Abstract
Esophageal adenocarcinoma develops in response to severe gastroesophageal reflux disease through the precursor lesion Barrett esophagus, in which the normal squamous epithelium is replaced by a columnar lining. The incidence of esophageal adenocarcinoma in the United States has increased by over 600% in the past 40 years and the overall survival rate remains less than 20% in the community. This review highlights some of the signaling pathways for which there is some evidence of a role in the development of esophageal adenocarcinoma. An increasingly detailed understanding of the biology of this cancer has emerged recently, revealing that in addition to the well-recognized alterations in single genes such as p53, p16, APC, and telomerase, there are interactions between the components of the reflux fluid, the homeobox gene Cdx2, and the Wnt, Notch, and Hedgehog signaling pathways.
Introduction
Esophageal adenocarcinoma is distinguished by having both a high case-fatality rate, with five year survival rates typically 15−20% in the community,Citation1-Citation3 and by having the fastest rising incidence of all cancers in many countries, with a more than 6-fold increase in incidence in the US in the past several decades.Citation4 Although recent data suggest that the incidence rise may have slowed,Citation5,Citation6 the increase in the number of patients with adenocarcinoma of the esophagus or gastroesophageal junction has stimulated a corresponding increase in research into the biology of this cancer. Here we use the initialism EAC to denote both esophageal and gastroesophageal junction adenocarcinomas although it is recognized that some tumors classified as junctional may be gastric cancers that have extended proximally.
EAC is often studied in conjunction with Barrett esophagus (BE), the condition in which the normal squamous epithelium in the distal esophagus is replaced by a metaplastic columnar mucosa, often containing goblet cells (intestinal metaplasia, IM) in response to chronic severe gastroesophageal reflux disease (GERD). BE is the main predisposing factor for EAC, through a generally accepted multistep process in which IM in a very small proportion of individuals (probably less than 0.5%/year)Citation7,Citation8 progresses through low grade dysplasia and high grade dysplasia stages to invasive EAC. This review includes findings for both BE and EAC although the focus is on cancer.
Molecular Pathogenesis of EAC
Mechanisms underlying molecular abnormalities in the pathogenesis of EAC
In the development of cancer, both genetic and epigenetic mechanisms contribute to the activation or inactivation of key signaling pathways and acquisition of the cancer phenotype “hallmarks”Citation9. It is generally accepted that Barrett multistep carcinogenesis is characterized by genomic instability,Citation10 which facilitates accumulation of lesions that target proto-oncogenes, tumor suppressor genes, mismatch repair genes, and mitotic checkpoint genes, thereby aiding tumorigenic progression.Citation11 In addition, reflux components have been shown to induce DNA damage in esophageal cells.Citation12-Citation14 Although there are no data showing that reflux causes more permanent genetic (e.g., mutations) or epigenetic alterations, recent Next Generation sequencing dataCitation15 show a high overall mutation rate in EAC that is only exceeded by lung cancer and melanoma, both of which are known to be largely driven by mutagens (smoking and UV light, respectively). Epigenetic studies focused on CpG island promoter hypermethylation suggest that there may be “high” and “low” methylation epigenotypes,Citation16 while genome-wide profiling not restricted to CpG sites indicates that the predominant epigenetic mechanism is widespread hypomethylation, which occurs before progression to HGD/EAC and acts in concert with gene amplification to upregulate expression of various genes.Citation17
Confirming which of the many molecular alterations are essential to driving progression to EAC has so far largely eluded researchers. This may be partly due to an emphasis on non-mechanistic studies to identify clinically relevant biomarkers of progression to EAC in patients with BE. Another factor may be differences in tumors arising in the tubular esophagus, the gastroesophageal junction, and the proximal stomach including cardia,Citation18-Citation20 as well as a large degree of heterogeneity found within both individual cancers and segments of Barrett esophagus.Citation21-Citation24
This gap in knowledge has contributed to our persisting inability to identify which patients with BE are most at risk of progression.Citation25 Baseline alterations including p16 and p53 loss combined with aneuploidy are strongly associated with the likelihood of progression to HGD/EAC in longitudinal studies (reviewed in refs. Citation20 and Citation25), but these findings require validation at other centers and are not currently suitable for routine use in clinical pathology laboratories. The lack of functional studies identifying drivers of disease has also hindered progress in the development of targeted therapies, including therapies aimed at preventing BE progression. While it is unlikely that all EAC will be treatable via inactivation of a single oncogene (as in the oncogene addiction model),Citation26 an effective approach may involve the collective targeting of a small number of molecules, possibly via a pathway approach.
Mechanistic studies on the molecular pathogenesis of EAC
There are relatively few studies examining the effect that abnormalities present in BE and EAC tissues have on the acquisition of tumorigenic phenotypes in experimental models. Genetically manipulable animal models have only recently been described,Citation27 and there is a paucity of appropriate cell lines.Citation28 Due to a lack of cell lines representing early stages of this disease, many studies have used adenocarcinoma cell lines to model events that are likely to have occurred earlier in the neoplastic sequence. Furthermore, the majority of in vitro studies to date, rather than modeling the effect of genetic alterations discovered in vivo, have focused on the ability of reflux components such as acid and bile to induce the expression of specific proteins and/or activate relevant pathways. While these effects may play a role in tumorigenesis in BE, it is likely that more permanent genetic or epigenetic changes are required in the evolution of EAC. More promisingly, the step-wise neoplastic transformation of a hTERT immortalised, non-dysplastic Barrett cell line using the defined genetic manipulations of p53 knockdown and expression of oncogenic H-Ras (G12V) has been reported.Citation29 These cells could prove useful to study the role of some of the molecular pathways (discussed below) in Barrett carcinogenesis and in the testing of novel therapeutic compounds targeting these pathways, particularly if combined with relevant in vitro 3-dimensional organotypicCitation30,Citation31 and organoid modelsCitation32 and in vivo tissue reconstitutionCitation33 or xenograft models.Citation34
In this review we highlight some of the signaling pathways for which there is evidence of a role in the development of EAC. Activation or inactivation of signaling pathways can occur at multiple levels from the growth factor/ligand that activates a pathway, to cell-surface receptors (often containing intracellular tyrosine kinase domains) and then to downstream kinases and intracellular effectors including transcription factors.
Growth factor and other cytokine-mediated signaling
Epidermal growth factor family
Epidermal growth factor (EGF) and the related family member transforming growth factor-α (TGFα) are two key ligands that have a stimulatory effect on epithelial cell proliferation via activation of the epidermal growth factor receptor (EGFR). There is evidence that signaling through EGFR may play a role in Barrett carcinogenesis to stimulate growth. Protein expression of EGF and TGFα is increased to similar levels in BE and EAC,Citation35,Citation36 suggesting that EGFR activation through these ligands via an autocrine signaling mechanism may be an early event in the BE metaplasia-dysplasia-EAC sequence. In BE, expression of TGFα was found to correlate with proliferation and TGFα immunoreactivity was found in the same areas as proliferating cells in BE glands showing high-grade dysplasia (HGD).Citation37 Altered EGF expression in some cases may be due to the presence of the EGF A61G polymorphism, which is associated with an increased risk of EAC.Citation38,Citation39
Increased signaling through the EGFR pathway could also be a consequence of changes in expression or function of EGFR family members (e.g., EGFR and c-erbB-2/Her2). EGFR protein expression is reportedly increased in up to two thirds of EAC and has been associated with tumor (T) stage, lymph node metastasis, and a trend toward worse disease-free and overall survival.Citation40-Citation44 The gene for EGFR is also amplified in HGD and around one third of EAC,Citation45,Citation46 and activating mutations in exons 18 and 21 of the EGFR gene have been identified in approximately 15% of BE and EAC.Citation47 Both EGFR overexpression and mutant p53 contribute to the enrichment of a subpopulation of human esophageal epithelial cells which, after negating the oncogene-induced senescence induced by EGFR overexpression, undergo epithelial to mesenchymal transition (EMT) on TGF-β stimulation.Citation48
The erbB-2/Her2 receptor is also amplified in approximately 10–50% of EAC with concomitant increased mRNA or protein expression.Citation49-Citation55 Amplification and overexpression of erbB-2 have been reported in HGD but not normal esophagus or BE with or without low grade dysplasia (LGD), suggesting that this lesion is a late stage event in BE carcinogenesis.Citation50,Citation52 Co-amplification of erb-B2 and EGFR occurs in approximately 15% of EAC in addition to increased immunoreactivity for erb-B2 in BE and EAC,Citation46 which suggests the possibility of ligand independent activation of this signaling pathway via receptor hetero-oligomerization and subsequent enhanced tumor cell survival.
Despite the evidence above, the results of clinical trials targeting EGFR in the treatment of EAC (reviewed by Mukherjee et al.Citation56) have not been very promising. This may be related to the presence of K-ras mutations, which are known to predict resistance to EGFR inhibition. These mutations are reported in up to a third of patients with HGD or EAC, but not in patients with non-dysplastic BE.Citation57 In contrast, targeting erbB-2 in patients with HER2+ metastatic esophago-gastric junctional adenocarcinoma has been more successful,Citation58 and is being tested further in a clinical trial with earlier stage disease (RTOG-1010, National Cancer Institute, USA).
Vascular endothelial growth factor family
Vascular endothelial growth factors (VEGFs) are crucial to the formation of new blood vessels (angiogenesis, particularly VEGF-A) and lymph vessels (lymphangiogenesis, particularly VEGF-C) through binding to their cognate VEGF receptors (VEGFR-1, -2, and -3). Angiogenesis is important for the continued growth of a tumor as it outgrows the existing blood supply, and lymphangiogenesis is thought to be important for the metastatic spread of tumors. Evidence suggests that activation of signaling through these pathways may be important early in the neoplastic progression of BE to EAC. A number of studies have reported an increase in the expression of VEGF-A across the sequence from non-dysplastic BE to dysplasia and EAC.Citation59-Citation62 However, correlation between VEGF-A expression, angiogenesis (in particular neovascularisation) and clinical outcome are unclear. Mobius et al.Citation59 showed that EAC with a high level of neovascularisation did not have significantly increased VEGF-A expression, although low tumor neovascularisation correlates with better survival.Citation63 In contrast, two other studies show a positive correlation between VEGF-A expression and high overall tumor vascularization,Citation60 which also correlated with lymph node metastasis in one study.Citation64 Co-expression of VEGF-C and VEGFR-3 on lymphatic vessels in EAC also suggests enhanced lymphangiogenesis and a potential facilitation of metastatic spread of this disease,Citation65 although VEGF-C expression does not correlate with survival.Citation66
There are a number of possible mechanisms for increased VEGF-A expression in Barrett carcinogenesis including induction by human chorionic gonadotropin,Citation67 which is increased in EAC,Citation68 by prostaglandinsCitation69-Citation73 or by bile acid.Citation74 In addition, polymorphisms in the VEGF-A gene, which are linked with increased VEGF expression, are associated with an increased risk of EAC, particularly in smokers.Citation75
These studies suggest that angiogenic properties are acquired early in disease progression, perhaps at the dysplasia stage. Inhibition of VEGF-A signaling as a therapeutic option in the treatment of this disease warrants further investigation, particularly since current clinical trials using a VEGF inhibitor, bevacizumab, are mainly aimed at junctional and gastric AC rather than EAC.Citation56
Insulin-like growth factor family
Obesity is associated with an increased risk of developing a number of cancers including EAC and may also contribute to development of BE.Citation76,Citation77 In Barrett carcinogenesis, obesity, particularly central adiposity, is thought to contribute through both GERD-related (e.g., mechanical promotion of GERD) and GERD-independent mechanisms.Citation78,Citation79 In addition, a large proportion of BE patients have metabolic syndrome, and approximately a quarter of these have hyperinsulinemia.Citation80 There is emerging evidence that GERD-independent mechanisms may include insulin-mediated production of insulin-like growth factor 1 (IGF-1) and decreased production of IGF binding proteins 1 and 3 (IGFBP-1 and -3).Citation81 As a consequence, increased bioavailability of IGF can potentially stimulate proliferation and cell survival by binding to the IGF receptor (IGFR) and subsequent activation of intracellular signal transduction pathways.
However, interpretation of IGF-1 bioavailability is complicated by differences reported in tissue vs. serum expression of the relevant molecules. Expression of IGFBP mRNA is increased in BE and EAC tissue compared with normal tissue and is also increased in BE tissue of EAC patients compared with BE tissue from tumor-free patients.Citation82 In contrast, Greer and colleaguesCitation83 found that serum insulin and IGF-1 levels are associated with an increased risk and serum IGFBP-1 and -3 with a decreased risk of BE compared with screening colonoscopy controls, but not when compared with GERD controls. The BE cases had a significantly higher waist-to-hip ratio but not BMI compared with colonoscopy controls, suggesting that central adiposity might be an important factor.Citation83 A longitudinal study of patients with BE also found no association with baseline serum IGF-1 and IGFBP-3 levels and risk of progression to EAC.Citation84
Activation of this pathway may also occur through modulation of the receptor, IGF-1R. Overactivation of IGF-1R has been implicated in several cancers in which it is thought to promote cell growth, survival, and angiogenesis, possibly via heterodimerization with EGFR. Protein expression of IGF-1R is increased in the sequence from BE to dysplasia and EAC, with around 80% of EAC showing positive expression.Citation85,Citation86 Increased expression may be a result of posttranscriptional regulation since there is no difference in IGF-1R mRNA expression between EAC and matched normal tissue, with the exception of individuals carrying a G1013A polymorphism in the igf-1r gene, suggesting that this polymorphism may enhance transcription or stabilize the transcript.Citation87 This same polymorphism increases the risk of developing BE and EAC in obese individuals by 3- and 5-fold respectively.Citation88 There is thus some evidence for a role of this complex signaling axis in EAC although the importance of tissue vs. serum bioavailability remains to be determined.
