. Furthermore, patients who initially respond to therapy invariably develop acquired resistance to treatment.
Figure 1. A schematic of the molecular pathways implicated in resistance to EGFR TKIs. Mutations in the EGFR kinase domain (predominantly T790M, which accounts for 50–60% of EGFR inhibitor resistance) abrogate the ability of first generation EGFR TKIs to inhibit mutant EGFR. Second generation EGFR TKIs BIBW2992, PF299804 and WZ4002 are currently in clinical trials and show promise as inhibitors of EGFR T790M. EGFR TKI resistance can also occur via upregulation or activation of other RTKs, such as AXL (20–25%), MET (5%) and HER2, which can bypass the inhibition of oncogenic EGFR signaling and activate downstream effector pathways. RTK-independent activation of some of these downstream effectors can also occur, leading to resistance. PTEN loss and activating PI3K mutations (i.e., E545K) have been observed, leading to constitutive AKT activation. Activating BRAF V600E mutations and MAPK1 amplification have been reported which lead to hyper-activation of MAPK signaling and resistance. NFκB pathway activation has been associated with resistance to EGFR TKI treatment. The Epithelial-Mesenchymal Transition (EMT) and transition to small-cell neuroendocrine phenotype have each been associated with resistance to EGFR TKIs, though the mechanistic underpinnings of these observations are unclear.
Secondary genetic alterations in EGFR
NSCLCs harboring mutant EGFR can exhibit de novo resistance to EGFR TKI therapy due to the nature of the primary activating lesion or the presence of a secondary drug-resistant EGFR mutation (). NSCLCs that are initially sensitive to EGFR TKI treatment have EGFR mutations primarily in exons 18, 19 and 21, whereas NSCLCs with infrequent exon 20 insertions or duplications are less responsive to EGFR TKI treatment both in vitroCitation18 and in patients.Citation29 Likewise, the T790M substitution in exon 20 is occasionally detected in a minor cell population in tumors displaying de novo drug resistanceCitation30 and can also be found at low frequency in circulating tumor cells captured from EGFR TKI naïve patients with metastatic disease.Citation31 Interestingly, the presence of a germline EGFR T790M mutation associated with familial NSCLC may enhance the effect of other EGFR activating mutations that occur in cis,Citation32 as well as increase the risk of developing NSCLC.Citation33-Citation35 In addition to T790M, other secondary mutations in EGFR may cause primary resistance. For example, the presence of D761Y in cis with the drug-sensitive L858R mutation reduces EGFR sensitivity to EGFR TKIs.Citation36 Altogether, these findings suggest that early detection of small fractions of second-site mutant alleles could be used for predicting response to NSCLCs that harbor drug-sensitive EGFR mutations. It is also becoming apparent that the intrinsic nature of different EGFR mutants elicits varying responses to EGFR TKI therapy,Citation11 and that successful treatment of lung cancers with drug-resistant mutations will require alternative strategies for EGFR kinase inhibition.
Table 1. EGFR alterations in cancer and mechanisms of resistance to EGFR TKIs
The second-site T790M mutation accounts for acquired EGFR TKI resistance in over 50% of NSCLC patients with EGFR-mutant tumors who initially respond to erlotinib or gefitinib. This amino acid change of a highly conserved “gatekeeper” residue near the kinase active site is frequently detected in TKI-resistant tumors. The T790M mutation in EGFR is analogous to the T315I substitution found in BCR-ABL in chronic myelogenous leukemia patients who have acquired resistance to the ABL kinase inhibitor imatinib.Citation37 While T315I causes a steric clash that blocks imatinib from binding in the ATP pocket of ABL kinase,Citation38 EGFR T790M most likely confers drug resistance primarily by enhancing the ATP affinity of EGFR L858R and thus reducing the efficacy of an ATP-competitive kinase inhibitor.Citation39 Although EGFR T790M mutations are rarely detected in EGFR TKI naïve patients, continuous treatment with erlotinib or gefitinib may promote positive selection and proliferation of even a small fraction of T790M-harboring cells present at the start of EGFR TKI therapy.Citation30
Other secondary mutations in EGFR have been reported in cases of acquired resistance, but at much lower frequency than EGFR T790M. These EGFR mutations include the exon 19 mutations L747SCitation40 and D761Y,Citation36 and the exon 21 mutation T854A.Citation41 Crystallographic studies indicate that L747S could affect the binding of ATP and small molecules or shift the conformational equilibrium of EGFR toward the active state, while D761Y is situated in the putative α-C helix and predicted to affect the kinase activation loop.Citation42 The T854A substitution in the ATP-binding site may cause loss of contact and binding affinity to EGFR TKIs due to the replacement of the bulky threonine with the smaller alanine. Alternatively, the T854A mutation could induce a local conformational change in the kinase that abrogates EGFR TKI efficacy.
Attempts have been made to inhibit EGFR T790M using second-generation irreversible quinazoline-based EGFR TKIs, including afatinib (BIBW2992), dacomitinib (PF299804), canertinib (CI-1033) and neratinib (HKI-272).Citation43-Citation45 While clinical studies showed that afatinib did not increase survival in NSCLC patients who developed resistance to erlotinib or gefitinib,Citation45 dual targeting of EGFR with afatinib and the monoclonal antibody cetuximab has recently been reported to be effective at targeting T790M-driven tumors in mouse models of T790M-mediated resistance and in some patients.Citation46,Citation47 Unfortunately, preclinical studies indicate that EGFR T790M mutant tumor cells quickly acquire resistance to dacomitinib through amplification of the T790M allele.Citation48 The limited clinical efficacy of these inhibitors may be attributed to the increased potency of these agents against both mutant and wild type EGFR. As the ATP affinity of EGFR T790M is similar to that of wild type EGFR, the inhibitor concentration needed to suppress EGFR T790M also inhibits the wild type EGFR protein, resulting in dose-limiting toxicity in the form of skin rash and diarrhea.Citation49 Consequently, targeting EGFR T790M with these irreversible inhibitors has had limited clinical efficacy thus far.
The toxicity ascribed to the concurrent inhibition of wild type and T790M-mutant EGFR proteins underscores the need to develop small molecule inhibitors of EGFR that selectively target tumor cells that express mutant EGFR while sparing cells that express wild type EGFR. In contrast to the quinazoline-based EGFR inhibitors, the pyrimidine-based EGFR TKI WZ4002 is approximately 100-fold more potent against EGFR T790M and 100-fold less effective against wild type EGFR.Citation49 This suggests that EGFR T790M can be sufficiently inhibited at physiological doses that will not target wild type EGFR and result in toxicity, making WZ4002 a promising therapeutic candidate.Citation4
Activation of PI3K/AKT signaling
Cell death in EGFR-mutant cancer cells is associated with a decrease in phosphoinositide-3-kinase (PI3K)/AKT signaling upon EGFR inhibition. The ability to survive in the face of inhibition of oncogenic EGFR often involves re-activation of the PI3K/AKT pathway through a variety of mechanisms in tumor cells. Loss of PTEN expression (found in approximately 4% of EGFR-mutant cases) is associated with resistance to EGFR TKI treatment through activation of the PI3K/AKT signaling pathway.Citation51,Citation52 In patients who develop EGFR TKI resistance, approximately 5% have been found to have a PIK3CA (p110alpha catalytic subunit of PI3K) mutation.Citation54 This is consistent with in vitro work showing that introduction of activated PIK3CA can confer resistance to NSCLC cell lines initially sensitive to EGFR TKI treatment.Citation53,Citation54 As we will discuss in detail below, many of the mechanisms that cause resistance to EGFR TKI treatment, including the EGFR T790M gatekeeper mutation and RTK bypass signaling, lead to activation of PI3K/AKT signaling.