Other receptor tyrosine kinases
C-Met is the tyrosine kinase receptor for hepatocyte growth factor (HGF) and is normally expressed by epithelial cells, where it is essential for morphogenesis and wound healing in adults. In cancer its abnormal activation has been associated with tumor growth, angiogenesis, and invasion. C-Met is overexpressed in dysplastic BE and EAC,Citation89-Citation91 although probably only in lesions where c-Met is amplified.Citation92 Stimulation of OE33 EAC cells with HGF results in reduced E-cadherin expression and stimulated β-catenin transcriptional activity leading to enhanced anchorage independent growth,Citation89 suggesting a role for c-Met signaling in the acquisition of an invasive phenotype in EAC.
The Axl receptor tyrosine kinase (RTK) was recently identified as being significantly upregulated in the progression of BE to EAC.Citation93 The Axl receptor has been implicated in mediating progression, metastasis and drug resistance in several other tumor types. Overexpression of Axl in EAC is inversely associated with survival and RNAi knockdown in 2 EAC cell lines reduced in vitro invasion, migration, and anchorage-independent growth and completely abrogated in vivo engraftment in immunocompromised mice.Citation93 This novel finding is intriguing given the recent development of small molecule inhibitors of Axl that have shown promising results in a mouse model of breast cancer.Citation94 Treatment of OE33 EAC cells with Axl inhibitors reduced anchorage-independent growth, invasion, and migration and blocked phosphorylation of ErbB-2, suggesting potential transactivation by Axl.Citation93
Overall, RTK signaling pathways are likely to play an important role in EAC and serve as attractive therapeutic targets due to the plethora of available approved and “in development” inhibitors. In particular, EGFR family members, cMET, fibroblast growth factor receptor (FGRF) family members, insulin receptor and IGF1R, collectively, were recently shown to be frequently hyper-activated in EAC and EAC cell lines,Citation95 although the mechanisms underlying RTK activation were not investigated in this study. In vitro studies indicated that using an individualized approach to target activated RTKs could be an effective tactic in the treatment of EAC, although many cell lines showed complex RTK profiles and combinations of inhibitors were required to induce cytotoxicity.Citation95
Leptin and adiponectin
In addition to IGF bioavailability, cytokines produced by adipocytes (adipokines) such as leptin and adiponectin may also contribute to obesity-mediated effects in Barrett carcinogenesis. Leptin is found at increased levels in the serum of obese people, while adiponectin is decreased. Leptin has been shown to have mitogenic effects on some tumor cell lines in vitro, including colon cancer.Citation96 In contrast, adiponectin is thought to induce apoptosisCitation97 and low plasma levels have been associated with an increased risk for a number of cancers including gastricCitation98and colon cancer.Citation99 Therefore, it has been hypothesized that altered adiponectin and leptin levels may contribute to the association between obesity and some cancers, including EAC.
While a role for leptin in the progression to EAC is undefined, there is evidence to suggest it may contribute to development of BE. Gastric leptin levels are increased in BE and are associated with increased risk of BE.Citation100 In contrast, the association between serum leptin and BE is unclear with studies showing either an association with BE in men but not women that is independent of both GERD and obesity,Citation101 or an association in women but not men.Citation102 Similarly, there are conflicting reports on the association between serum adiponectin levels and BE, which may also be gender dependentCitation101-Citation103. Serum adiponectin levels are lower in patients with EAC compared with controls,Citation104 which may contribute to increased tumor cell survival. As with IGF-1 and IGFBPs, expression of leptin and adiponectin in BE and EAC patients deserves further investigation.
There are limited data on alterations in the receptors for leptin and adiponection in the development of EAC. Receptors for leptin are highly expressed in normal and inflamed esophagus and BE,Citation100 but expression in EAC has not been reported. In keeping with adiponectin playing a protective role in carcinogenesis, expression of adiponectin receptors is decreased in BE at mRNA level.Citation105
While the studies described above may not provide a compelling case for the involvement of adipokines in Barrett carcinogenesis, there is additional support from in vitro studies. Leptin induces proliferation and inhibits apoptosis via activation of COX-2, leading to prostaglandin E2-mediated transactivation of EGFR and JNK activation in OE33 EAC cells.Citation106,Citation107 Increased proliferation may also be partly due to leptin-induced HB-EGF and TGFα expression and secretion leading to subsequent EGFR transactivation.Citation106 In contrast, adiponectin attenuates leptin induced proliferation in EAC cell lines, at least partly by inhibiting AKT activation,Citation108 and may induce apoptosis via modulating expression of pro- and anti-apoptotic Bcl-2 family members.Citation105
Transforming growth factor β
Transforming growth factor β (TGF β) is central to epithelial homeostasis by regulating both proliferation and differentiation. Dysregulated response to TGFβ has been associated with a range of epithelial cancers. In normal cells, one of the functions of TGFβ is to induce a reversible cell cycle arrest and many epithelial tumors are refractory to this response. In contrast, TGFβ is implicated in an epithelial to mesenchymal transition (EMT) in tumor cells, particularly at the invasive edges, where this change in phenotype is thought to aid invasion and metastasis. Both of these mechanisms have been implicated in the progression of BE to EAC. Expression of TGFβ is upregulated in EAC compared with normal esophagus and BE and is associated with advanced stage.Citation109,Citation110 In addition, increased expression of TGFβ at the invasive margins of EAC correlates with markers of EMT.Citation111
TGFβ signaling can be impaired through modulation of the downstream transcriptional mediators, particularly SMAD2 and 4. Loss of heterozygosity (LOH) at chromosome 18q, location of SMAD2 and 4 genes, occurs in BE carcinogenesisCitation112 and SMAD4 is mutated in approximately 8% of EAC.Citation15 In addition, expression of SMAD2 and SMAD4 is decreased in BE and EAC, possibly via promoter methylation in the case of SMAD4,Citation113 suggesting that response to anti-proliferative signaling by TGFβ is impaired. This was confirmed in ex vivo organ culture of normal, BE, and EAC biopsy tissue via measuring p21/WAF1 and MCM2 (a proliferation marker) expression in response to TGFβ113. Interestingly, expression of Ski and SnoN, negative regulators of SMAD transcriptional function, is also increased in BE, but is then decreased or lost in dysplasia and EAC,Citation114 suggesting a further level of regulation of this pathway in the progression from BE to EAC.
Ligand/death-receptor mediated apoptotic pathways
Apoptosis induced through the tumor necrosis factor receptor (TNFR) superfamily by ligands such as FasL/CD95L and TNF-related apoptosis inducing ligand (TRAIL) is important in the regulation of the immune system. Signaling via these pathways is also often downregulated in cancer. In addition, some cancers upregulate expression of ligands, which is thought to have an effect against immune surveillance. The evidence that modulation of pro-apoptotic ligand/receptor signaling complexes plays a role in Barrett carcinogenesis is unclear. In fact, both increased proliferation index and apoptosis rate are linked with progression to EAC, suggesting that suppression of apoptosis may be less critical in Barrett carcinogenesis compared with other cancers.Citation115
FasL expression may be increased in BE and further increased in dysplasia and EACCitation116,Citation117 and correlates with depletion of CD45+ tumor infiltrating lymphocytes,Citation118 suggesting that BE progression is associated with FasL-mediated avoidance of immune surveillance. In contrast, FasL is not expressed on the cell surface or secreted into the medium by EAC cell lines,Citation119 which is inconsistent with a role in establishing immune privilege. In contrast to FasL, TRAIL is expressed in BE but is rarely and weakly expressed in dysplasia and EAC.Citation120
Similarly, the evidence for receptor modulation is unclear. Fas expression may be either increasedCitation117,Citation121 or decreasedCitation122 in dysplasia and EAC. In vitro data show that bile salts preferentially upregulate Fas expression in the normal squamous derived Het1A cell line but not in BE-derived BAR-T or EAC-derived FLO-1 cell lines, which may suggest that bile reflux could play a role in the selection of cells that have developed apoptosis resistance via dysregulation of Fas-mediated immune surveillance.Citation123 In contrast the TRAIL receptor, DR5, is upregulated in up to 90% of EAC compared with matched normal tissue,Citation124 which would be expected to sensitize tumors to TRAIL-induced apoptosis. Thus, there is little evidence that regulation of apoptosis at the level of ligands or TNFR family members is a major mechanism driving Barrett carcinogenesis.
Apoptosis signaling downstream of both extrinsic (e.g., death receptor ligands) and intrinsic (e.g., mitochondrial centric) stimuli is regulated by a number of proteins including caspases and the pro- and anti-apoptotic members of the Bcl-2 family of proteins. Polymorphisms in the genes for caspase-7 and caspase-9 are significantly associated with an increased risk of EAC,Citation125 and polymorphisms in caspase-7 and Bcl-2 modify the risk of EAC in smokers.Citation126 Data regarding expression of Bcl-2 family members is controversial, with studies suggesting that anti-apoptotic Bcl-2 is either not expressed in BE, dysplasia or EAC,Citation127 or that it is increased in BE and LGD but decreases in HGD and EAC.Citation128-Citation130 This suggests that increased Bcl-2 may have a role early in the development of BE but not in progression to EAC. Indeed, loss of Bcl-2 in dysplastic BE and EAC has been associated with tumor progression and poor survival.Citation131 In contrast, increased anti-apoptotic Bcl-XL and decreased pro-apoptotic Bax expression have been described in the progression of BE to EAC, possibly indicative of a switch to a more anti-apoptotic state.Citation85,Citation132 Together, these data suggest that the balance of pro- and anti-apoptotic signaling may impact on the effect of environmental factors in the development of EAC. For example, a more anti-apoptotic intracellular environment may result in the survival of potential neoplastic cells in the DNA damaging and potential mutagenic environments provided by GERD and smoking.
Kinases, transcription factors, and other effectors
RAS/RAF/MAPK and PI3-kinase /AKT pathways
RAS/RAF/MAPK and PI3-kinase (PI3K)/AKT are central downstream mediators of a number of signaling pathways, particularly tyrosine kinase receptors. Together, they control a myriad of cellular processes including cell growth, proliferation, differentiation and motility, all of which are involved in tumorigenesis. In particular, MAPK pathway components were found to be upregulated in around 40% of EAC,Citation95 suggesting that using MEK inhibitors to target MAPK activation could be an effective treatment, possibly in combination with RTK inhibitors. Aside from the modulation of ligand/RTK activation as described above, there is evidence that alterations to these downstream mediators may also contribute to the progression of BE to EAC.
Expression of mutant oncogenic ras (K-ras or H-ras) is rarely found in non-dysplastic BE but is detected in up to 40% of dysplasia and EAC samples,Citation57,Citation133-Citation135 suggesting that acquisition of this mutation is important in progression. Mutation of BRAF, downstream of Ras, is also found at low frequency (5–10%) in dysplasia and EAC, although never in combination with Ras mutation,Citation135 and thus represents an alternative mechanism for activating downstream signaling. Introduction of H-ras together with RNAi knockdown of p53 in p16-deficient non-dysplastic BAR-T Barrett cells leads to tumorigenic transformation,Citation29 demonstrating a mechanistic role for Ras activation in BE carcinogenesis. However, H-Ras or p53 knockdown alone were not sufficient,Citation29 highlighting the need for multiple steps in the development of EAC.
PIK3CA, the gene that encodes for the p110α catalytic subunit of PI3K is mutated in approximately 6% of EAC but no activating mutations have been reported in BE.Citation136 PIK3CA is also amplified in a small proportion of EAC,Citation137 suggesting that acquisition of PIK3CA lesions may be involved in the progression to EAC in a small subset of patients. Phospho-Akt, an indicator of active Akt signaling, is increased along the progression from normal esophagus to BE, dysplasia, and EAC and is associated with tumor progression.Citation138,Citation139 However, this is likely due to increased upstream signaling since activating mutations in Akt have not been reported in this disease.
Perhaps not surprisingly, Ras/ERK/MAPK and PI3K/Akt activation also appear to be central to signaling pathways activated by a number of factors relevant to BE, including acid, bile, leptin, and gastrin, resulting in enhanced proliferation, inhibition of apoptosis, and upregulation of MUC1, 4 and 5AC and COX-2.Citation106,Citation138,Citation140-Citation146 Thus, there is a putative role for these central molecules in mediating signaling by multiple effectors relevant to Barrett carcinogenesis.
COX-2
Cycloxygenase-2 (COX-2) is a key enzyme in the arachidonic acid pathway that acts to produce prostaglandin as part of the inflammatory response. Chronic inflammation is believed to potentiate neoplastic development at least partially due to mediators such as prostaglandins. It is in this context that COX-2 is thought to contribute to Barrett carcinogenesis. Increased COX-2 expression is detected in the progression from BE to EAC,Citation69,Citation72,Citation147,Citation148 and is associated with proliferation and reduced survival.Citation149 However, COX-2 expression appears to be independent of the degree of inflammation, although it is highest in the distal part compared with proximal BE,Citation150 which is also the most frequent location of EAC.
COX-2 expression may also be increased as a direct effect of reflux components. Acid and bile are well established to induce COX-2 expression and prostaglandin production in vitro in EAC cell lines, in ex vivo organ cultures of BE tissue and in animal models, via a mechanism that involves reactive oxygen species-mediated PI3K/AKT and ERK/MAPK activation.Citation72,Citation142,Citation146,Citation151,Citation152 COX-2 may also be upregulated by p53 via a NFκB-dependent mechanism.Citation153
There is controversy surrounding the presence of two polymorphisms in the promoter of the COX-2 gene, which have been linked with increased expression and activity of COX-2 as well as the risk of developing EAC. Two separate studies each found different haplotypes of the same polymorphisms as being more common in EAC than controls.Citation154,Citation155 An intragenic polymorphism has also been associated with an increased risk of EAC.Citation156
There are functional data indicating a causative role for COX-2 mediated inflammation in Barrett carcinogenesis. Selective COX-2 inhibition in primary cultures of BE cells and ex vivo organ cultures of BE reduces COX-2 activity, prostaglandin production and proliferation and in primary cultures this could be reversed by addition of prostaglandin E2.Citation157,Citation158 Similar effects are seen in EAC cell lines,Citation159-Citation161 suggesting a dependence of EAC on COX-2-mediated prostaglandin production. A xenograft model study suggests that targeting COX-2 may also be a viable therapeutic option in the treatment of established EAC.Citation162
Use of aspirin and NSAIDs is associated with reduced esophageal cancer risk in population-based studies. Taken together, these data highlight the possibility of COX-2 inhibition as a chemopreventive strategy. Unfortunately, a small (100 patients) celecoxib COX-2 inhibition trial failed to show a benefit in preventing progression of dysplasia to EACCitation163 and the large AspECT chemoprevention trial also seems to have found no clear benefit from daily aspirin to prevent esophageal cancer.Citation164
NFκB
NFκB controls the transcription of a large number of genes in response to a range of stimuli including intracellular stresses and cytokine mediated activation of receptor signaling pathways. NFκB is intimately linked with regulation of the host inflammatory and immune response by regulating the expression of a number of key cytokines including TNFα, IL-1β, IL-6 and IL-8, which themselves can activate NFκB. Overactivation of NFκB has been linked to neoplasia, including EAC, through promoting cell survival, particularly in the context of chronic inflammation.
NFκB is located on chromosome 4, which is frequently amplified in Barrett carcinogenesis,Citation165 and is frequently expressed in the progression from BE to EAC.Citation166,Citation167 NFκB is activated by acid and bile in EAC cell lines, possibly via production of reactive oxygen species,Citation166,Citation168,Citation169 and thereby provides evidence for over-activation of NFκB in the progression to EAC as a consequence of GERD. This is supported by recent data showing that bile induced activation of NFκB in non-dysplastic BE cells leads to apoptosis resistance in the face of concomitant bile-induced DNA damage.Citation170,Citation171 Upregulation of COX-2 by acid and bile is also thought to be mediated by NFκB,Citation153,Citation172,Citation173 which may further enhance esophageal tumorigenesis via upregulation of additional inflammatory mediators.
Cell cycle regulators
Control of progression through the cell cycle is pivotal to regulating cellular proliferation. Much of that control is exerted through the action of cyclins and cyclin dependent kinases (CDKs) that act at different stages of the cycle. Dysregulation of cell cycle mediators appears to be central to development of EAC. Cyclin D is expressed in response to extracellular signals that promote cell proliferation, such as growth factors and forms a complex with CDK4 to phosphorylate and inactivate Rb. Nuclear cyclin D1 expression is increased in BE and is even more frequent in dysplasia and EAC.Citation174-Citation176 This may be at least partly due to the G870A polymorphism in the gene, which results in protein stabilization and a longer half-life. However, there are conflicting results regarding the presence of this polymorphism in EAC. Studies have demonstrated an association between this polymorphism and the risk of reflux disease, BE, and EAC,Citation177 as well as earlier age of onset of EAC, poorer survival and distant metastasis.Citation178,Citation179 However, these associations have not been observed in other studies.Citation180,Citation181 Cyclin E expression is also increased in a proportion of dysplastic BE and EAC and correlates with amplification of 19q12, the location of the gene for cyclin E.Citation182,Citation183
Rb, p27 and p21/WAF1 are tumor suppressor genes that block progression through the cell cycle by inhibiting cyclin-CDK complexes. Loss of heterozygosity (LOH) of the Rb locus and loss of Rb protein expression is common in EACCitation121,Citation184-Citation186 and is thought to represent a target for inactivation in the latter stages of EAC development.Citation187 In contrast, inactivation of p16, which indirectly negatively regulates the function of Rb, occurs frequently in non-dysplastic BECitation188-Citation190 or at the non-dysplasia to LGD interface,Citation191 and seems to represent one of the key early molecular events driving BE carcinogenesis. p27 expression is downregulated in the majority of EAC.Citation192 In a mouse surgically induced reflux model, development of both BE and EAC are increased in p27 knockout mice compared with wild-type,Citation193 demonstrating that loss of p27 can enhance Barrett carcinogenesis, possibly at an early stage. In contrast, p21 is increased in dysplastic BE and EAC but not non-dysplastic BE.Citation194
p53
P53 is a well-known tumor suppressor that is frequently inactivated in most cancers. The function of p53 is central to controlling both cell cycle progression and initiation of apoptosis in response to extrinsic and intrinsic signals. p53 LOH and p53 gene mutations, occur in the majority of EAC casesCitation195-Citation198 and these lesions are associated with poor outcome.Citation199-Citation202 Mutations often result in stabilized p53 protein and increased staining for p53 has been detected in non-dysplastic BE and more frequently in dysplasia and EAC.Citation196,Citation203-Citation206 However, mutations do not appear to account for the majority of p53 protein accumulation in BE carcinogenesis.Citation207,Citation208 Increased p53 expression is correlated with increased proliferation in the progression of BE to EAC in some studies,Citation209,Citation210 and may be a valuable biomarker predicting increased risk of disease progression in patients with BE.Citation211
Wnt, Notch, and Shh
The Wnt, Notch, and sonic hedgehog (Shh) signaling pathways are important for regulating cellular differentiation and proliferation during embryogenesis and normal tissue homeostasis in adults. These signaling pathways have also been implicated in tumorigenesis including development of BE and EAC.
Central to the Wnt pathway is stabilization of β-catenin and nuclear relocalization to form the β-catenin/TCF transcription complex. Nuclear accumulation of β-catenin has been commonly described in Barrett carcinogenesis and is independent of activating mutations in exon 3.Citation212-Citation214 Nuclear accumulation may be a consequence of APC LOH, which is a frequent late event in EAC,Citation215,Citation216 via APC promoter methylationCitation217-Citation219 or via upregulation of Wnt ligands and epigenetic silencing of Wnt inhibitory factor (WIF1).Citation220,Citation221 Significantly, increased Wnt signaling in organotypic cultures of squamous esophageal cells promoted expression of intestinal-type proteins that are also expressed in BE.Citation30
β-catenin can also be found in complex with cadherins, such as E-cadherin and downregulation of E-cadherin can lead to increased signaling through β-catenin. Reduced membranous E-cadherin is common in BE and EAC,Citation222,Citation223 possibly due to promoter methylation.Citation224 Stimulation of OE33 EAC cells with HGF induces nuclear β-catenin, possibly as a consequence of E-cadherin downregulation.Citation89 Similar findings have been described following stimulation with TNFα, which is upregulated in the progression from BE to EAC and also results in β-catenin mediated c-myc transcription.Citation225 Loss of E-cadherin in EAC may also be a consequence of overexpression of the transcriptional repressor, Slug.Citation226
In the intestine, the Notch pathway controls intestinal cell fate determination through promoting expression of the Hes1 transcription factor. Hes1 negatively regulates expression of Hath1/Atoh1, which in the absence of Hes1 promotes differentiation of intestinal progenitor cells into secretory cell lineages, including goblet cells.Citation227,Citation228 Notch signaling appears to play a similar role in the development of BE. Intestinal-type BE with goblet cells show lower expression of Hes1 and upregulation of Hath1/Atoh1 and MUC2 compared with the non-goblet cell, proliferative BE crypts.Citation229,Citation230 Interestingly, the bile acid DCA suppresses Hes1 in EAC cell lines, possibly via upregulation of Cdx2, leading to increased Hath1/Atoh1 expression and expression of Muc2.Citation230,Citation231 In contrast, progression of BE to EAC is associated with activation of Notch signaling and expression of Hath1 in patient tissue,Citation232 cell linesCitation229,Citation231 and a mouse model of BE/EAC.Citation27 This activation of Notch signaling, with increased SOX9 expression, is associated with dysfunctional TGFβ signaling through loss of TGFβ adaptor β2SP.Citation233
Shh signaling is important in the embryonic development of the gastrointestinal epithelium, including the esophageal epithelium and in intestinal epithelial homeostasis, but is not active in the normal adult esophagus.Citation234,Citation235 Abnormal activation of Shh signaling by acid and bile reflux has been implicated in the pathogenesis of BE,Citation236,Citation237 possibly through activation of the bone morphogenic protein-4 (BMP-4) signaling pathwayCitation238,Citation239 and the downstream transcription factor SOX9.Citation240 Hedgehog signaling and upregulation of the downstream GLI1 transcription factor may also contribute to EAC tumorigenesis,Citation241,Citation242 including through interaction with the mammalian target of rapamycin (mTOR) pathway,Citation243 which is itself activated by chronic inflammation in the esophagus.Citation244 Therefore, targeting this pathway could be an effective approach to treat BE and/or EAC, especially in combination with mTOR inhibitors.Citation243
C-myc
The c-myc transcription factor is a proto-oncogene important for regulating the expression of several genes with roles in cell proliferation and thus over-activation of c-myc has been implicated in tumorigenesis, including development of EAC. Upregulated c-myc expression increases in the progression of BE to EAC,Citation245,Citation246 possibly as a result of c-myc gene amplification, although this is not found in non-dysplastic BE.Citation45,Citation187,Citation247,Citation248 Acidified bile, but not bile or acid alone, can induce c-myc expression in OE33 EAC cells,Citation168,Citation246 demonstrating that non-genetic mechanisms may also activate c-myc-mediated transcription in Barrett carcinogenesis. Interestingly, c-myc, cooperating with caudal-type homeobox 1 (cdx1), has been implicated in the development of metaplasia,Citation249 suggesting it may also act early in Barrett carcinogenesis.
Summary
The signaling pathways reviewed above are shown schematically in , with the RTK pathways in and non-RTK pathways in . Much of the data for these pathways is descriptive, there has been a deficit of suitable cell lines and animal models and human studies have tended to compare separate cohorts of individuals rather than the same cohorts followed longitudinally. Partly for these reasons, the critical driver aberrations involved in BE/EAC pathogenesis have not been confirmed. However, activation of Notch and Hedgehog pathways, through mechanisms that include aberrant TGFβ signaling, cdx2 activation by the bile components of the refluxate and interactions with the TNFα/mTOR pathway, seem increasingly important. Obesity, especially central obesity due to visceral adipose tissue, is a highly important risk factor for EACCitation250 and BE.Citation251 With one of the strongest associations with obesity of all human cancers, EAC provides a valuable opportunity to investigate the causal relationship of adiposity with cancer. Many of the pathways reviewed here are also activated in obesity but the results for obesity-related areas such as the IGF family and adipokines have so far been mixed or conflicting.
Figure 1. Signaling pathways in the development of esophageal adenocarcinoma. Both receptor tyrosine kinase (A) and non-receptor tyrosine kinase (B) signaling pathways have been implicated in the progression of Barrett esophagus to esophageal adenocarcinoma. Black up or down arrows indicate changes in expression (usually at the protein level), Pol indicates gene polymorphism implicated in disease, Mut indicates gene mutation (usually activating), Amp indicates gene amplification, LOH indicates loss of heterozygosity, and Met indicates promoter methylation. Blue (dotted) arrows indicate effects on expression, red (dashed) arrows indicate effects on activity, gray (solid) arrows indicate translocation. ROS, reactive oxygen species.