Activation of MAPK signaling
Although many studies implicate PI3K/AKT as a critical pathway mediating EGFR-mutant NSCLC cell survival and drug resistance, the role of the MAPK signaling pathway in EGFR TKI resistance has been less clear until recently. However, MAPK1 amplification was recently observed in EGFR-mutant lung cancer cells that developed resistance to the irreversible EGFR inhibitor WZ4002 and also in one patient specimen with acquired resistance to erlotinib.Citation50 In these drug resistant models, treatment with a MEK or ERK inhibitor alone was not able to induce apoptosis, but required combination therapy with an EGFR inhibitor. Importantly, in these studies, combining PI3K or AKT inhibition with WZ4002 was also unable to induce apoptosis in EGFR-mutant NSCLC cells resistant to WZ4002 treatment, suggesting that therapeutic strategies must be personalized according to the specific molecular event through which resistance occurs. Additionally, there have been recent reports of patients whose resistant tumors harbor an activating BRAF V600E or G469A mutation.Citation55 Together, these findings suggest that resistance to EGFR TKI treatment can occur through reactivation of MAPK signaling.Citation50,Citation56
“Bypass Signaling” via Receptor Tyrosine Kinases (RTKs)
Upregulation of specific RTKs can promote lung tumorigenesis and drive therapeutic resistance in several cancers, including NSCLCs harboring mutant EGFR or the EML4-ALK fusion, CRC, HNSCC, breast cancer and others. Here, we review the roles of several of these RTKs, including MET, IGF1R, AXL and HER2 (ERbB2) in resistance to EGFR inhibitor treatment in NSCLC.
MET amplification
Approximately 5% of lung cancer specimens from patients who develop resistance to gefitinib or erlotinib harbor amplification of the MET RTK.Citation57 MET (also known as hepatocyte growth factor receptor, HGF-R) activation bypasses EGFR inhibition through sustained activation of the PI3K/AKT signaling pathway.Citation53,Citation58 Interestingly, MET amplification and the EGFR T790M resistance mutation can coexist in resistant tumor specimens.Citation53 EGFR-mutant lung cancer cells exposed to HGF, the ligand for MET, are resistant to EGFR TKI treatment, suggesting that MET activation is sufficient to confer resistance in otherwise drug-sensitive cells.Citation59 Interestingly, these studies have shown that MET can also act through ErbB3-mediated activation of PI3K. Further reports have shown that MET-mediated resistance to EGFR TKI treatment can be overcome by inhibition of PI3K alone or in combination with erlotinib in vitro and in vivo.Citation60 It is unclear whether EGFR-mutant cells actively “acquire” MET amplification or whether a pre-existent, minor subpopulation of tumor cells with MET amplification are selected for during EGFR TKI treatment. The latter hypothesis may be supported by the clinical observation that MET amplification is seen at low frequency in EGFR-mutant lung cancers prior to treatment and was associated with resistance to EGFR TKI treatment in these patients.Citation61
Presently, it is uncertain whether MET inhibition will be effective in overcoming acquired EGFR TKI resistance in the clinic; however, clinical trials are underway. Recently, the MET inhibitor ARQ197 showed a statistically significant increase in progression free survival in combination with erlotinib in a Phase III trial, but the trial was ultimately discontinued due to failure to satisfy the primary endpoint of overall survival benefit in this patient cohort.Citation62,Citation63 Additionally, a Phase I clinical trial combining erlotinib and the MET/VEGFR2/RET inhibitor XL184 has been completed.Citation64 In this trial, one NSCLC patient with acquired resistance to erlotinib associated with MET copy number gain was able to achieve a partial response to the combination.
IGF1R
Insulin-like growth factor receptor 1 (IGF1R) is a RTK that is known to engage in cross-talk with EGFR and activates both the PI3K/AKT and MAPK pathway.Citation65 Combined inhibition of IGF1R and EGFR induces apoptosis in a subset of EGFR-mutant lung cancer cell lines whereas inhibition of IGF1R or EGFR alone induces only cell cycle arrest.Citation66 Furthermore, loss of the inhibitory IGF-binding proteins (leading to increased IGF1R signaling) has been associated with acquired resistance to EGFR TKI treatment in squamous cell lung cancer models that overexpress wild type EGFR. Thus, IGF1R may contribute to resistance to EGFR TKI treatment in some NSCLCs. However, IGF1R has yet to be validated as a bona fide driver of resistance to EGFR TKI treatment in human clinical samples.
Activation of AXL kinase
AXL is a RTK previously implicated in resistance to imatinib in some KIT-driven gastrointestinal stromal tumor cells and in resistance to lapatinib treatment in some breast cancer cells.(Citation67,Citation68) More recently, activation of AXL has been shown to drive resistance to EGFR TKI treatment in EGFR-mutant NSCLC as well. Preclinical studies using multiple in vitro and in vivo EGFR-mutant NSCLC models of acquired EGFR TKI resistance revealed that activation of AXL either through overexpression or upregulation of its ligand GAS6 conferred drug resistance.Citation69 AXL activation resulted in a restoration of MAPK, PI3K/AKT and NFκB signaling in the EGFR-mutant NSCLC models with acquired erlotinib resistance. Combined treatment with erlotinib and an AXL inhibitor restored sensitivity in the resistant models by inducing cell death, whereas single agent treatment was ineffective. Conversely, forced overexpression of AXL in an EGFR TKI-sensitive EGFR-mutant NSCLC cell line was sufficient to induce erlotinib resistance. Increased expression of AXL and GAS6 was observed in approximately 25% of EGFR-mutant NSCLCs obtained from patients that developed acquired resistance to erlotinib or gefitinib.Citation69 The data showed that AXL upregulation is a clinically important mechanism of acquired resistance to EGFR inhibitor treatment. AXL is a novel rational therapeutic target that when inhibited may enhance response to EGFR TKI treatment in selected NSCLC patients. Current efforts are focused on the development of specific AXL inhibitors and the design of clinical trials testing AXL and EGFR inhibition in the clinic.
HER2 amplification
Studies examining the mechanisms of acquired resistance to the anti-EGFR monoclonal antibody cetuximab have led to the identification of upregulation of other ErbB family members as a potential mechanism of resistance to EGFR targeted therapies.Citation70 In these studies, initially sensitive NSCLC cell lines that developed resistance to cetuximab in vitro harbored amplification of HER2. HER2 overexpression or knockdown conferred resistance or sensitivity, respectively, to cetuximab in some NSCLC models. Interestingly, inhibition or disruption of HER2/HER3 hetero-dimerization via treatment with the monoclonal antibody pertuzumab was sufficient to restore sensitivity to cetuximab both in vitro and in xenograft models.Citation71 Furthermore, using FISH analysis, Takezawa et al. revealed that HER2 was amplified in approximately 12% of tumors with acquired resistance to EGFR TKI treatment, whereas HER2 amplification was present in only 1% of EGFR TKI naïve tumors.Citation70 Of note, HER2 amplification was not found to coexist with the EGFR T790M gatekeeper mutation in these clinical specimens.
Activation of NFκB signaling
The NFκB signaling pathway is implicated in a wide spectrum of biological functions, including immune and inflammatory responses and cancer.Citation72 In a search to better define the molecular determinants of EGFR oncogene dependence and of response and resistance to EGFR targeted therapy in NSCLC, a novel genetic approach demonstrated that NFκB pathway activation can promote primary resistance to EGFR TKI treatment in EGFR-mutant NSCLCs.Citation73 This study applied a RNA interference based screening approach using a pooled shRNA screening library to identify genes that, when knocked down, restored sensitivity to an erlotinib-resistant EGFR-mutant NSCLC cell line not harboring the EGFR T790M resistance mutation. Unexpectedly, 18 of the 36 screening hits were previously implicated in NFkB signaling, including CD95/FAS. Furthermore, NFκB activation levels were shown to be a specific predictive biomarker of response to treatment with an EGFR TKI, but not chemotherapy, in EGFR-mutant NSCLC patients. High NFκB activity as indicated by low levels of IκB, a negative regulator of NFκB signaling, identified EGFR-mutant NSCLC patients that responded poorly to EGFR TKI treatment. These results indicate that the NFκB pathway plays an important role in modulating response to EGFR inhibition by enabling EGFR-mutant tumor cells to bypass dependence on oncogenic EGFR. Combined therapy with an EGFR TKI and an inhibitor of NFκB signaling can enhance responses in some EGFR-mutant NSCLC models and is currently being pursued in clinical trials.Citation73,Citation74
The DNA Damage Response and Efficacy of EGFR TKI Treatment
Recent data suggest a role for the DNA damage response (DDR) in modulating sensitivity to EGFR inhibitor treatment. In breast cancer, it has been observed that low BRCA1 levels can be used as a biomarker for sensitivity to PARP inhibitor treatment.Citation75,Citation76 Additionally, erlotinib has been shown to suppress DNA repair in some breast cancer lines and cetuximab treatment can modulate the expression of DDR genes and sensitize to PARP inhibitor treatment in some HNSCC cell lines.Citation77 These data suggest that there could be a link between EGFR inhibition and the DDR in some EGFR-driven tumors.