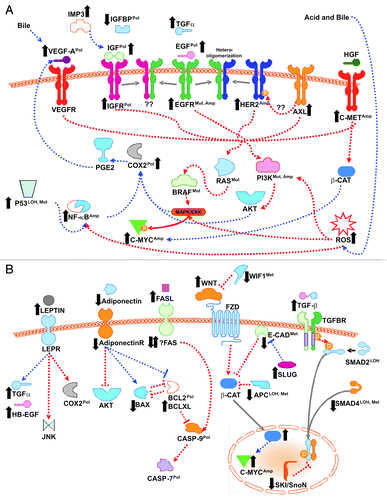
In conclusion, this review demonstrates that considerable recent progress has been made to unravel the pathways involved in EAC pathogenesis. As for other cancers, EAC research is entering an exciting era of discovery searching for associations between variations in massive scale data and disease, as exemplified by several completedCitation15 and ongoing next generation sequencing (NGS) studies and the BE genome-wide association study (GWAS).Citation252 Ultimately, the functional importance of these variations will need to be assessed. It has been shown that computational algorithms and metaanalysis can identify perturbed signaling pathways in disease, but laboratory-based pathway studies such as those reviewed here remain essential.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
References
- Crane SJ, Locke GR 3rd, Harmsen WS, Zinsmeister AR, Romero Y, Talley NJ. Survival trends in patients with gastric and esophageal adenocarcinomas: a population-based study. Mayo Clin Proc 2008; 83:1087 - 94; http://dx.doi.org/10.4065/83.10.1087; PMID: 18828967
- Bouvier AM, Binquet C, Gagnaire A, Jouve JL, Faivre J, Bedenne L. Management and prognosis of esophageal cancers: has progress been made?. Eur J Cancer 2006; 42:228 - 33; http://dx.doi.org/10.1016/j.ejca.2005.08.038; PMID: 16337786
- Younes M, Henson DE, Ertan A, Miller CC. Incidence and survival trends of esophageal carcinoma in the United States: racial and gender differences by histological type. Scand J Gastroenterol 2002; 37:1359 - 65; http://dx.doi.org/10.1080/003655202762671215; PMID: 12523583
- Pohl H, Welch HG. The role of overdiagnosis and reclassification in the marked increase of esophageal adenocarcinoma incidence. J Natl Cancer Inst 2005; 97:142 - 6; http://dx.doi.org/10.1093/jnci/dji024; PMID: 15657344
- Lagergren J, Mattsson F. No further increase in the incidence of esophageal adenocarcinoma in Sweden. Int J Cancer 2011; 129:513 - 6; http://dx.doi.org/10.1002/ijc.25701; PMID: 20878977
- Pohl H, Sirovich B, Welch HG. Esophageal adenocarcinoma incidence: are we reaching the peak?. Cancer Epidemiol Biomarkers Prev 2010; 19:1468 - 70; http://dx.doi.org/10.1158/1055-9965.EPI-10-0012; PMID: 20501776
- Bhat S, Coleman HG, Yousef F, Johnston BT, McManus DT, Gavin AT, et al. Risk of malignant progression in Barrett esophagus patients: results from a large population-based study. J Natl Cancer Inst 2011; 103:1049 - 57; http://dx.doi.org/10.1093/jnci/djr203; PMID: 21680910
- Hvid-Jensen F, Pedersen L, Drewes AM, Sørensen HT, Funch-Jensen P. Incidence of adenocarcinoma among patients with Barrett esophagus. N Engl J Med 2011; 365:1375 - 83; http://dx.doi.org/10.1056/NEJMoa1103042; PMID: 21995385
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646 - 74; http://dx.doi.org/10.1016/j.cell.2011.02.013; PMID: 21376230
- Rabinovitch PS, Reid BJ, Haggitt RC, Norwood TH, Rubin CE. Progression to cancer in Barrett esophagus is associated with genomic instability. Lab Invest 1989; 60:65 - 71; PMID: 2911184
- Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature 1998; 396:643 - 9; http://dx.doi.org/10.1038/25292; PMID: 9872311
- Zhang HY, Hormi-Carver K, Zhang X, Spechler SJ, Souza RF. In Benign Barrett's Epithelial Cells, Acid Exposure Generates Reactive Oxygen Species That Cause DNA Double-Strand Breaks. Cancer Res 2009.
- Dvorak K, Payne CM, Chavarria M, Ramsey L, Dvorakova B, Bernstein H, et al. Bile acids in combination with low pH induce oxidative stress and oxidative DNA damage: relevance to the pathogenesis of Barrett oesophagus. Gut 2007; 56:763 - 71; http://dx.doi.org/10.1136/gut.2006.103697; PMID: 17145738
- Clemons NJ, McColl KE, Fitzgerald RC. Nitric oxide and acid induce double-strand DNA breaks in Barrett esophagus carcinogenesis via distinct mechanisms. Gastroenterology 2007; 133:1198 - 209; http://dx.doi.org/10.1053/j.gastro.2007.06.061; PMID: 17919494
- Dulak AM, Stojanov P, Peng S, Lawrence MS, Fox C, Stewart C, et al. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat Genet 2013; 45:478 - 86; http://dx.doi.org/10.1038/ng.2591; PMID: 23525077
- Kaz AM, Wong CJ, Luo Y, Virgin JB, Washington MK, Willis JE, et al. DNA methylation profiling in Barrett esophagus and esophageal adenocarcinoma reveals unique methylation signatures and molecular subclasses. Epigenetics 2011; 6:1403 - 12; http://dx.doi.org/10.4161/epi.6.12.18199; PMID: 22139570
- Alvarez H, Opalinska J, Zhou L, Sohal D, Fazzari MJ, Yu Y, et al. Widespread hypomethylation occurs early and synergizes with gene amplification during esophageal carcinogenesis. PLoS Genet 2011; 7:e1001356; http://dx.doi.org/10.1371/journal.pgen.1001356; PMID: 21483804
- El-Rifai W, Frierson HF Jr., Moskaluk CA, Harper JC, Petroni GR, Bissonette EA, et al. Genetic differences between adenocarcinomas arising in Barrett esophagus and gastric mucosa. Gastroenterology 2001; 121:592 - 8; http://dx.doi.org/10.1053/gast.2001.27215; PMID: 11522743
- Lehmann K, Schneider PM. Differences in the molecular biology of adenocarcinoma of the esophagus, gastric cardia, and upper gastric third. Recent Results Cancer Res 2010; 182:65 - 72; http://dx.doi.org/10.1007/978-3-540-70579-6_5; PMID: 20676871
- Reid BJ. Early events during neoplastic progression in Barrett esophagus. Cancer Biomark 2010; 9:307 - 24; PMID: 22112482
- Maley CC, Galipeau PC, Finley JC, Wongsurawat VJ, Li X, Sanchez CA, et al. Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat Genet 2006; 38:468 - 73; http://dx.doi.org/10.1038/ng1768; PMID: 16565718
- Merlo LM, Shah NA, Li X, Blount PL, Vaughan TL, Reid BJ, et al. A comprehensive survey of clonal diversity measures in Barrett esophagus as biomarkers of progression to esophageal adenocarcinoma. Cancer Prev Res (Phila) 2010; 3:1388 - 97; http://dx.doi.org/10.1158/1940-6207.CAPR-10-0108; PMID: 20947487
- Leedham SJ, Preston SL, McDonald SA, Elia G, Bhandari P, Poller D, et al. Individual crypt genetic heterogeneity and the origin of metaplastic glandular epithelium in human Barrett oesophagus. Gut 2008; 57:1041 - 8; http://dx.doi.org/10.1136/gut.2007.143339; PMID: 18305067
- Eads CA, Lord RV, Kurumboor SK, Wickramasinghe K, Skinner ML, Long TI, et al. Fields of aberrant CpG island hypermethylation in Barrett esophagus and associated adenocarcinoma. Cancer Res 2000; 60:5021 - 6; PMID: 11016622
- Phillips WA, Lord RV, Nancarrow DJ, Watson DI, Whiteman DC. Barrett esophagus. J Gastroenterol Hepatol 2011; 26:639 - 48; http://dx.doi.org/10.1111/j.1440-1746.2010.06602.x; PMID: 21166712
- Weinstein IB, Joe A. Oncogene addiction. Cancer Res 2008; 68:3077 - 80, discussion 3080; http://dx.doi.org/10.1158/0008-5472.CAN-07-3293; PMID: 18451130
- Quante M, Bhagat G, Abrams JA, Marache F, Good P, Lee MD, et al. Bile acid and inflammation activate gastric cardia stem cells in a mouse model of Barrett-like metaplasia. Cancer Cell 2012; 21:36 - 51; http://dx.doi.org/10.1016/j.ccr.2011.12.004; PMID: 22264787
- Boonstra JJ, van Marion R, Beer DG, Lin L, Chaves P, Ribeiro C, et al. Verification and unmasking of widely used human esophageal adenocarcinoma cell lines. J Natl Cancer Inst 2010; 102:271 - 4; http://dx.doi.org/10.1093/jnci/djp499; PMID: 20075370
- Zhang X, Yu C, Wilson K, Zhang HY, Melton SD, Huo X, et al. Malignant transformation of non-neoplastic Barrett epithelial cells through well-defined genetic manipulations. PLoS One 2010; 5:5; PMID: 20927195
- Kong J, Crissey MA, Stairs DB, Sepulveda AR, Lynch JP. Cox2 and β-catenin/T-cell factor signaling intestinalize human esophageal keratinocytes when cultured under organotypic conditions. Neoplasia 2011; 13:792 - 805; PMID: 21969813
- Kosoff RE, Gardiner KL, Merlo LM, Pavlov K, Rustgi AK, Maley CC. Development and characterization of an organotypic model of Barrett esophagus. J Cell Physiol 2012; 227:2654 - 9; http://dx.doi.org/10.1002/jcp.23007; PMID: 21882191
- Sato T, Stange DE, Ferrante M, Vries RGJ, Van Es JH, Van den Brink S, et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett epithelium. Gastroenterology 2011; 141:1762 - 72; http://dx.doi.org/10.1053/j.gastro.2011.07.050; PMID: 21889923
- Croagh D, Cheng S, Tikoo A, Nandurkar S, Thomas RJ, Kaur P, et al. Reconstitution of stratified murine and human oesophageal epithelia in an in vivo transplant culture system. Scand J Gastroenterol 2008; 43:1158 - 68; http://dx.doi.org/10.1080/00365520802102489; PMID: 18609138
- Gros SJ. Orthotopic models of esophageal carcinoma and their use in drug discovery. Curr Protoc Pharmacol 2011; Chapter 14:Unit14 20.