In NSCLC, emerging evidence indicates that the DDR could be an important regulator of response to EGFR inhibitor treatment in some patients. Recent clinical evidence showed that EGFR-mutant NSCLC patients whose tumors harbored low BRCA1 levels achieve significantly longer progression free survival during erlotinib treatment.Citation78 A possible explanation for these clinical observations is that EGFR may activate elements of the DDR. EGFR inhibition may therefore decrease activation of the DDR, create a potential synthetically lethal relationship between EGFR inhibition and the DDR in tumor cells in which elements of the DDR, such as BRCA1, are suppressed through EGFR-independent mechanisms. Furthermore, a genetic screen identified several DDR genes that, when knocked down, can confer erlotinib sensitivity in EGFR-mutant cell lines that are otherwise resistant to the drug.Citation73 These observations provided rationale for the initiation of clinical trials assessing the efficacy of PARP inhibitor treatment in NSCLC patients with EGFR TKI-resistant tumors. Further investigation is required to explain mechanistically the clinical response observed in EGFR-mutant NSCLC patients whose tumors harbor low BRCA1 levels and to understand the molecular basis for the interplay between components of the DDR and EGFR oncogene dependence. The studies to date provide rationale to determine whether additional DDR components regulate EGFR oncogene dependence and EGFR inhibitor sensitivity.
Morphological Changes, Cellular Reprogramming and Other Factors Associated with EGFR TKI Resistance
Histological transition to small cell lung cancer (SCLC)
Whether drug resistance mechanisms observed in NSCLCs occur due to the selection of minor resistant sub-clones that exist prior to therapy, focal genetic alterations induced during therapy or more global epigenetic reprogramming events that occur in response to therapy remains unclear in most cases. Interesting insight into this unresolved issue comes from the recent demonstration that some EGFR-mutant lung adenocarcinomas can transform to small cell lung cancer histology and aberrantly express neuroendocrine biomarkers during the acquisition of resistance to EGFR TKI therapy.Citation54,Citation62,Citation79-Citation81 This histological transformation could have resulted from an epigenetic reprogramming event that occurred in the context of the original genetic landscape of these tumors because each of the resistant specimens harbored the EGFR-mutant allele prior to therapy. Additionally, no specimen had the EGFR T790M gatekeeper mutation or MET amplification. The histological transformation was clinically important because the EGFR TKI resistant tumors with SCLC transformation were sensitive to standard SCLC treatments.Citation54 To our knowledge, a small-cell transition has not been demonstrated in in vitro models of acquired EGFR TKI resistance. However, pulmonary alveolar cells can transition to a neuroendocrine morphology upon p53 and Rb1 loss, although at low frequency.Citation82
Epithelial-Mesenchymal Transition (EMT)
In addition to SCLC transformation, another histological transformation, EMT, has also been reported in human tumors with acquired resistance to EGFR TKI treatment.Citation83 EMT is implicated in enhanced migratory capacity, resistance to apoptosis and a stem cell like phenotype. However, the function and mechanistic underpinnings of EMT in the setting of EGFR TKI resistance are incompletely understood. High expression of Slug, a key transcriptional regulator of the EMT program, has been correlated with increased recurrence and decreased survival in NSCLC patients.Citation84,Citation85 Furthermore, E-cadherin expression was shown to be a biomarker predicting erlotinib response in some NSCLC patients.Citation86 Similar observations have been made in NSCLC in vitro models as well. NSCLC cells initially sensitive to erlotinib that acquire resistance in vitro express biomarkers of an EMT including loss of E-Cadherin and upregulation of vimentin, Twist, Slug and enhanced migration. Interestingly, vimentin has been shown to modulate AXL expression in erlotinib-resistant EGFR-mutant NSCLC cell lines driven by AXL.Citation69 Further studies have shown that Slug overexpression alone can induce EMT in EGFR-mutant NSCLC lines and protect cells from gefitinib-induced apoptosis. Conversely, ectopic expression of E-cadherin enhances sensitivity to gefitinib.Citation87 Likewise, low levels of miR-200, a suppressor of EMT, can promote EGFR inhibitor resistance.Citation88 The role of EMT in acquired EGFR TKI resistance remains to be fully clarified. Altogether, the data raise the intriguing possibility that therapies targeting Slug or other EMT regulators in combination with EGFR may overcome EGFR TKI resistance in some NSCLCs.
Whether the emergence of EMT and SCLC phenotypes in acquired resistance is the result of broad epigenetic changes or whether inhibition of oncogenic EGFR regulates epigenetic modifiers themselves, is unclear. However, emerging evidence has linked epigenetic alterations such as CpG methylation to drug resistance in multiple models.Citation89 In support of this, EGFR-mutant NSCLC cells that persist after EGFR TKI treatment exhibit a distinctive chromatin state that is regulated by histone demethylase and other epigenetic modifiers.Citation90 Overall, the broader genomic, epigenetic and proteomic changes occurring during acquired resistance have been challenging to study and remain poorly defined. Thus, further investigation is necessary to clarify how acquired resistance evolves in EGFR-mutant NSCLC, particularly in the context of histologically transformed phenotypes. Furthermore, recent data indicate that the tumor microenvironment and paracrine signaling may contribute to drug resistance in NSCLC and some other tumor types.Citation91,Citation92 Given the abundance of RTK bypass mechanisms and the previously unappreciated role of NFκB signaling in EGFR TKI resistance, factors produced by neighboring cells, stromal elements, the ECM and infiltrating immune cells could all potentially promote resistance to EGFR targeted therapies in patients.
The Role of BIM in Drug Resistance
Upregulation of the pro-apoptotic B-cell CLL/lymphoma 2 (BCL2) family member, BIM, is required for some TKIs to induce apoptosis in kinase-dependent malignancies such as EGFR-mutant NSCLC. One group found that cell lines sharing a common oncogenic driver and treated with the corresponding kinase inhibitor exhibited varying degrees of TKI-mediated apoptosis despite comparable inhibition of the target kinase and downstream effectors.Citation54 Interestingly, BIM RNA levels prior to TKI treatment correlated positively with the ability of the appropriate TKIs to induce apoptosis in EGFR-mutant, HER2-amplified and PIK3CA-mutant cancers. Similarly, BIM RNA levels in EGFR-mutant lung cancer primary specimens were also predictive of clinical response to EGFR inhibitors. In addition, paired-end DNA sequencing recently identified a common intronic deletion in BIM that results in expression of BIM isoforms that lack the pro-apoptotic BCL2-homology domain 3 (BH3).Citation93 In NSCLC cell lines, this polymorphism was shown to be sufficient to confer EGFR TKI resistance that was overcome by treatment with small molecule BH3 mimetics. Notably, NSCLC patients harboring the BIM intronic deletion exhibited poorer responses to EGFR TKI treatment. These data provide rationale for the clinical testing of BH3 mimetics to overcome drug resistance mediated by the BIM polymorphism. Altogether, these recent studies suggest impaired function of BIM by various mechanisms could account for some of the heterogeneity of response to TKI treatment in patients.
EGFR as a Target Beyond Lung Cancers
In addition to NSCLC, EGFR has been implicated as an oncogenic driver in a number of other cancers, including glioma, colorectal cancer, head and neck squamous cell carcinoma and pancreatic cancer.