- Jankowski J, Coghill G, Tregaskis B, Hopwood D, Wormsley KG. Epidermal growth factor in the oesophagus. Gut 1992; 33:1448 - 53; http://dx.doi.org/10.1136/gut.33.11.1448; PMID: 1452065
- Jankowski J, McMenemin R, Hopwood D, Penston J, Wormsley KG. Abnormal expression of growth regulatory factors in Barrett oesophagus. Clin Sci (Lond) 1991; 81:663 - 8; PMID: 1661653
- Jankowski J, McMenemin R, Yu C, Hopwood D, Wormsley KG. Proliferating cell nuclear antigen in oesophageal diseases; correlation with transforming growth factor alpha expression. Gut 1992; 33:587 - 91; http://dx.doi.org/10.1136/gut.33.5.587; PMID: 1351861
- Menke V, Pot RG, Moons LM, van Zoest KP, Hansen B, van Dekken H, et al. Functional single-nucleotide polymorphism of epidermal growth factor is associated with the development of Barrett esophagus and esophageal adenocarcinoma. J Hum Genet 2012; 57:26 - 32; http://dx.doi.org/10.1038/jhg.2011.124; PMID: 22129558
- Lanuti M, Liu G, Goodwin JM, Zhai R, Fuchs BC, Asomaning K, et al. A functional epidermal growth factor (EGF) polymorphism, EGF serum levels, and esophageal adenocarcinoma risk and outcome. Clin Cancer Res 2008; 14:3216 - 22; http://dx.doi.org/10.1158/1078-0432.CCR-07-4932; PMID: 18483390
- Yacoub L, Goldman H, Odze RD. Transforming growth factor-alpha, epidermal growth factor receptor, and MiB-1 expression in Barrett-associated neoplasia: correlation with prognosis. Mod Pathol 1997; 10:105 - 12; PMID: 9127315
- Wilkinson NW, Black JD, Roukhadze E, Driscoll D, Smiley S, Hoshi H, et al. Epidermal growth factor receptor expression correlates with histologic grade in resected esophageal adenocarcinoma. J Gastrointest Surg 2004; 8:448 - 53; http://dx.doi.org/10.1016/j.gassur.2004.01.006; PMID: 15120370
- Friess H, Fukuda A, Tang WH, Eichenberger A, Furlan N, Zimmermann A, et al. Concomitant analysis of the epidermal growth factor receptor family in esophageal cancer: overexpression of epidermal growth factor receptor mRNA but not of c-erbB-2 and c-erbB-3. World J Surg 1999; 23:1010 - 8; http://dx.doi.org/10.1007/s002689900616; PMID: 10512940
- Wang KL, Wu TT, Choi IS, Wang H, Resetkova E, Correa AM, et al. Expression of epidermal growth factor receptor in esophageal and esophagogastric junction adenocarcinomas: association with poor outcome. Cancer 2007; 109:658 - 67; http://dx.doi.org/10.1002/cncr.22445; PMID: 17211865
- Gibson MK, Abraham SC, Wu TT, Burtness B, Heitmiller RF, Heath E, et al. Epidermal growth factor receptor, p53 mutation, and pathological response predict survival in patients with locally advanced esophageal cancer treated with preoperative chemoradiotherapy. Clin Cancer Res 2003; 9:6461 - 8; PMID: 14695149
- Rygiel AM, Milano F, Ten Kate FJ, Schaap A, Wang KK, Peppelenbosch MP, et al. Gains and amplifications of c-myc, EGFR, and 20.q13 loci in the no dysplasia-dysplasia-adenocarcinoma sequence of Barrett esophagus. Cancer Epidemiol Biomarkers Prev 2008; 17:1380 - 5; http://dx.doi.org/10.1158/1055-9965.EPI-07-2734; PMID: 18559552
- al-Kasspooles M, Moore JH, Orringer MB, Beer DG. Amplification and over-expression of the EGFR and erbB-2 genes in human esophageal adenocarcinomas. Int J Cancer 1993; 54:213 - 9; http://dx.doi.org/10.1002/ijc.2910540209; PMID: 8098013
- Kwak EL, Jankowski J, Thayer SP, Lauwers GY, Brannigan BW, Harris PL, et al. Epidermal growth factor receptor kinase domain mutations in esophageal and pancreatic adenocarcinomas. Clin Cancer Res 2006; 12:4283 - 7; http://dx.doi.org/10.1158/1078-0432.CCR-06-0189; PMID: 16857803
- Ohashi S, Natsuizaka M, Wong GS, Michaylira CZ, Grugan KD, Stairs DB, et al. Epidermal growth factor receptor and mutant p53 expand an esophageal cellular subpopulation capable of epithelial-to-mesenchymal transition through ZEB transcription factors. Cancer Res 2010; 70:4174 - 84; http://dx.doi.org/10.1158/0008-5472.CAN-09-4614; PMID: 20424117
- Dahlberg PS, Jacobson BA, Dahal G, Fink JM, Kratzke RA, Maddaus MA, et al. ERBB2 amplifications in esophageal adenocarcinoma. Ann Thorac Surg 2004; 78:1790 - 800; http://dx.doi.org/10.1016/j.athoracsur.2004.05.037; PMID: 15511476
- Walch A, Specht K, Braselmann H, Stein H, Siewert JR, Hopt U, et al. Coamplification and coexpression of GRB7 and ERBB2 is found in high grade intraepithelial neoplasia and in invasive Barrett carcinoma. Int J Cancer 2004; 112:747 - 53; http://dx.doi.org/10.1002/ijc.20411; PMID: 15386389
- Reichelt U, Duesedau P, Tsourlakis MCh, Quaas A, Link BC, Schurr PG, et al. Frequent homogeneous HER-2 amplification in primary and metastatic adenocarcinoma of the esophagus. Mod Pathol 2007; 20:120 - 9; http://dx.doi.org/10.1038/modpathol.3800712; PMID: 17143264
- Rossi E, Villanacci V, Bassotti G, Casa DD, Missale G, Minelli L, et al. Her-2/neu in barrett esophagus: a comparative study between histology, immunohistochemistry, and fluorescence in situ hybridization. Diagn Mol Pathol 2006; 15:125 - 30; http://dx.doi.org/10.1097/01.pdm.0000213455.22527.f7; PMID: 16932066
- Brien TP, Odze RD, Sheehan CE, McKenna BJ, Ross JS. HER-2/neu gene amplification by FISH predicts poor survival in Barrett esophagus-associated adenocarcinoma. Hum Pathol 2000; 31:35 - 9; http://dx.doi.org/10.1016/S0046-8177(00)80195-1; PMID: 10665910
- Thompson SK, Sullivan TR, Davies R, Ruszkiewicz AR. Her-2/neu gene amplification in esophageal adenocarcinoma and its influence on survival. Ann Surg Oncol 2011; 18:2010 - 7; http://dx.doi.org/10.1245/s10434-011-1554-1; PMID: 21267790
- Hu Y, Bandla S, Godfrey TE, Tan D, Luketich JD, Pennathur A, et al. HER2 amplification, overexpression and score criteria in esophageal adenocarcinoma. Mod Pathol 2011; 24:899 - 907; http://dx.doi.org/10.1038/modpathol.2011.47; PMID: 21460800
- Mukherjee K, Chakravarthy AB, Goff LW, El-Rifai W. Esophageal adenocarcinoma: treatment modalities in the era of targeted therapy. Dig Dis Sci 2010; 55:3304 - 14; http://dx.doi.org/10.1007/s10620-010-1187-4; PMID: 20300841
- Lord RV, O’Grady R, Sheehan C, Field AF, Ward RL. K-ras codon 12 mutations in Barrett oesophagus and adenocarcinomas of the oesophagus and oesophagogastric junction. J Gastroenterol Hepatol 2000; 15:730 - 6; http://dx.doi.org/10.1046/j.1440-1746.2000.02163.x; PMID: 10937677
- Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, et al, ToGA Trial Investigators. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet 2010; 376:687 - 97; http://dx.doi.org/10.1016/S0140-6736(10)61121-X; PMID: 20728210
- Möbius C, Stein HJ, Becker I, Feith M, Theisen J, Gais P, et al. The ‘angiogenic switch’ in the progression from Barrett metaplasia to esophageal adenocarcinoma. Eur J Surg Oncol 2003; 29:890 - 4; http://dx.doi.org/10.1016/j.ejso.2003.07.002; PMID: 14624783
- Couvelard A, Paraf F, Gratio V, Scoazec JY, Hénin D, Degott C, et al. Angiogenesis in the neoplastic sequence of Barrett oesophagus. Correlation with VEGF expression. J Pathol 2000; 192:14 - 8; http://dx.doi.org/10.1002/1096-9896(2000)9999:9999<::AID-PATH709>3.0.CO;2-F; PMID: 10951394
- Lord RV, Park JM, Wickramasinghe K, DeMeester SR, Oberg S, Salonga D, et al. Vascular endothelial growth factor and basic fibroblast growth factor expression in esophageal adenocarcinoma and Barrett esophagus. J Thorac Cardiovasc Surg 2003; 125:246 - 53; http://dx.doi.org/10.1067/mtc.2003.203; PMID: 12579092
- Griffiths EA, Pritchard SA, McGrath SM, Valentine HR, Price PM, Welch IM, et al. Increasing expression of hypoxia-inducible proteins in the Barrett metaplasia-dysplasia-adenocarcinoma sequence. Br J Cancer 2007; 96:1377 - 83; PMID: 17437013
- Möbius C, Stein HJ, Becker I, Feith M, Theisen J, Gais P, et al. Vascular endothelial growth factor expression and neovascularization in Barrett carcinoma. World J Surg 2004; 28:675 - 9; http://dx.doi.org/10.1007/s00268-004-7286-7; PMID: 15175900
- Millikan KW, Mall JW, Myers JA, Hollinger EF, Doolas A, Saclarides TJ. Do angiogenesis and growth factor expression predict prognosis of esophageal cancer?. Am Surg 2000; 66:401 - 5, discussion 405-6; PMID: 10776879
- Auvinen MI, Sihvo EI, Ruohtula T, Salminen JT, Koivistoinen A, Siivola P, et al. Incipient angiogenesis in Barrett epithelium and lymphangiogenesis in Barrett adenocarcinoma. J Clin Oncol 2002; 20:2971 - 9; http://dx.doi.org/10.1200/JCO.2002.09.011; PMID: 12089227
- Möbius C, Freire J, Becker I, Feith M, Brücher BL, Hennig M, et al. VEGF-C expression in squamous cell carcinoma and adenocarcinoma of the esophagus. World J Surg 2007; 31:1768 - 72, discussion 1773-4; http://dx.doi.org/10.1007/s00268-006-0373-1; PMID: 17354029
- Neulen J, Yan Z, Raczek S, Weindel K, Keck C, Weich HA, et al. Human chorionic gonadotropin-dependent expression of vascular endothelial growth factor/vascular permeability factor in human granulosa cells: importance in ovarian hyperstimulation syndrome. J Clin Endocrinol Metab 1995; 80:1967 - 71; http://dx.doi.org/10.1210/jc.80.6.1967; PMID: 7775647
- Couvelard A, Paraf F, Vidaud D, Dubois S, Vidaud M, Fléjou JF, et al. Human chorionic gonadotrophin beta expression in malignant Barrett oesophagus. Virchows Arch 2004; 445:279 - 84; http://dx.doi.org/10.1007/s00428-004-1078-1; PMID: 15309632
- Wilson KT, Fu S, Ramanujam KS, Meltzer SJ. Increased expression of inducible nitric oxide synthase and cyclooxygenase-2 in Barrett esophagus and associated adenocarcinomas. Cancer Res 1998; 58:2929 - 34; PMID: 9679948
- von Rahden BH, Stein HJ, Hartl SA, Theisen J, Stigler B, Siewert JR, et al. Expression of prostaglandin E synthase in Barrett cancer. Dis Esophagus 2008; 21:304 - 8; http://dx.doi.org/10.1111/j.1442-2050.2007.00801.x; PMID: 18477251
- von Rahden BH, Stein HJ, Pühringer F, Koch I, Langer R, Piontek G, et al. Coexpression of cyclooxygenases (COX-1, COX-2) and vascular endothelial growth factors (VEGF-A, VEGF-C) in esophageal adenocarcinoma. Cancer Res 2005; 65:5038 - 44; http://dx.doi.org/10.1158/0008-5472.CAN-04-1107; PMID: 15958546
- Shirvani VN, Ouatu-Lascar R, Kaur BS, Omary MB, Triadafilopoulos G. Cyclooxygenase 2 expression in Barrett esophagus and adenocarcinoma: Ex vivo induction by bile salts and acid exposure. Gastroenterology 2000; 118:487 - 96; http://dx.doi.org/10.1016/S0016-5085(00)70254-X; PMID: 10702199
- Lanas A, Ortego J, Sopeña F, Alcedo J, Barrio E, Bujanda L, et al. Effects of long-term cyclo-oxygenase 2 selective and acid inhibition on Barrett oesophagus. Aliment Pharmacol Ther 2007; 26:913 - 23; http://dx.doi.org/10.1111/j.1365-2036.2007.03429.x; PMID: 17767476
- Burnat G, Rau T, Elshimi E, Hahn EG, Konturek PC. Bile acids induce overexpression of homeobox gene CDX-2 and vascular endothelial growth factor (VEGF) in human Barrett esophageal mucosa and adenocarcinoma cell line. Scand J Gastroenterol 2007; 42:1460 - 5; http://dx.doi.org/10.1080/00365520701452209; PMID: 17852856
- Zhai R, Liu G, Asomaning K, Su L, Kulke MH, Heist RS, et al. Genetic polymorphisms of VEGF, interactions with cigarette smoking exposure and esophageal adenocarcinoma risk. Carcinogenesis 2008; 29:2330 - 4; http://dx.doi.org/10.1093/carcin/bgn210; PMID: 18780893
- Chow WH, Blot WJ, Vaughan TL, Risch HA, Gammon MD, Stanford JL, et al. Body mass index and risk of adenocarcinomas of the esophagus and gastric cardia. J Natl Cancer Inst 1998; 90:150 - 5; http://dx.doi.org/10.1093/jnci/90.2.150; PMID: 9450576
- Kamat P, Wen S, Morris J, Anandasabapathy S. Exploring the association between elevated body mass index and Barrett esophagus: a systematic review and meta-analysis. Ann Thorac Surg 2009; 87:655 - 62; http://dx.doi.org/10.1016/j.athoracsur.2008.08.003; PMID: 19161814
- Corley DA, Kubo A, Levin TR, Block G, Habel L, Zhao W, et al. Abdominal obesity and body mass index as risk factors for Barrett esophagus. Gastroenterology 2007; 133:34 - 41, quiz 311; http://dx.doi.org/10.1053/j.gastro.2007.04.046; PMID: 17631128
- Edelstein ZR, Farrow DC, Bronner MP, Rosen SN, Vaughan TL. Central adiposity and risk of Barrett esophagus. Gastroenterology 2007; 133:403 - 11; http://dx.doi.org/10.1053/j.gastro.2007.05.026; PMID: 17681161
- Ryan AM, Healy LA, Power DG, Byrne M, Murphy S, Byrne PJ, et al. Barrett esophagus: prevalence of central adiposity, metabolic syndrome, and a proinflammatory state. Ann Surg 2008; 247:909 - 15; http://dx.doi.org/10.1097/SLA.0b013e3181612cac; PMID: 18520215
- Renehan AG, Frystyk J, Flyvbjerg A. Obesity and cancer risk: the role of the insulin-IGF axis. Trends Endocrinol Metab 2006; 17:328 - 36; http://dx.doi.org/10.1016/j.tem.2006.08.006; PMID: 16956771
- Di Martino E, Wild CP, Rotimi O, Darnton JS, Olliver RJ, Hardie LJ. IGFBP-3 and IGFBP-10 (CYR61) up-regulation during the development of Barrett oesophagus and associated oesophageal adenocarcinoma: potential biomarkers of disease risk. Biomarkers 2006; 11:547 - 61; http://dx.doi.org/10.1080/13547500600896791; PMID: 17056474
- Greer KB, Thompson CL, Brenner L, Bednarchik B, Dawson D, Willis J, et al. Association of insulin and insulin-like growth factors with Barrett oesophagus. Gut 2012; 61:665 - 72; http://dx.doi.org/10.1136/gutjnl-2011-300641; PMID: 21930730
- Siahpush SH, Vaughan TL, Lampe JN, Freeman R, Lewis S, Odze RD, et al. Longitudinal study of insulin-like growth factor, insulin-like growth factor binding protein-3, and their polymorphisms: risk of neoplastic progression in Barrett esophagus. Cancer Epidemiol Biomarkers Prev 2007; 16:2387 - 95; http://dx.doi.org/10.1158/1055-9965.EPI-06-0986; PMID: 18006928
- Iravani S, Zhang HQ, Yuan ZQ, Cheng JQ, Karl RC, Jove R, et al. Modification of insulin-like growth factor 1 receptor, c-Src, and Bcl-XL protein expression during the progression of Barrett neoplasia. Hum Pathol 2003; 34:975 - 82; http://dx.doi.org/10.1053/S0046-8177(03)00354-X; PMID: 14608530
- Kalinina T, Bockhorn M, Kaifi JT, Thieltges S, Güngör C, Effenberger KE, et al. Insulin-like growth factor-1 receptor as a novel prognostic marker and its implication as a cotarget in the treatment of human adenocarcinoma of the esophagus. Int J Cancer 2010; 127:1931 - 40; http://dx.doi.org/10.1002/ijc.25196; PMID: 20104520
- Zhao R, Macdonald K, Casson AG. Insulin-like growth factor type I receptor gene expression and obesity in esophageal adenocarcinoma. Mol Carcinog 2009; 48:982 - 8; http://dx.doi.org/10.1002/mc.20562; PMID: 19582762
- MacDonald K, Porter GA, Guernsey DL, Zhao R, Casson AG. A polymorphic variant of the insulin-like growth factor type I receptor gene modifies risk of obesity for esophageal adenocarcinoma. Cancer Epidemiol 2009; 33:37 - 40; http://dx.doi.org/10.1016/j.canep.2009.04.014; PMID: 19679045
- Anderson MR, Harrison R, Atherfold PA, Campbell MJ, Darnton SJ, Obszynska J, et al. Met receptor signaling: a key effector in esophageal adenocarcinoma. Clin Cancer Res 2006; 12:5936 - 43; http://dx.doi.org/10.1158/1078-0432.CCR-06-1208; PMID: 17062664
- Herrera LJ, El-Hefnawy T, Queiroz de Oliveira PE, Raja S, Finkelstein S, Gooding W, et al. The HGF receptor c-Met is overexpressed in esophageal adenocarcinoma. Neoplasia 2005; 7:75 - 84; http://dx.doi.org/10.1593/neo.04367; PMID: 15720819
- Tuynman JB, Lagarde SM, Ten Kate FJ, Richel DJ, van Lanschot JJ. Met expression is an independent prognostic risk factor in patients with oesophageal adenocarcinoma. Br J Cancer 2008; 98:1102 - 8; http://dx.doi.org/10.1038/sj.bjc.6604251; PMID: 18349821
- Miller CT, Lin L, Casper AM, Lim J, Thomas DG, Orringer MB, et al. Genomic amplification of MET with boundaries within fragile site FRA7G and upregulation of MET pathways in esophageal adenocarcinoma. Oncogene 2006; 25:409 - 18; PMID: 16186806
- Hector A, Montgomery EA, Karikari C, Canto M, Dunbar KB, Wang JS, et al. The Axl receptor tyrosine kinase is an adverse prognostic factor and a therapeutic target in esophageal adenocarcinoma. Cancer Biol Ther 2010; 10:1009 - 18; http://dx.doi.org/10.4161/cbt.10.10.13248; PMID: 20818175
- Holland SJ, Pan A, Franci C, Hu Y, Chang B, Li W, et al. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res 2010; 70:1544 - 54; http://dx.doi.org/10.1158/0008-5472.CAN-09-2997; PMID: 20145120
- Paterson AL, Shannon NB, Lao-Sirieix P, Ong CA, Peters CJ, O’Donovan M, et al. A systematic approach to therapeutic target selection in oesophago-gastric cancer. Gut 2012; http://dx.doi.org/10.1136/gutjnl-2012-302039; PMID: 22773546
- Hardwick JC, Van Den Brink GR, Offerhaus GJ, Van Deventer SJ, Peppelenbosch MP. Leptin is a growth factor for colonic epithelial cells. Gastroenterology 2001; 121:79 - 90; http://dx.doi.org/10.1053/gast.2001.25490; PMID: 11438496
- Bråkenhielm E, Veitonmäki N, Cao R, Kihara S, Matsuzawa Y, Zhivotovsky B, et al. Adiponectin-induced antiangiogenesis and antitumor activity involve caspase-mediated endothelial cell apoptosis. Proc Natl Acad Sci U S A 2004; 101:2476 - 81; http://dx.doi.org/10.1073/pnas.0308671100; PMID: 14983034
- Ishikawa M, Kitayama J, Kazama S, Hiramatsu T, Hatano K, Nagawa H. Plasma adiponectin and gastric cancer. Clin Cancer Res 2005; 11:466 - 72; PMID: 15701829
- Otake S, Takeda H, Suzuki Y, Fukui T, Watanabe S, Ishihama K, et al. Association of visceral fat accumulation and plasma adiponectin with colorectal adenoma: evidence for participation of insulin resistance. Clin Cancer Res 2005; 11:3642 - 6; http://dx.doi.org/10.1158/1078-0432.CCR-04-1868; PMID: 15897559
- Francois F, Roper J, Goodman AJ, Pei Z, Ghumman M, Mourad M, et al. The association of gastric leptin with oesophageal inflammation and metaplasia. Gut 2008; 57:16 - 24; http://dx.doi.org/10.1136/gut.2007.131672; PMID: 17761783
- Kendall BJ, Macdonald GA, Hayward NK, Prins JB, Brown I, Walker N, et al, Study of Digestive Health. Leptin and the risk of Barrett oesophagus. Gut 2008; 57:448 - 54; http://dx.doi.org/10.1136/gut.2007.131243; PMID: 18178609
- Thompson OM, Beresford SA, Kirk EA, Bronner MP, Vaughan TL. Serum leptin and adiponectin levels and risk of Barrett esophagus and intestinal metaplasia of the gastroesophageal junction. Obesity (Silver Spring) 2010; 18:2204 - 11; http://dx.doi.org/10.1038/oby.2009.508; PMID: 20111023
- Rubenstein JH, Dahlkemper A, Kao JY, Zhang M, Morgenstern H, McMahon L, et al. A pilot study of the association of low plasma adiponectin and Barrett esophagus. Am J Gastroenterol 2008; 103:1358 - 64; http://dx.doi.org/10.1111/j.1572-0241.2008.01823.x; PMID: 18510610
- Yildirim A, Bilici M, Cayir K, Yanmaz V, Yildirim S, Tekin SB. Serum adiponectin levels in patients with esophageal cancer. Jpn J Clin Oncol 2009; 39:92 - 6; http://dx.doi.org/10.1093/jjco/hyn143; PMID: 19116211
- Konturek PC, Burnat G, Rau T, Hahn EG, Konturek S. Effect of adiponectin and ghrelin on apoptosis of Barrett adenocarcinoma cell line. Dig Dis Sci 2008; 53:597 - 605; http://dx.doi.org/10.1007/s10620-007-9922-1; PMID: 17763959
- Ogunwobi O, Mutungi G, Beales IL. Leptin stimulates proliferation and inhibits apoptosis in Barrett esophageal adenocarcinoma cells by cyclooxygenase-2-dependent, prostaglandin-E2-mediated transactivation of the epidermal growth factor receptor and c-Jun NH2-terminal kinase activation. Endocrinology 2006; 147:4505 - 16; http://dx.doi.org/10.1210/en.2006-0224; PMID: 16740977
- Beales IL, Ogunwobi OO. Leptin synergistically enhances the anti-apoptotic and growth-promoting effects of acid in OE33 oesophageal adenocarcinoma cells in culture. Mol Cell Endocrinol 2007; 274:60 - 8; http://dx.doi.org/10.1016/j.mce.2007.05.017; PMID: 17618045
- Ogunwobi OO, Beales IL. Globular adiponectin, acting via adiponectin receptor-1, inhibits leptin-stimulated oesophageal adenocarcinoma cell proliferation. Mol Cell Endocrinol 2008; 285:43 - 50; http://dx.doi.org/10.1016/j.mce.2008.01.023; PMID: 18313838
- Koliopanos A, Friess H, di Mola FF, Tang WH, Kubulus D, Brigstock D, et al. Connective tissue growth factor gene expression alters tumor progression in esophageal cancer. World J Surg 2002; 26:420 - 7; http://dx.doi.org/10.1007/s00268-001-0242-x; PMID: 11910473
- von Rahden BH, Stein HJ, Feith M, Pühringer F, Theisen J, Siewert JR, et al. Overexpression of TGF-beta1 in esophageal (Barrett) adenocarcinoma is associated with advanced stage of disease and poor prognosis. Mol Carcinog 2006; 45:786 - 94; http://dx.doi.org/10.1002/mc.20259; PMID: 16921482
- Rees JR, Onwuegbusi BA, Save VE, Alderson D, Fitzgerald RC. In vivo and in vitro evidence for transforming growth factor-beta1-mediated epithelial to mesenchymal transition in esophageal adenocarcinoma. Cancer Res 2006; 66:9583 - 90; http://dx.doi.org/10.1158/0008-5472.CAN-06-1842; PMID: 17018615
- Barrett MT, Galipeau PC, Sanchez CA, Emond MJ, Reid BJ. Determination of the frequency of loss of heterozygosity in esophageal adenocarcinoma by cell sorting, whole genome amplification and microsatellite polymorphisms. Oncogene 1996; 12:1873 - 8; PMID: 8649847
- Onwuegbusi BA, Aitchison A, Chin SF, Kranjac T, Mills I, Huang Y, et al. Impaired transforming growth factor beta signalling in Barrett carcinogenesis due to frequent SMAD4 inactivation. Gut 2006; 55:764 - 74; http://dx.doi.org/10.1136/gut.2005.076430; PMID: 16368780
- Villanacci V, Bellone G, Battaglia E, Rossi E, Carbone A, Prati A, et al. Ski/SnoN expression in the sequence metaplasia-dysplasia-adenocarcinoma of Barrett esophagus. Hum Pathol 2008; 39:403 - 9; http://dx.doi.org/10.1016/j.humpath.2007.07.009; PMID: 18261624
- Soslow RA, Remotti H, Baergen RN, Altorki NK. Suppression of apoptosis does not foster neoplastic growth in Barrett esophagus. Mod Pathol 1999; 12:239 - 50; PMID: 10102608
- Younes M, Schwartz MR, Finnie D, Younes A. Overexpression of Fas ligand (FasL) during malignant transformation in the large bowel and in Barrett metaplasia of the esophagus. Hum Pathol 1999; 30:1309 - 13; http://dx.doi.org/10.1016/S0046-8177(99)90061-8; PMID: 10571510
- Younes M, Lechago J, Ertan A, Finnie D, Younes A. Decreased expression of Fas (CD95/APO1) associated with goblet cell metaplasia in Barrett esophagus. Hum Pathol 2000; 31:434 - 8; http://dx.doi.org/10.1053/hp.2000.6715; PMID: 10821489
- Bennett MW, O’Connell J, O’Sullivan GC, Brady C, Roche D, Collins JK, et al. The Fas counterattack in vivo: apoptotic depletion of tumor-infiltrating lymphocytes associated with Fas ligand expression by human esophageal carcinoma. J Immunol 1998; 160:5669 - 75; PMID: 9605174
- Watson GA, Naran S, Zhang X, Stang MT, Queiroz de Oliveira PE, Hughes SJ. Cytoplasmic overexpression of CD95L in esophageal adenocarcinoma cells overcomes resistance to CD95-mediated apoptosis. Neoplasia 2011; 13:198 - 205; PMID: 21390183