Malignant glioma is commonly characterized by amplification of a truncated EGFR protein, EGFRvIII, that lacks part of the extracellular ligand-binding domain due to genomic deletion of exons 2 to 7 and is thus ligand-independent and constitutively active.Citation94-Citation96 Unlike NSCLC patients who harbor mutant EGFR, glioma patients with EGFRvIII generally respond poorly to EGFR TKI treatment despite evidence indicating that erlotinib is able to cross the blood–brain barrier.Citation97 This disparity in response has been attributed to mutant-specific differences in the kinetics of kinase site occupancy by EGFR TKIs. Relative to wild type EGFR, the glioma-derived EGFRvIII releases erlotinib more quickly from its active site, while the EGFR L858R and EGFR del746–750 mutants typically found in NSCLC release erlotinib more slowly. These data suggest that sensitivity to EGFR TKI therapy may depend at least in part on the nature of the EGFR activating mutation.Citation98 Furthermore, studies have found that the common loss of PTEN in gliomas is associated with significantly diminished responsiveness to EGFR kinase inhibitors.Citation99
In contrast to lung cancer and glioma, EGFR mutations are uncommon in colorectal cancers.Citation100,Citation101 Instead, overexpression of EGFR is commonly detected in colorectal cancer cell lines and tumors.Citation102 EGFR expression is positively correlated with increased malignant properties in colorectal cancers,Citation103-Citation105 and is increased in most colorectal cancer metastases.Citation106,Citation107 The clinical significance of EGFR overexpression and its influence on patient prognosis, however, remains unclear.Citation103,Citation108-Citation110 Treatment with the anti-EGFR antibodies cetuximab and panitumumab has shown anti-tumor effects and blocking of EGFR signaling in some colorectal cancer patients with metastatic disease, and is clinically approved for use in those patients whose tumors harbor wild type KRAS.Citation111-Citation113 However, response to EGFR-directed antibody therapy does not uniformly correlate with EGFR expression levels in colorectal cancers.Citation114,Citation115
EGFR is overexpressed in 80–90% of HNSCC cases and increased EGFR expression is associated with decreased survival.Citation116-Citation120 Thus, the EGFR signaling pathway is a therapeutic target in HNSCC. Clinical trials employing both anti-EGFR monoclonal antibodies and TKIs have produced promising results. Although these inhibitors have had limited success in HNSCC as single agents, there is evidence that they can act synergistically with radiotherapy in locally advanced disease and with platinum-based chemotherapy in metastatic or recurrent disease.Citation23,Citation121,Citation122 Thus far only cetuximab has been approved for the treatment of HNSCC, but the use of other monoclonal antibodies and EGFR TKIs (including erlotinib and gefitinib) is currently being investigated.Citation123
Recent studies indicate that EGFR plays a critical role in KRAS-induced tumorigenesis in pancreatic ductal adenocarcinoma (PDAC). Current models suggest that oncogenic driver mutations within an individual signaling pathway do not co-exist in a given cancer cell. Consistent with this theory, KRAS and EGFR mutations are purported to be mutually exclusive in NSCLC tumors.Citation124 Similarly, clinical trials have shown that EGFR inhibitors are only effective in CRC patients harboring non-mutant RAS.Citation125 EGFR mutations occur in less than 3% of human pancreatic cancers. Intriguingly, however, these mutations have been observed to co-occur with KRAS mutations in PDAC.Citation126 Furthermore, the presence of oncogenic KRAS mutations in non-tumorigenic human pancreata suggests that secondary lesions are required for PDAC tumorigenesis.Citation127 Injury-induced inflammation or activation of KRAS induces acinar-to-ductal metaplasia (ADM), an early step in PDAC in which damaged acini are replaced with duct-like cells.Citation128 Pancreatic overexpression of the EGFR ligand, TGFα, has been shown to induce ADM and tumorigenesis in mouse models of PDACCitation129 by a mechanism dependent on the EGFR ligand sheddase ADAM17.Citation130 Furthermore, upregulation of EGFR expression in ADM has been reported in KRAS-driven mouse models and human pancreatitis samples.Citation131,Citation132 Studies indicate that EGFR is required for both tumor initiation and maintenance of early neoplastic lesions and that genetic ablation or pharmacologic inhibition of EGFR can prevent KRAS-driven PDAC development.Citation130,Citation132 These results support a model in which EGFR signaling is necessary to sustain the threshold of RAS activity needed for tumorigenesis.Citation130,Citation133 In 2007, a clinical trial combining erlotinib with gemcitabine provided only modest benefit to a limited number of unselected PDAC patients.Citation134 This clinical evidence and the recent data from preclinical models of PDAC suggest that appropriately selected patients whose PDAC harbors activation of EGFR could benefit from EGFR inhibitor treatment.
Conclusion
In recent years, tremendous advances in our genetic and biological understanding of cancer, the development of numerous targeted cancer therapies, and the pace of translational research has led to a paradigm shift in oncology toward a targeted, precision approach to diagnosis and treatment. An increased understanding of the molecular basis of mechanisms of primary and acquired resistance to EGFR-targeted therapy in NSCLC and other cancers has contributed to this remarkable transformation in oncology. This knowledge has also led to the development of effective and promising therapeutics for the clinical subset of patients with mutant EGFR-driven lung cancers. The challenge now is to characterize completely the molecular events that regulate response and resistance to EGFR inhibition in patients and to translate this knowledge into the clinic to optimize outcomes for patients by preventing or overcoming drug resistance. While NSCLC has served as the archetype for EGFR-driven cancer, ongoing studies will hopefully shed light on the biological mechanisms by which EGFR promotes the growth of other tumor types, perhaps in cooperation with other molecular lesions. This information will allow us to use EGFR-directed therapies more effectively to improve outcomes for a wider spectrum of cancer patients.
Acknowledgments
The authors apologize to those investigators whose work in the field was not incorporated into this review due to space constraints. The authors acknowledge the members of the Bivona lab for helpful discussions and funding support (to T.G.B) from the following sources: NIH Director’s New Innovator Award, Howard Hughes Medical Institute, Doris Duke Charitable Foundation, American Lung Association, National Lung Cancer Partnership, Uniting Against Lung Cancer, Sidney Kimmel Foundation for Cancer Research.
Disclosure of Potential Conflicts of Interest
T.G.B. is a consultant and advisory board member of the Cancer Therapeutics Innovation Group. The other authors have no disclosures to report.
Related Research Data
References
- Siegel R, Ward E, Brawley O, Jemal A. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin 2011; 61:212 - 36; http://dx.doi.org/10.3322/caac.20121; PMID: 21685461
- Goldstraw P, Crowley J, Chansky K, Giroux DJ, Groome PA, Rami-Porta R, et al, International Association for the Study of Lung Cancer International Staging Committee, Participating Institutions. The IASLC Lung Cancer Staging Project: proposals for the revision of the TNM stage groupings in the forthcoming (seventh) edition of the TNM Classification of malignant tumours. J Thorac Oncol 2007; 2:706 - 14; http://dx.doi.org/10.1097/JTO.0b013e31812f3c1a; PMID: 17762336
- Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin 2005; 55:74 - 108; http://dx.doi.org/10.3322/canjclin.55.2.74; PMID: 15761078
- Pao W, Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer 2010; 10:760 - 74; http://dx.doi.org/10.1038/nrc2947; PMID: 20966921
- Weinstein IB. Cancer. Addiction to oncogenes--the Achilles heal of cancer. Science 2002; 297:63 - 4; http://dx.doi.org/10.1126/science.1073096; PMID: 12098689
- Saintigny P, Burger JA. Recent advances in non-small cell lung cancer biology and clinical management. Discov Med 2012; 13:287 - 97; PMID: 22541616
- Zandi R, Larsen AB, Andersen P, Stockhausen MT, Poulsen HS. Mechanisms for oncogenic activation of the epidermal growth factor receptor. Cell Signal 2007; 19:2013 - 23; http://dx.doi.org/10.1016/j.cellsig.2007.06.023; PMID: 17681753
- Chan SK, Gullick WJ, Hill ME. Mutations of the epidermal growth factor receptor in non-small cell lung cancer -- search and destroy. Eur J Cancer 2006; 42:17 - 23; http://dx.doi.org/10.1016/j.ejca.2005.07.031; PMID: 16364841
- Shigematsu H, Gazdar AF. Somatic mutations of epidermal growth factor receptor signaling pathway in lung cancers. Int J Cancer 2006; 118:257 - 62; http://dx.doi.org/10.1002/ijc.21496; PMID: 16231326
- Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer 2007; 7:169 - 81; http://dx.doi.org/10.1038/nrc2088; PMID: 17318210
- Yun CH, Boggon TJ, Li Y, Woo MS, Greulich H, Meyerson M, et al. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell 2007; 11:217 - 27; http://dx.doi.org/10.1016/j.ccr.2006.12.017; PMID: 17349580
- Jura N, Endres NF, Engel K, Deindl S, Das R, Lamers MH, et al. Mechanism for activation of the EGF receptor catalytic domain by the juxtamembrane segment. Cell 2009; 137:1293 - 307; http://dx.doi.org/10.1016/j.cell.2009.04.025; PMID: 19563760
- Thiel KW, Carpenter G. Epidermal growth factor receptor juxtamembrane region regulates allosteric tyrosine kinase activation. Proc Natl Acad Sci U S A 2007; 104:19238 - 43; http://dx.doi.org/10.1073/pnas.0703854104; PMID: 18042729
- Red Brewer M, Choi SH, Alvarado D, Moravcevic K, Pozzi A, Lemmon MA, et al. The juxtamembrane region of the EGF receptor functions as an activation domain. Mol Cell 2009; 34:641 - 51; http://dx.doi.org/10.1016/j.molcel.2009.04.034; PMID: 19560417
- Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009; 361:947 - 57; http://dx.doi.org/10.1056/NEJMoa0810699; PMID: 19692680
- Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, Tsurutani J, et al, West Japan Oncology Group. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol 2010; 11:121 - 8; http://dx.doi.org/10.1016/S1470-2045(09)70364-X; PMID: 20022809
- Rosell R, Moran T, Queralt C, Porta R, Cardenal F, Camps C, et al, Spanish Lung Cancer Group. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 2009; 361:958 - 67; http://dx.doi.org/10.1056/NEJMoa0904554; PMID: 19692684
- Greulich H, Chen TH, Feng W, Jänne PA, Alvarez JV, Zappaterra M, et al. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med 2005; 2:e313; http://dx.doi.org/10.1371/journal.pmed.0020313; PMID: 16187797
- Mukohara T, Engelman JA, Hanna NH, Yeap BY, Kobayashi S, Lindeman N, et al. Differential effects of gefitinib and cetuximab on non-small-cell lung cancers bearing epidermal growth factor receptor mutations. J Natl Cancer Inst 2005; 97:1185 - 94; http://dx.doi.org/10.1093/jnci/dji238; PMID: 16106023
- Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A 2004; 101:13306 - 11; http://dx.doi.org/10.1073/pnas.0405220101; PMID: 15329413
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004; 350:2129 - 39; http://dx.doi.org/10.1056/NEJMoa040938; PMID: 15118073
- Carter CA, Giaccone G. Treatment of nonsmall cell lung cancer: overcoming the resistance to epidermal growth factor receptor inhibitors. Curr Opin Oncol 2012; 24:123 - 9; http://dx.doi.org/10.1097/CCO.0b013e32834ec6a7; PMID: 22314615
- Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM, Cohen RB, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med 2006; 354:567 - 78; http://dx.doi.org/10.1056/NEJMoa053422; PMID: 16467544
- Jonker DJ, O’Callaghan CJ, Karapetis CS, Zalcberg JR, Tu D, Au HJ, et al. Cetuximab for the treatment of colorectal cancer. N Engl J Med 2007; 357:2040 - 8; http://dx.doi.org/10.1056/NEJMoa071834; PMID: 18003960
- Mendelsohn J, Baselga J. Epidermal growth factor receptor targeting in cancer. Semin Oncol 2006; 33:369 - 85; http://dx.doi.org/10.1053/j.seminoncol.2006.04.003; PMID: 16890793
- Yan L, Hsu K, Beckman RA. Antibody-based therapy for solid tumors. Cancer J 2008; 14:178 - 83; http://dx.doi.org/10.1097/PPO.0b013e318172d71a; PMID: 18536557
- Yang XD, Jia XC, Corvalan JR, Wang P, Davis CG. Development of ABX-EGF, a fully human anti-EGF receptor monoclonal antibody, for cancer therapy. Crit Rev Oncol Hematol 2001; 38:17 - 23; http://dx.doi.org/10.1016/S1040-8428(00)00134-7; PMID: 11255078
- Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell 2005; 7:301 - 11; http://dx.doi.org/10.1016/j.ccr.2005.03.003; PMID: 15837620
- Wu JY, Wu SG, Yang CH, Gow CH, Chang YL, Yu CJ, et al. Lung cancer with epidermal growth factor receptor exon 20 mutations is associated with poor gefitinib treatment response. Clin Cancer Res 2008; 14:4877 - 82; http://dx.doi.org/10.1158/1078-0432.CCR-07-5123; PMID: 18676761
- Inukai M, Toyooka S, Ito S, Asano H, Ichihara S, Soh J, et al. Presence of epidermal growth factor receptor gene T790M mutation as a minor clone in non-small cell lung cancer. Cancer Res 2006; 66:7854 - 8; http://dx.doi.org/10.1158/0008-5472.CAN-06-1951; PMID: 16912157
- Maheswaran S, Sequist LV, Nagrath S, Ulkus L, Brannigan B, Collura CV, et al. Detection of mutations in EGFR in circulating lung-cancer cells. N Engl J Med 2008; 359:366 - 77; http://dx.doi.org/10.1056/NEJMoa0800668; PMID: 18596266
- Prudkin L, Tang X, Wistuba II. Germ-line and somatic presentations of the EGFR T790M mutation in lung cancer. J Thorac Oncol 2009; 4:139 - 41; http://dx.doi.org/10.1097/JTO.0b013e3181915f92; PMID: 19096324
- Bell DW, Gore I, Okimoto RA, Godin-Heymann N, Sordella R, Mulloy R, et al. Inherited susceptibility to lung cancer may be associated with the T790M drug resistance mutation in EGFR. Nat Genet 2005; 37:1315 - 6; http://dx.doi.org/10.1038/ng1671; PMID: 16258541
- Girard N, Lou E, Azzoli CG, Reddy R, Robson M, Harlan M, et al. Analysis of genetic variants in never-smokers with lung cancer facilitated by an Internet-based blood collection protocol: a preliminary report. Clin Cancer Res 2010; 16:755 - 63; http://dx.doi.org/10.1158/1078-0432.CCR-09-2437; PMID: 20068085
- Vikis H, Sato M, James M, Wang D, Wang Y, Wang M, et al. EGFR-T790M is a rare lung cancer susceptibility allele with enhanced kinase activity. Cancer Res 2007; 67:4665 - 70; http://dx.doi.org/10.1158/0008-5472.CAN-07-0217; PMID: 17510392
- Balak MN, Gong Y, Riely GJ, Somwar R, Li AR, Zakowski MF, et al. Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin Cancer Res 2006; 12:6494 - 501; http://dx.doi.org/10.1158/1078-0432.CCR-06-1570; PMID: 17085664
- Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 2001; 293:876 - 80; http://dx.doi.org/10.1126/science.1062538; PMID: 11423618
- Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell 2002; 2:117 - 25; http://dx.doi.org/10.1016/S1535-6108(02)00096-X; PMID: 12204532
- Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A 2008; 105:2070 - 5; http://dx.doi.org/10.1073/pnas.0709662105; PMID: 18227510
- Costa DB, Schumer ST, Tenen DG, Kobayashi S. Differential responses to erlotinib in epidermal growth factor receptor (EGFR)-mutated lung cancers with acquired resistance to gefitinib carrying the L747S or T790M secondary mutations. J Clin Oncol 2008; 26:1182 - 4, author reply 1184-6; http://dx.doi.org/10.1200/JCO.2007.14.9039; PMID: 18309959
- Bean J, Riely GJ, Balak M, Marks JL, Ladanyi M, Miller VA, et al. Acquired resistance to epidermal growth factor receptor kinase inhibitors associated with a novel T854A mutation in a patient with EGFR-mutant lung adenocarcinoma. Clin Cancer Res 2008; 14:7519 - 25; http://dx.doi.org/10.1158/1078-0432.CCR-08-0151; PMID: 19010870
- Costa DB, Halmos B, Kumar A, Schumer ST, Huberman MS, Boggon TJ, et al. BIM mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung cancers with oncogenic EGFR mutations. PLoS Med 2007; 4:1669 - 79, discussion 1680; http://dx.doi.org/10.1371/journal.pmed.0040315; PMID: 17973572
- Engelman JA, Zejnullahu K, Gale CM, Lifshits E, Gonzales AJ, Shimamura T, et al. PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res 2007; 67:11924 - 32; http://dx.doi.org/10.1158/0008-5472.CAN-07-1885; PMID: 18089823
- Jänne PA, von Pawel J, Cohen RB, Crino L, Butts CA, Olson SS, et al. Multicenter, randomized, phase II trial of CI-1033, an irreversible pan-ERBB inhibitor, for previously treated advanced non small-cell lung cancer. J Clin Oncol 2007; 25:3936 - 44; http://dx.doi.org/10.1200/JCO.2007.11.1336; PMID: 17761977
- Miller VA, Hirsh V, Cadranel J, Chen YM, Park K, Kim SW, et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trial. Lancet Oncol 2012; 13:528 - 38; http://dx.doi.org/10.1016/S1470-2045(12)70087-6; PMID: 22452896
- Regales L, Gong Y, Shen R, de Stanchina E, Vivanco I, Goel A, et al. Dual targeting of EGFR can overcome a major drug resistance mutation in mouse models of EGFR mutant lung cancer. J Clin Invest 2009; 119:3000 - 10; PMID: 19759520
- Janjigian, Y.Y., Groen, H. J., Horn, L., Smit, E. F., Fu, Y., Wang, F., Shahidi, M., Denis, L. J., Pao, W., Miller, V. A., Activity and tolerability of afatinib (BIBW 2992) and cetuximab in NSCLC patients with acquired resistance to erlotinib or gefitinib. 2011.