- Popnikolov NK, Gatalica Z, Adegboyega PA, Norris BA, Pasricha PJ. Downregulation of TNF-related apoptosis-inducing ligand (TRAIL)/Apo2L in Barrett esophagus with dysplasia and adenocarcinoma. Appl Immunohistochem Mol Morphol 2006; 14:161 - 5; http://dx.doi.org/10.1097/01.pai.0000157905.30872.9f; PMID: 16785783
- Coppola D, Schreiber RH, Mora L, Dalton W, Karl RC. Significance of Fas and retinoblastoma protein expression during the progression of Barrett metaplasia to adenocarcinoma. Ann Surg Oncol 1999; 6:298 - 304; http://dx.doi.org/10.1007/s10434-999-0298-7; PMID: 10340890
- Hughes SJ, Nambu Y, Soldes OS, Hamstra D, Rehemtulla A, Iannettoni MD, et al. Fas/APO-1 (CD95) is not translocated to the cell membrane in esophageal adenocarcinoma. Cancer Res 1997; 57:5571 - 8; PMID: 9407969
- Naran S, Abrams P, de Oliveira PQ, Hughes SJ. Bile salts differentially sensitize esophageal squamous cells to CD95 (Fas/Apo-1 receptor) mediated apoptosis. J Surg Res 2011; 171:504 - 9; http://dx.doi.org/10.1016/j.jss.2010.05.001; PMID: 20934723
- Peng D, Sheta EA, Powell SM, Moskaluk CA, Washington K, Goldknopf IL, et al. Alterations in Barrett-related adenocarcinomas: a proteomic approach. Int J Cancer 2008; 122:1303 - 10; http://dx.doi.org/10.1002/ijc.23258; PMID: 18000824
- Liu CY, Wu MC, Chen F, Ter-Minassian M, Asomaning K, Zhai R, et al. A Large-scale genetic association study of esophageal adenocarcinoma risk. Carcinogenesis 2010; 31:1259 - 63; http://dx.doi.org/10.1093/carcin/bgq092; PMID: 20453000
- Wu I-C, Zhao Y, Zhai R, Liu CY, Chen F, Ter-Minassian M, et al. Interactions between genetic polymorphisms in the apoptotic pathway and environmental factors on esophageal adenocarcinoma risk. Carcinogenesis 2011; 32:502 - 6; http://dx.doi.org/10.1093/carcin/bgq287; PMID: 21212151
- Goldblum JR, Rice TW. bcl-2 protein expression in the Barrett metaplasia-dysplasia-carcinoma sequence. Mod Pathol 1995; 8:866 - 9; PMID: 8552577
- Katada N, Hinder RA, Smyrk TC, Hirabayashi N, Perdikis G, Lund RJ, et al. Apoptosis is inhibited early in the dysplasia-carcinoma sequence of Barrett esophagus. Arch Surg 1997; 132:728 - 33; http://dx.doi.org/10.1001/archsurg.1997.01430310042007; PMID: 9230856
- Rioux-Leclercq N, Turlin B, Sutherland F, Heresbach N, Launois B, Campion JP, et al. Analysis of Ki-67, p53 and Bcl-2 expression in the dysplasia-carcinoma sequence of Barrett esophagus. Oncol Rep 1999; 6:877 - 82; PMID: 10373674
- Lauwers GY, Kandemir O, Kubilis PS, Scott GV. Cellular kinetics in Barrett epithelium carcinogenic sequence: roles of apoptosis, bcl-2 protein, and cellular proliferation. Mod Pathol 1997; 10:1201 - 8; PMID: 9436964
- Raouf AA, Evoy DA, Carton E, Mulligan E, Griffin MM, Reynolds JV. Loss of Bcl-2 expression in Barrett dysplasia and adenocarcinoma is associated with tumor progression and worse survival but not with response to neoadjuvant chemoradiation. Dis Esophagus 2003; 16:17 - 23; http://dx.doi.org/10.1046/j.1442-2050.2003.00281.x; PMID: 12581249
- van der Woude CJ, Jansen PL, Tiebosch AT, Beuving A, Homan M, Kleibeuker JH, et al. Expression of apoptosis-related proteins in Barrett metaplasia-dysplasia-carcinoma sequence: a switch to a more resistant phenotype. Hum Pathol 2002; 33:686 - 92; http://dx.doi.org/10.1053/hupa.2002.124908; PMID: 12196918
- Galiana C, Lozano JC, Bancel B, Nakazawa H, Yamasaki H. High frequency of Ki-ras amplification and p53 gene mutations in adenocarcinomas of the human esophagus. Mol Carcinog 1995; 14:286 - 93; http://dx.doi.org/10.1002/mc.2940140409; PMID: 8519418
- Trautmann B, Wittekind C, Strobel D, Meixner H, Keymling J, Gossner L, et al. K-ras point mutations are rare events in premalignant forms of Barrett oesophagus. Eur J Gastroenterol Hepatol 1996; 8:799 - 804; PMID: 8864678
- Sommerer F, Vieth M, Markwarth A, Röhrich K, Vomschloss S, May A, et al. Mutations of BRAF and KRAS2 in the development of Barrett adenocarcinoma. Oncogene 2004; 23:554 - 8; http://dx.doi.org/10.1038/sj.onc.1207189; PMID: 14724583
- Phillips WA, Russell SE, Ciavarella ML, Choong DY, Montgomery KG, Smith K, et al. Mutation analysis of PIK3CA and PIK3CB in esophageal cancer and Barrett esophagus. Int J Cancer 2006; 118:2644 - 6; http://dx.doi.org/10.1002/ijc.21706; PMID: 16380997
- Miller CT, Moy JR, Lin L, Schipper M, Normolle D, Brenner DE, et al. Gene amplification in esophageal adenocarcinomas and Barrett with high-grade dysplasia. Clin Cancer Res 2003; 9:4819 - 25; PMID: 14581353
- Beales IL, Ogunwobi O, Cameron E, El-Amin K, Mutungi G, Wilkinson M. Activation of Akt is increased in the dysplasia-carcinoma sequence in Barrett oesophagus and contributes to increased proliferation and inhibition of apoptosis: a histopathological and functional study. BMC Cancer 2007; 7:97; http://dx.doi.org/10.1186/1471-2407-7-97; PMID: 17559672
- Sagatys E, Garrett CR, Boulware D, Kelley S, Malafa M, Cheng JQ, et al. Activation of the serine/threonine protein kinase Akt during the progression of Barrett neoplasia. Hum Pathol 2007; 38:1526 - 31; http://dx.doi.org/10.1016/j.humpath.2007.03.003; PMID: 17640711
- Mariette C, Piessen G, Leteurtre E, Hémon B, Triboulet JP, Van Seuningen I. Activation of MUC1 mucin expression by bile acids in human esophageal adenocarcinomatous cells and tissues is mediated by the phosphatidylinositol 3-kinase. Surgery 2008; 143:58 - 71; http://dx.doi.org/10.1016/j.surg.2007.07.043; PMID: 18154934
- Mariette C, Perrais M, Leteurtre E, Jonckheere N, Hémon B, Pigny P, et al. Transcriptional regulation of human mucin MUC4 by bile acids in oesophageal cancer cells is promoter-dependent and involves activation of the phosphatidylinositol 3-kinase signalling pathway. Biochem J 2004; 377:701 - 8; http://dx.doi.org/10.1042/BJ20031132; PMID: 14583090
- Zhang F, Altorki NK, Wu YC, Soslow RA, Subbaramaiah K, Dannenberg AJ. Duodenal reflux induces cyclooxygenase-2 in the esophageal mucosa of rats: evidence for involvement of bile acids. Gastroenterology 2001; 121:1391 - 9; http://dx.doi.org/10.1053/gast.2001.29781; PMID: 11729118
- Song S, Byrd JC, Guha S, Liu K-F, Koul D, Bresalier RS. Induction of MUC5AC mucin by conjugated bile acids in the esophagus involves the phosphatidylinositol 3-kinase/protein kinase C/activator protein-1 pathway. Cancer 2010; 117:2386 - 97; http://dx.doi.org/10.1002/cncr.25796; PMID: 21157954
- Ogunwobi OO, Beales IL. Glycine-extended gastrin stimulates proliferation via JAK2- and Akt-dependent NF-kappaB activation in Barrett oesophageal adenocarcinoma cells. Mol Cell Endocrinol 2008; 296:94 - 102; http://dx.doi.org/10.1016/j.mce.2008.08.004; PMID: 18771702
- Harris JC, Clarke PA, Awan A, Jankowski J, Watson SA. An antiapoptotic role for gastrin and the gastrin/CCK-2 receptor in Barrett esophagus. Cancer Res 2004; 64:1915 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-03-2713; PMID: 15026323
- Song S, Guha S, Liu K, Buttar NS, Bresalier RS. COX-2 induction by unconjugated bile acids involves reactive oxygen species-mediated signalling pathways in Barrett oesophagus and oesophageal adenocarcinoma. Gut 2007; 56:1512 - 21; http://dx.doi.org/10.1136/gut.2007.121244; PMID: 17604323
- Morris CD, Armstrong GR, Bigley G, Green H, Attwood SE. Cyclooxygenase-2 expression in the Barrett metaplasia-dysplasia-adenocarcinoma sequence. Am J Gastroenterol 2001; 96:990 - 6; PMID: 11316217
- Botelho NK, Schneiders FI, Lord SJ, Freeman AK, Tyagi S, Nancarrow DJ, et al. Gene expression alterations in formalin-fixed, paraffin-embedded Barrett esophagus and esophageal adenocarcinoma tissues. Cancer Biol Ther 2010; 10:172 - 9; http://dx.doi.org/10.4161/cbt.10.2.12166; PMID: 20543560
- Möbius C, Stein HJ, Spiess C, Becker I, Feith M, Theisen J, et al. COX2 expression, angiogenesis, proliferation and survival in Barrett cancer. Eur J Surg Oncol 2005; 31:755 - 9; http://dx.doi.org/10.1016/j.ejso.2005.01.006; PMID: 15979837
- Abdalla SI, Sanderson IR, Fitzgerald RC. Effect of inflammation on cyclooxygenase (COX)-2 expression in benign and malignant oesophageal cells. Carcinogenesis 2005; 26:1627 - 33; http://dx.doi.org/10.1093/carcin/bgi114; PMID: 15878911
- Zhang F, Subbaramaiah K, Altorki N, Dannenberg AJ. Dihydroxy bile acids activate the transcription of cyclooxygenase-2. J Biol Chem 1998; 273:2424 - 8; http://dx.doi.org/10.1074/jbc.273.4.2424; PMID: 9442092
- Looby E, Abdel-Latif MM, Athié-Morales V, Duggan S, Long A, Kelleher D. Deoxycholate induces COX-2 expression via Erk1/2-, p38-MAPK and AP-1-dependent mechanisms in esophageal cancer cells. BMC Cancer 2009; 9:190; http://dx.doi.org/10.1186/1471-2407-9-190; PMID: 19534809
- Benoit V, de Moraes E, Dar NA, Taranchon E, Bours V, Hautefeuille A, et al. Transcriptional activation of cyclooxygenase-2 by tumor suppressor p53 requires nuclear factor-kappaB. Oncogene 2006; 25:5708 - 18; http://dx.doi.org/10.1038/sj.onc.1209579; PMID: 16682957
- Moons LM, Kuipers EJ, Rygiel AM, Groothuismink AZ, Geldof H, Bode WA, et al. COX-2 CA-haplotype is a risk factor for the development of esophageal adenocarcinoma. Am J Gastroenterol 2007; 102:2373 - 9; http://dx.doi.org/10.1111/j.1572-0241.2007.01373.x; PMID: 17581270
- Kristinsson JO, van Westerveld P, te Morsche RH, Roelofs HM, Wobbes T, Witteman BJ, et al. Cyclooxygenase-2 polymorphisms and the risk of esophageal adeno- or squamous cell carcinoma. World J Gastroenterol 2009; 15:3493 - 7; http://dx.doi.org/10.3748/wjg.15.3493; PMID: 19630103
- Ferguson HR, Wild CP, Anderson LA, Murphy SJ, Johnston BT, Murray LJ, et al. Cyclooxygenase-2 and inducible nitric oxide synthase gene polymorphisms and risk of reflux esophagitis, Barrett esophagus, and esophageal adenocarcinoma. Cancer Epidemiol Biomarkers Prev 2008; 17:727 - 31; http://dx.doi.org/10.1158/1055-9965.EPI-07-2570; PMID: 18349295
- Buttar NS, Wang KK, Anderson MA, Dierkhising RA, Pacifico RJ, Krishnadath KK, et al. The effect of selective cyclooxygenase-2 inhibition in Barrett esophagus epithelium: an in vitro study. J Natl Cancer Inst 2002; 94:422 - 9; http://dx.doi.org/10.1093/jnci/94.6.422; PMID: 11904314
- Kaur BS, Khamnehei N, Iravani M, Namburu SS, Lin O, Triadafilopoulos G. Rofecoxib inhibits cyclooxygenase 2 expression and activity and reduces cell proliferation in Barrett esophagus. Gastroenterology 2002; 123:60 - 7; http://dx.doi.org/10.1053/gast.2002.34244; PMID: 12105834
- Cheong E, Ivory K, Doleman J, Parker ML, Rhodes M, Johnson IT. Synthetic and naturally occurring COX-2 inhibitors suppress proliferation in a human oesophageal adenocarcinoma cell line (OE33) by inducing apoptosis and cell cycle arrest. Carcinogenesis 2004; 25:1945 - 52; http://dx.doi.org/10.1093/carcin/bgh184; PMID: 15155531
- Souza RF, Shewmake K, Beer DG, Cryer B, Spechler SJ. Selective inhibition of cyclooxygenase-2 suppresses growth and induces apoptosis in human esophageal adenocarcinoma cells. Cancer Res 2000; 60:5767 - 72; PMID: 11059772
- Piazuelo E, Jiminez P, Strunk M, Santander S, Garcia A, Esteva F, et al. Effects of selective PGE2 receptor antagonists in esophageal adenocarcinoma cells derived from Barrett's esophagus. Prostaglandins &. Other Lipid Mediators 2006; 81:150 - 61; http://dx.doi.org/10.1016/j.prostaglandins.2006.09.002