- Ercan D, Zejnullahu K, Yonesaka K, Xiao Y, Capelletti M, Rogers A, et al. Amplification of EGFR T790M causes resistance to an irreversible EGFR inhibitor. Oncogene 2010; 29:2346 - 56; http://dx.doi.org/10.1038/onc.2009.526; PMID: 20118985
- Zhou W, Ercan D, Chen L, Yun CH, Li D, Capelletti M, et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 2009; 462:1070 - 4; http://dx.doi.org/10.1038/nature08622; PMID: 20033049
- Ercan D, Xu C, Yanagita M, Monast CS, Pratilas CA, Montero J, et al. Reactivation of ERK signaling causes resistance to EGFR kinase inhibitors. Cancer Discov 2012; 2:934 - 47; http://dx.doi.org/10.1158/2159-8290.CD-12-0103; PMID: 22961667
- Sos ML, Koker M, Weir BA, Heynck S, Rabinovsky R, Zander T, et al. PTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of Akt and EGFR. Cancer Res 2009; 69:3256 - 61; http://dx.doi.org/10.1158/0008-5472.CAN-08-4055; PMID: 19351834
- Vivanco I, Rohle D, Versele M, Iwanami A, Kuga D, Oldrini B, et al. The phosphatase and tensin homolog regulates epidermal growth factor receptor (EGFR) inhibitor response by targeting EGFR for degradation. Proc Natl Acad Sci U S A 2010; 107:6459 - 64; http://dx.doi.org/10.1073/pnas.0911188107; PMID: 20308550
- Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007; 316:1039 - 43; http://dx.doi.org/10.1126/science.1141478; PMID: 17463250
- Faber AC, Corcoran RB, Ebi H, Sequist LV, Waltman BA, Chung E, et al. BIM expression in treatment-naive cancers predicts responsiveness to kinase inhibitors. Cancer Discov 2011; 1:352 - 65; http://dx.doi.org/10.1158/2159-8290.CD-11-0106; PMID: 22145099
- Ohashi K, Sequist LV, Arcila ME, Moran T, Chmielecki J, Lin YL, et al. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc Natl Acad Sci U S A 2012; 109:E2127 - 33; http://dx.doi.org/10.1073/pnas.1203530109; PMID: 22773810
- Nunes-Xavier C, Romá-Mateo C, Ríos P, Tárrega C, Cejudo-Marín R, Tabernero L, et al. Dual-specificity MAP kinase phosphatases as targets of cancer treatment. Anticancer Agents Med Chem 2011; 11:109 - 32; http://dx.doi.org/10.2174/187152011794941190; PMID: 21288197
- West L, Vidwans SJ, Campbell NP, Shrager J, Simon GR, Bueno R, et al. A novel classification of lung cancer into molecular subtypes. PLoS One 2012; 7:e31906; http://dx.doi.org/10.1371/journal.pone.0031906; PMID: 22363766
- Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A 2007; 104:20932 - 7; http://dx.doi.org/10.1073/pnas.0710370104; PMID: 18093943
- Yano S, Wang W, Li Q, Matsumoto K, Sakurama H, Nakamura T, et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations. Cancer Res 2008; 68:9479 - 87; http://dx.doi.org/10.1158/0008-5472.CAN-08-1643; PMID: 19010923
- Donev IS, Wang W, Yamada T, Li Q, Takeuchi S, Matsumoto K, et al. Transient PI3K inhibition induces apoptosis and overcomes HGF-mediated resistance to EGFR-TKIs in EGFR mutant lung cancer. Clin Cancer Res 2011; 17:2260 - 9; http://dx.doi.org/10.1158/1078-0432.CCR-10-1993; PMID: 21220474
- Turke AB, Zejnullahu K, Wu YL, Song Y, Dias-Santagata D, Lifshits E, et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell 2010; 17:77 - 88; http://dx.doi.org/10.1016/j.ccr.2009.11.022; PMID: 20129249
- Tatematsu A, Shimizu J, Murakami Y, Horio Y, Nakamura S, Hida T, et al. Epidermal growth factor receptor mutations in small cell lung cancer. Clin Cancer Res 2008; 14:6092 - 6; http://dx.doi.org/10.1158/1078-0432.CCR-08-0332; PMID: 18829487
- Scagliotti GV, Novello S, Schiller JH, Hirsh V, Sequist LV, Soria JC, et al. Rationale and design of MARQUEE: a phase III, randomized, double-blind study of tivantinib plus erlotinib versus placebo plus erlotinib in previously treated patients with locally advanced or metastatic, nonsquamous, non-small-cell lung cancer. Clin Lung Cancer 2012; 13:391 - 5; http://dx.doi.org/10.1016/j.cllc.2012.01.003; PMID: 22440336
- Sharma N, Adjei AA. In the clinic: ongoing clinical trials evaluating c-MET-inhibiting drugs. Ther Adv Med Oncol 2011; 3:Suppl S37 - 50; http://dx.doi.org/10.1177/1758834011423403; PMID: 22128287
- Belani CP, Goss G, Blumenschein G Jr.. Recent clinical developments and rationale for combining targeted agents in non-small cell lung cancer (NSCLC). Cancer Treat Rev 2012; 38:173 - 84; http://dx.doi.org/10.1016/j.ctrv.2011.05.009; PMID: 21715100
- Gong Y, Yao E, Shen R, Goel A, Arcila M, Teruya-Feldstein J, et al. High expression levels of total IGF-1R and sensitivity of NSCLC cells in vitro to an anti-IGF-1R antibody (R1507). PLoS One 2009; 4:e7273; http://dx.doi.org/10.1371/journal.pone.0007273; PMID: 19806209
- Liu L, Greger J, Shi H, Liu Y, Greshock J, Annan R, et al. Novel mechanism of lapatinib resistance in HER2-positive breast tumor cells: activation of AXL. Cancer Res 2009; 69:6871 - 8; http://dx.doi.org/10.1158/0008-5472.CAN-08-4490; PMID: 19671800
- Mahadevan D, Cooke L, Riley C, Swart R, Simons B, Della Croce K, et al. A novel tyrosine kinase switch is a mechanism of imatinib resistance in gastrointestinal stromal tumors. Oncogene 2007; 26:3909 - 19; http://dx.doi.org/10.1038/sj.onc.1210173; PMID: 17325667
- Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet 2012; 44:852 - 60; http://dx.doi.org/10.1038/ng.2330; PMID: 22751098
- Takezawa K, Pirazzoli V, Arcila ME, Nebhan CA, Song X, de Stanchina E, et al. HER2 amplification: a potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov 2012; 2:922 - 33; http://dx.doi.org/10.1158/2159-8290.CD-12-0108; PMID: 22956644
- Yonesaka K, Zejnullahu K, Okamoto I, Satoh T, Cappuzzo F, Souglakos J, et al. Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci Transl Med 2011; 3:99ra86; http://dx.doi.org/10.1126/scitranslmed.3002442; PMID: 21900593
- Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat Immunol 2011; 12:715 - 23; http://dx.doi.org/10.1038/ni.2060; PMID: 21772280
- Bivona TG, Hieronymus H, Parker J, Chang K, Taron M, Rosell R, et al. FAS and NF-κB signalling modulate dependence of lung cancers on mutant EGFR. Nature 2011; 471:523 - 6; http://dx.doi.org/10.1038/nature09870; PMID: 21430781
- Workman P, Clarke PA. Resisting targeted therapy: fifty ways to leave your EGFR. Cancer Cell 2011; 19:437 - 40; http://dx.doi.org/10.1016/j.ccr.2011.03.020; PMID: 21481786
- Plummer R. Poly(ADP-ribose) polymerase inhibition: a new direction for BRCA and triple-negative breast cancer?. Breast Cancer Res 2011; 13:218; http://dx.doi.org/10.1186/bcr2877; PMID: 21884642
- Nowsheen S, Cooper T, Bonner JA, LoBuglio AF, Yang ES. HER2 overexpression renders human breast cancers sensitive to PARP inhibition independently of any defect in homologous recombination DNA repair. Cancer Res 2012; 72:4796 - 806; http://dx.doi.org/10.1158/0008-5472.CAN-12-1287; PMID: 22987487
- Nowsheen S, Bonner JA, Lobuglio AF, Trummell H, Whitley AC, Dobelbower MC, et al. Cetuximab augments cytotoxicity with poly (adp-ribose) polymerase inhibition in head and neck cancer. PLoS One 2011; 6:e24148; http://dx.doi.org/10.1371/journal.pone.0024148; PMID: 21912620
- Rosell R, Molina MA, Costa C, Simonetti S, Gimenez-Capitan A, Bertran-Alamillo J, et al. Pretreatment EGFR T790M mutation and BRCA1 mRNA expression in erlotinib-treated advanced non-small-cell lung cancer patients with EGFR mutations. Clin Cancer Res 2011; 17:1160 - 8; http://dx.doi.org/10.1158/1078-0432.CCR-10-2158; PMID: 21233402
- Arcila ME, Oxnard GR, Nafa K, Riely GJ, Solomon SB, Zakowski MF, et al. Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay. Clin Cancer Res 2011; 17:1169 - 80; http://dx.doi.org/10.1158/1078-0432.CCR-10-2277; PMID: 21248300
- Zakowski MF, Ladanyi M, Kris MG, Memorial Sloan-Kettering Cancer Center Lung Cancer OncoGenome Group. EGFR mutations in small-cell lung cancers in patients who have never smoked. N Engl J Med 2006; 355:213 - 5; http://dx.doi.org/10.1056/NEJMc053610; PMID: 16837691
- Alam N, Gustafson KS, Ladanyi M, Zakowski MF, Kapoor A, Truskinovsky AM, et al. Small-cell carcinoma with an epidermal growth factor receptor mutation in a never-smoker with gefitinib-responsive adenocarcinoma of the lung. Clin Lung Cancer 2010; 11:E1 - 4; http://dx.doi.org/10.3816/CLC.2010.n.046; PMID: 20837450
- Sutherland KD, Proost N, Brouns I, Adriaensen D, Song JY, Berns A. Cell of origin of small cell lung cancer: inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell 2011; 19:754 - 64; http://dx.doi.org/10.1016/j.ccr.2011.04.019; PMID: 21665149
- Suda K, Tomizawa K, Fujii M, Murakami H, Osada H, Maehara Y, et al. Epithelial to mesenchymal transition in an epidermal growth factor receptor-mutant lung cancer cell line with acquired resistance to erlotinib. J Thorac Oncol 2011; 6:1152 - 61; http://dx.doi.org/10.1097/JTO.0b013e318216ee52; PMID: 21597390
- Bhat-Nakshatri P, Appaiah H, Ballas C, Pick-Franke P, Goulet R Jr., Badve S, et al. SLUG/SNAI2 and tumor necrosis factor generate breast cells with CD44+/CD24- phenotype. BMC Cancer 2010; 10:411; http://dx.doi.org/10.1186/1471-2407-10-411; PMID: 20691079
- Chen YC, Hsu HS, Chen YW, Tsai TH, How CK, Wang CY, et al. Oct-4 expression maintained cancer stem-like properties in lung cancer-derived CD133-positive cells. PLoS One 2008; 3:e2637; http://dx.doi.org/10.1371/journal.pone.0002637; PMID: 18612434
- Yauch RL, Januario T, Eberhard DA, Cavet G, Zhu W, Fu L, et al. Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin Cancer Res 2005; 11:8686 - 98; http://dx.doi.org/10.1158/1078-0432.CCR-05-1492; PMID: 16361555
- Witta SE, Gemmill RM, Hirsch FR, Coldren CD, Hedman K, Ravdel L, et al. Restoring E-cadherin expression increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res 2006; 66:944 - 50; http://dx.doi.org/10.1158/0008-5472.CAN-05-1988; PMID: 16424029
- Mongroo PS, Rustgi AK. The role of the miR-200 family in epithelial-mesenchymal transition. Cancer Biol Ther 2010; 10:219 - 22; http://dx.doi.org/10.4161/cbt.10.3.12548; PMID: 20592490
- Ogawa T, Liggett TE, Melnikov AA, Monitto CL, Kusuke D, Shiga K, et al. Methylation of death-associated protein kinase is associated with cetuximab and erlotinib resistance. Cell Cycle 2012; 11:1656 - 63; http://dx.doi.org/10.4161/cc.20120; PMID: 22487682
- Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010; 141:69 - 80; http://dx.doi.org/10.1016/j.cell.2010.02.027; PMID: 20371346
- Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012; 487:500 - 4; http://dx.doi.org/10.1038/nature11183; PMID: 22763439
- Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 2012; 487:505 - 9; http://dx.doi.org/10.1038/nature11249; PMID: 22763448
- Ng KP, Hillmer AM, Chuah CT, Juan WC, Ko TK, Teo AS, et al. A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat Med 2012; 18:521 - 8; http://dx.doi.org/10.1038/nm.2713; PMID: 22426421
- Frederick L, Wang XY, Eley G, James CD. Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res 2000; 60:1383 - 7; PMID: 10728703
- Huang HS, Nagane M, Klingbeil CK, Lin H, Nishikawa R, Ji XD, et al. The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. J Biol Chem 1997; 272:2927 - 35; http://dx.doi.org/10.1074/jbc.272.5.2927; PMID: 9006938
- Sugawa N, Ekstrand AJ, James CD, Collins VP. Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc Natl Acad Sci U S A 1990; 87:8602 - 6; http://dx.doi.org/10.1073/pnas.87.21.8602; PMID: 2236070
- Porta R, Sánchez-Torres JM, Paz-Ares L, Massutí B, Reguart N, Mayo C, et al. Brain metastases from lung cancer responding to erlotinib: the importance of EGFR mutation. Eur Respir J 2011; 37:624 - 31; http://dx.doi.org/10.1183/09031936.00195609; PMID: 20595147
- Barkovich KJ, Hariono S, Garske AL, Zhang J, Blair JA, Fan QW, et al. Kinetics of inhibitor cycling underlie therapeutic disparities between EGFR-driven lung and brain cancers. Cancer Discov 2012; 2:450 - 7; http://dx.doi.org/10.1158/2159-8290.CD-11-0287; PMID: 22588882
- Mellinghoff IK, Wang MY, Vivanco I, Haas-Kogan DA, Zhu S, Dia EQ, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med 2005; 353:2012 - 24; http://dx.doi.org/10.1056/NEJMoa051918; PMID: 16282176
- Barber TD, Vogelstein B, Kinzler KW, Velculescu VE. Somatic mutations of EGFR in colorectal cancers and glioblastomas. N Engl J Med 2004; 351:2883; http://dx.doi.org/10.1056/NEJM200412303512724; PMID: 15625347
- Lee JW, Soung YH, Kim SY, Park WS, Nam SW, Lee JY, et al. Absence of EGFR mutation in the kinase domain in common human cancers besides non-small cell lung cancer. Int J Cancer 2005; 113:510 - 1; http://dx.doi.org/10.1002/ijc.20591; PMID: 15455340
- Ciardiello F, Kim N, Saeki T, Dono R, Persico MG, Plowman GD, et al. Differential expression of epidermal growth factor-related proteins in human colorectal tumors. Proc Natl Acad Sci U S A 1991; 88:7792 - 6; http://dx.doi.org/10.1073/pnas.88.17.7792; PMID: 1715580
- Goldstein NS, Armin M. Epidermal growth factor receptor immunohistochemical reactivity in patients with American Joint Committee on Cancer Stage IV colon adenocarcinoma: implications for a standardized scoring system. Cancer 2001; 92:1331 - 46; http://dx.doi.org/10.1002/1097-0142(20010901)92:5<1331::AID-CNCR1455>3.0.CO;2-M; PMID: 11571750
- Hemming AW, Davis NL, Kluftinger A, Robinson B, Quenville NF, Liseman B, et al. Prognostic markers of colorectal cancer: an evaluation of DNA content, epidermal growth factor receptor, and Ki-67. J Surg Oncol 1992; 51:147 - 52; http://dx.doi.org/10.1002/jso.2930510304; PMID: 1434639
- Steele RJ, Kelly P, Ellul B, Eremin O. Epidermal growth factor receptor expression in colorectal cancer. Br J Surg 1990; 77:1352 - 4; http://dx.doi.org/10.1002/bjs.1800771211; PMID: 2276016
- Chee CE, Sinicrope FA. Targeted therapeutic agents for colorectal cancer. Gastroenterol Clin North Am 2010; 39:601 - 13; http://dx.doi.org/10.1016/j.gtc.2010.08.017; PMID: 20951919
- Ciardiello F, Tortora G. Epidermal growth factor receptor (EGFR) as a target in cancer therapy: understanding the role of receptor expression and other molecular determinants that could influence the response to anti-EGFR drugs. Eur J Cancer 2003; 39:1348 - 54; http://dx.doi.org/10.1016/S0959-8049(03)00235-1; PMID: 12826036
- McKay JA, Murray LJ, Curran S, Ross VG, Clark C, Murray GI, et al. Evaluation of the epidermal growth factor receptor (EGFR) in colorectal tumours and lymph node metastases. Eur J Cancer 2002; 38:2258 - 64; http://dx.doi.org/10.1016/S0959-8049(02)00234-4; PMID: 12441262
- Resnick MB, Routhier J, Konkin T, Sabo E, Pricolo VE. Epidermal growth factor receptor, c-MET, beta-catenin, and p53 expression as prognostic indicators in stage II colon cancer: a tissue microarray study. Clin Cancer Res 2004; 10:3069 - 75; http://dx.doi.org/10.1158/1078-0432.CCR-03-0462; PMID: 15131045
- Spano JP, Fagard R, Soria JC, Rixe O, Khayat D, Milano G. Epidermal growth factor receptor signaling in colorectal cancer: preclinical data and therapeutic perspectives. Ann Oncol 2005; 16:189 - 94; http://dx.doi.org/10.1093/annonc/mdi057; PMID: 15668269
- Holubec L, Liska V, Matejka VM, Fiala O, Dreslerova J, Mrazkova P, et al. The role of cetuximab in the treatment of metastatic colorectal cancer. Anticancer Res 2012; 32:4007 - 11; PMID: 22993351
- Kim GP, Grothey A. Targeting colorectal cancer with human anti-EGFR monoclonocal antibodies: focus on panitumumab. Biologics 2008; 2:223 - 8; PMID: 19707356
- Krasinskas AM. EGFR Signaling in Colorectal Carcinoma. Patholog Res Int 2011; 2011:932932; http://dx.doi.org/10.4061/2011/932932; PMID: 21403829
- Chung KY, Shia J, Kemeny NE, Shah M, Schwartz GK, Tse A, et al. Cetuximab shows activity in colorectal cancer patients with tumors that do not express the epidermal growth factor receptor by immunohistochemistry. J Clin Oncol 2005; 23:1803 - 10; http://dx.doi.org/10.1200/JCO.2005.08.037; PMID: 15677699
- Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med 2004; 351:337 - 45; http://dx.doi.org/10.1056/NEJMoa033025; PMID: 15269313
- Grandis JR, Tweardy DJ. Elevated levels of transforming growth factor alpha and epidermal growth factor receptor messenger RNA are early markers of carcinogenesis in head and neck cancer. Cancer Res 1993; 53:3579 - 84; PMID: 8339264
- He Y, Zeng Q, Drenning SD, Melhem MF, Tweardy DJ, Huang L, et al. Inhibition of human squamous cell carcinoma growth in vivo by epidermal growth factor receptor antisense RNA transcribed from the U6 promoter. J Natl Cancer Inst 1998; 90:1080 - 7; http://dx.doi.org/10.1093/jnci/90.14.1080; PMID: 9672256
- Rubin Grandis J, Melhem MF, Barnes EL, Tweardy DJ. Quantitative immunohistochemical analysis of transforming growth factor-alpha and epidermal growth factor receptor in patients with squamous cell carcinoma of the head and neck. Cancer 1996; 78:1284 - 92; http://dx.doi.org/10.1002/(SICI)1097-0142(19960915)78:6<1284::AID-CNCR17>3.0.CO;2-X; PMID: 8826952
- Thomas SM, Zeng Q, Epperly MW, Gooding WE, Pastan I, Wang QC, et al. Abrogation of head and neck squamous cell carcinoma growth by epidermal growth factor receptor ligand fused to pseudomonas exotoxin transforming growth factor alpha-PE38. Clin Cancer Res 2004; 10:7079 - 87; http://dx.doi.org/10.1158/1078-0432.CCR-04-0587; PMID: 15501988
- Udayachander M, Dean CJ, Meenakshi AN, Sivakumar N, Babu PB, Sivakumar J. Anti-tumor activity of monoclonal antibody CIBCNSH3 generated to the human EGF receptor. Hum Antibodies 1997; 8:60 - 4; PMID: 9289389
- Vermorken JB, Mesia R, Rivera F, Remenar E, Kawecki A, Rottey S, et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med 2008; 359:1116 - 27; http://dx.doi.org/10.1056/NEJMoa0802656; PMID: 18784101
- Vermorken JB, Trigo J, Hitt R, Koralewski P, Diaz-Rubio E, Rolland F, et al. Open-label, uncontrolled, multicenter phase II study to evaluate the efficacy and toxicity of cetuximab as a single agent in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck who failed to respond to platinum-based therapy. J Clin Oncol 2007; 25:2171 - 7; http://dx.doi.org/10.1200/JCO.2006.06.7447; PMID: 17538161
- Kundu SK, Nestor M. Targeted therapy in head and neck cancer. Tumour Biol 2012; 33:707 - 21; http://dx.doi.org/10.1007/s13277-012-0350-2; PMID: 22373581
- Shigematsu H, Lin L, Takahashi T, Nomura M, Suzuki M, Wistuba II, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst 2005; 97:339 - 46; http://dx.doi.org/10.1093/jnci/dji055; PMID: 15741570
- Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 2008; 359:1757 - 65; http://dx.doi.org/10.1056/NEJMoa0804385; PMID: 18946061
- Oliveira-Cunha M, Hadfield KD, Siriwardena AK, Newman W. EGFR and KRAS mutational analysis and their correlation to survival in pancreatic and periampullary cancer. Pancreas 2012; 41:428 - 34; http://dx.doi.org/10.1097/MPA.0b013e3182327a03; PMID: 22422135
- Lüttges J, Reinecke-Lüthge A, Möllmann B, Menke MA, Clemens A, Klimpfinger M, et al. Duct changes and K-ras mutations in the disease-free pancreas: analysis of type, age relation and spatial distribution. Virchows Arch 1999; 435:461 - 8; http://dx.doi.org/10.1007/s004280050428; PMID: 10592048
- Reichert M, Rustgi AK. Pancreatic ductal cells in development, regeneration, and neoplasia. J Clin Invest 2011; 121:4572 - 8; http://dx.doi.org/10.1172/JCI57131; PMID: 22133881
- De Lisle RC, Logsdon CD. Pancreatic acinar cells in culture: expression of acinar and ductal antigens in a growth-related manner. Eur J Cell Biol 1990; 51:64 - 75; PMID: 2184038
- Ardito CM, Grüner BM, Takeuchi KK, Lubeseder-Martellato C, Teichmann N, Mazur PK, et al. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell 2012; 22:304 - 17; http://dx.doi.org/10.1016/j.ccr.2012.07.024; PMID: 22975374
- Korc M, Friess H, Yamanaka Y, Kobrin MS, Buchler M, Beger HG. Chronic pancreatitis is associated with increased concentrations of epidermal growth factor receptor, transforming growth factor alpha, and phospholipase C gamma. Gut 1994; 35:1468 - 73; http://dx.doi.org/10.1136/gut.35.10.1468; PMID: 7959207
- Navas C, Hernández-Porras I, Schuhmacher AJ, Sibilia M, Guerra C, Barbacid M. EGF receptor signaling is essential for k-ras oncogene-driven pancreatic ductal adenocarcinoma. Cancer Cell 2012; 22:318 - 30; http://dx.doi.org/10.1016/j.ccr.2012.08.001; PMID: 22975375
- Ji B, Tsou L, Wang H, Gaiser S, Chang DZ, Daniluk J, et al. Ras activity levels control the development of pancreatic diseases. Gastroenterology 2009; 137:1072 - 82, 1082, e1-6; http://dx.doi.org/10.1053/j.gastro.2009.05.052; PMID: 19501586
- Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, et al, National Cancer Institute of Canada Clinical Trials Group. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 2007; 25:1960 - 6; http://dx.doi.org/10.1200/JCO.2006.07.9525; PMID: 17452677