- Santander S, Cebrián C, Esquivias P, Conde B, Esteva F, Jiménez P, et al. Cyclooxygenase inhibitors decrease the growth and induce regression of human esophageal adenocarcinoma xenografts in nude mice. Int J Oncol 2012; 40:527 - 34; PMID: 21971589
- Heath EI, Canto MI, Piantadosi S, Montgomery E, Weinstein WM, Herman JG, et al, Chemoprevention for Barrett Esophagus Trial Research Group. Secondary chemoprevention of Barrett esophagus with celecoxib: results of a randomized trial. J Natl Cancer Inst 2007; 99:545 - 57; http://dx.doi.org/10.1093/jnci/djk112; PMID: 17405999
- Jankowski J, Barr H, deCaestecker J, Watson P, Attwood S, Moayyedi P. Aspirin in the prevention of cancer. Lancet 2011; 377:1649 - 50, author reply 1651-2; http://dx.doi.org/10.1016/S0140-6736(11)60666-1; PMID: 21571137
- Doak SH, Jenkins GJ, Parry EM, D’Souza FR, Griffiths AP, Toffazal N, et al. Chromosome 4 hyperploidy represents an early genetic aberration in premalignant Barrett oesophagus. Gut 2003; 52:623 - 8; http://dx.doi.org/10.1136/gut.52.5.623; PMID: 12692043
- Abdel-Latif MM, O’Riordan J, Windle HJ, Carton E, Ravi N, Kelleher D, et al. NF-kappaB activation in esophageal adenocarcinoma: relationship to Barrett metaplasia, survival, and response to neoadjuvant chemoradiotherapy. Ann Surg 2004; 239:491 - 500; http://dx.doi.org/10.1097/01.sla.0000118751.95179.c6; PMID: 15024310
- O’Riordan JM, Abdel-latif MM, Ravi N, McNamara D, Byrne PJ, McDonald GS, et al. Proinflammatory cytokine and nuclear factor kappa-B expression along the inflammation-metaplasia-dysplasia-adenocarcinoma sequence in the esophagus. Am J Gastroenterol 2005; 100:1257 - 64; http://dx.doi.org/10.1111/j.1572-0241.2005.41338.x; PMID: 15929754
- Jenkins GJ, Harries K, Doak SH, Wilmes A, Griffiths AP, Baxter JN, et al. The bile acid deoxycholic acid (DCA) at neutral pH activates NF-kappaB and induces IL-8 expression in oesophageal cells in vitro. Carcinogenesis 2004; 25:317 - 23; http://dx.doi.org/10.1093/carcin/bgh032; PMID: 14656946
- Jenkins GJS, Cronin J, Alhamdani A, Rawat N, D’Souza F, Thomas T, et al. The bile acid deoxycholic acid has a non-linear dose response for DNA damage and possibly NF-kappaB activation in oesophageal cells, with a mechanism of action involving ROS. Mutagenesis 2008; 23:399 - 405; http://dx.doi.org/10.1093/mutage/gen029; PMID: 18515815
- Huo X, Juergens S, Zhang X, Rezaei D, Yu C, Strauch ED, et al. Deoxycholic acid causes DNA damage while inducing apoptotic resistance through NF-κB activation in benign Barrett epithelial cells. Am J Physiol Gastrointest Liver Physiol 2011; 301:G278 - 86; http://dx.doi.org/10.1152/ajpgi.00092.2011; PMID: 21636532
- Hormi-Carver K, Zhang X, Zhang HY, Whitehead RH, Terada LS, Spechler SJ, et al. Unlike esophageal squamous cells, Barrett epithelial cells resist apoptosis by activating the nuclear factor-kappaB pathway. Cancer Res 2009; 69:672 - 7; http://dx.doi.org/10.1158/0008-5472.CAN-08-3703; PMID: 19147583
- Awan AK, Iftikhar SY, Morris TM, Clarke PA, Grabowska AM, Waraich N, et al. Androgen receptors may act in a paracrine manner to regulate oesophageal adenocarcinoma growth. Eur J Surg Oncol 2007; 33:561 - 8; http://dx.doi.org/10.1016/j.ejso.2006.12.001; PMID: 17254742
- Konturek PC, Nikiforuk A, Kania J, Raithel M, Hahn EG, Mühldorfer S. Activation of NFkappaB represents the central event in the neoplastic progression associated with Barrett esophagus: a possible link to the inflammation and overexpression of COX-2, PPARgamma and growth factors. Dig Dis Sci 2004; 49:1075 - 83; http://dx.doi.org/10.1023/B:DDAS.0000037790.11724.70; PMID: 15387324
- Arber N, Gammon MD, Hibshoosh H, Britton JA, Zhang Y, Schonberg JB, et al. Overexpression of cyclin D1 occurs in both squamous carcinomas and adenocarcinomas of the esophagus and in adenocarcinomas of the stomach. Hum Pathol 1999; 30:1087 - 92; http://dx.doi.org/10.1016/S0046-8177(99)90227-7; PMID: 10492044
- Arber N, Lightdale C, Rotterdam H, Han KH, Sgambato A, Yap E, et al. Increased expression of the cyclin D1 gene in Barrett esophagus. Cancer Epidemiol Biomarkers Prev 1996; 5:457 - 9; PMID: 8781742
- Bani-Hani K, Martin IG, Hardie LJ, Mapstone N, Briggs JA, Forman D, et al. Prospective study of cyclin D1 overexpression in Barrett esophagus: association with increased risk of adenocarcinoma. J Natl Cancer Inst 2000; 92:1316 - 21; http://dx.doi.org/10.1093/jnci/92.16.1316; PMID: 10944553
- Casson AG, Zheng Z, Evans SC, Geldenhuys L, van Zanten SV, Veugelers PJ, et al. Cyclin D1 polymorphism (G870A) and risk for esophageal adenocarcinoma. Cancer 2005; 104:730 - 9; http://dx.doi.org/10.1002/cncr.21229; PMID: 15971196
- Izzo JG, Wu T-T, Wu X, Ensor J, Luthra R, Pan J, et al. Cyclin D1 guanine/adenine 870 polymorphism with altered protein expression is associated with genomic instability and aggressive clinical biology of esophageal adenocarcinoma. J Clin Oncol 2007; 25:698 - 707; http://dx.doi.org/10.1200/JCO.2006.08.0283; PMID: 17308274
- Izzo JG, Malhotra U, Wu TT, Ensor J, Babenko IM, Swisher SG, et al. Impact of cyclin D1 A870G polymorphism in esophageal adenocarcinoma tumorigenesis. Semin Oncol 2005; 32:Suppl 9 S11 - 5; http://dx.doi.org/10.1053/j.seminoncol.2005.04.023; PMID: 16399423
- Liu G, Cescon DW, Zhai R, Zhou W, Kulke MH, Ma C, et al. p53 Arg72Pro, MDM2 T309G and CCND1 G870A polymorphisms are not associated with susceptibility to esophageal adenocarcinoma. Dis Esophagus 2010; 23:36 - 9; http://dx.doi.org/10.1111/j.1442-2050.2009.00960.x; PMID: 19302219
- Geddert H, Kiel S, Zotz RB, Zhang J, Willers R, Gabbert HE, et al. Polymorphism of p16 INK4A and cyclin D1 in adenocarcinomas of the upper gastrointestinal tract. J Cancer Res Clin Oncol 2005; 131:803 - 8; http://dx.doi.org/10.1007/s00432-005-0021-4; PMID: 16163549
- Sarbia M, Bektas N, Müller W, Heep H, Borchard F, Gabbert HE. Expression of cyclin E in dysplasia, carcinoma, and nonmalignant lesions of Barrett esophagus. Cancer 1999; 86:2597 - 601; http://dx.doi.org/10.1002/(SICI)1097-0142(19991215)86:12<2597::AID-CNCR3>3.0.CO;2-0; PMID: 10594854
- Lin L, Prescott MS, Zhu Z, Singh P, Chun SY, Kuick RD, et al. Identification and characterization of a 19q12 amplicon in esophageal adenocarcinomas reveals cyclin E as the best candidate gene for this amplicon. Cancer Res 2000; 60:7021 - 7; PMID: 11156406
- Boynton RF, Huang Y, Blount PL, Reid BJ, Raskind WH, Haggitt RC, et al. Frequent loss of heterozygosity at the retinoblastoma locus in human esophageal cancers. Cancer Res 1991; 51:5766 - 9; PMID: 1913694
- Morgan RJ, Newcomb PV, Bailey M, Hardwick RH, Alderson D. Loss of heterozygosity at microsatellite marker sites for tumour suppressor genes in oesophageal adenocarcinoma. [EJSO] Eur J Surg Oncol 1998; 24:34 - 7; http://dx.doi.org/10.1016/S0748-7983(98)80122-4; PMID: 9542513
- Huang Y, Meltzer SJ, Yin J, Tong Y, Chang EH, Srivastava S, et al. Altered messenger RNA and unique mutational profiles of p53 and Rb in human esophageal carcinomas. Cancer Res 1993; 53:1889 - 94; PMID: 8467510
- Sarbia M, Arjumand J, Wolter M, Reifenberger G, Heep H, Gabbert HE. Frequent c-myc amplification in high-grade dysplasia and adenocarcinoma in Barrett esophagus. Am J Clin Pathol 2001; 115:835 - 40; http://dx.doi.org/10.1309/MXXH-25N3-UAL2-G7XX; PMID: 11392879
- Klump B, Hsieh CJ, Holzmann K, Gregor M, Porschen R. Hypermethylation of the CDKN2/p16 promoter during neoplastic progression in Barrett esophagus. Gastroenterology 1998; 115:1381 - 6; http://dx.doi.org/10.1016/S0016-5085(98)70016-2; PMID: 9834265
- Wong DJ, Paulson TG, Prevo LJ, Galipeau PC, Longton G, Blount PL, et al. p16(INK4a) lesions are common, early abnormalities that undergo clonal expansion in Barrett metaplastic epithelium. Cancer Res 2001; 61:8284 - 9; PMID: 11719461
- Maley CC, Galipeau PC, Li X, Sanchez CA, Paulson TG, Reid BJ. Selectively advantageous mutations and hitchhikers in neoplasms: p16 lesions are selected in Barrett esophagus. Cancer Res 2004; 64:3414 - 27; http://dx.doi.org/10.1158/0008-5472.CAN-03-3249; PMID: 15150093
- Schulmann K, Sterian A, Berki A, Yin J, Sato F, Xu Y, et al. Inactivation of p16, RUNX3, and HPP1 occurs early in Barrett-associated neoplastic progression and predicts progression risk. Oncogene 2005; 24:4138 - 48; http://dx.doi.org/10.1038/sj.onc.1208598; PMID: 15824739
- Singh SP, Lipman J, Goldman H, Ellis FH Jr., Aizenman L, Cangi MG, et al. Loss or altered subcellular localization of p27 in Barrett associated adenocarcinoma. Cancer Res 1998; 58:1730 - 5; PMID: 9563491
- Ellis FH Jr., Xu X, Kulke MH, LoCicero J 3rd, Loda M. Malignant transformation of the esophageal mucosa is enhanced in p27 knockout mice. J Thorac Cardiovasc Surg 2001; 122:809 - 14; http://dx.doi.org/10.1067/mtc.2001.116471; PMID: 11581618
- Hanas JS, Lerner MR, Lightfoot SA, Raczkowski C, Kastens DJ, Brackett DJ, et al. Expression of the cyclin-dependent kinase inhibitor p21(WAF1/CIP1) and p53 tumor suppressor in dysplastic progression and adenocarcinoma in Barrett esophagus. Cancer 1999; 86:756 - 63; http://dx.doi.org/10.1002/(SICI)1097-0142(19990901)86:5<756::AID-CNCR9>3.0.CO;2-X; PMID: 10463972
- Huang Y, Boynton RF, Blount PL, Silverstein RJ, Yin J, Tong Y, et al. Loss of heterozygosity involves multiple tumor suppressor genes in human esophageal cancers. Cancer Res 1992; 52:6525 - 30; PMID: 1423299
- Hamelin R, Fléjou JF, Muzeau F, Potet F, Laurent-Puig P, Fékété F, et al. TP53 gene mutations and p53 protein immunoreactivity in malignant and premalignant Barrett esophagus. Gastroenterology 1994; 107:1012 - 8; PMID: 7523212
- Schneider PM, Casson AG, Levin B, Garewal HS, Hoelscher AH, Becker K, et al. Mutations of p53 in Barrett esophagus and Barrett cancer: a prospective study of ninety-eight cases. J Thorac Cardiovasc Surg 1996; 111:323 - 31, discussion 331-3; http://dx.doi.org/10.1016/S0022-5223(96)70441-5; PMID: 8583805
- Blount PL, Galipeau PC, Sanchez CA, Neshat K, Levine DS, Yin J, et al. 17p allelic losses in diploid cells of patients with Barrett esophagus who develop aneuploidy. Cancer Res 1994; 54:2292 - 5; PMID: 8162566
- Ribeiro U Jr., Finkelstein SD, Safatle-Ribeiro AV, Landreneau RJ, Clarke MR, Bakker A, et al. p53 sequence analysis predicts treatment response and outcome of patients with esophageal carcinoma. Cancer 1998; 83:7 - 18; http://dx.doi.org/10.1002/(SICI)1097-0142(19980701)83:1<7::AID-CNCR2>3.0.CO;2-R; PMID: 9655287
- Schneider PM, Stoeltzing O, Roth JA, Hoelscher AH, Wegerer S, Mizumoto S, et al. P53 mutational status improves estimation of prognosis in patients with curatively resected adenocarcinoma in Barrett esophagus. Clin Cancer Res 2000; 6:3153 - 8; PMID: 10955797
- Casson AG, Evans SC, Gillis A, Porter GA, Veugelers P, Darnton SJ, et al. Clinical implications of p53 tumor suppressor gene mutation and protein expression in esophageal adenocarcinomas: results of a ten-year prospective study. J Thorac Cardiovasc Surg 2003; 125:1121 - 31; http://dx.doi.org/10.1067/mtc.2003.176; PMID: 12771886
- Ireland AP, Shibata DK, Chandrasoma P, Lord RV, Peters JH, DeMeester TR. Clinical significance of p53 mutations in adenocarcinoma of the esophagus and cardia. Ann Surg 2000; 231:179 - 87; http://dx.doi.org/10.1097/00000658-200002000-00005; PMID: 10674608
- Blount PL, Ramel S, Raskind WH, Haggitt RC, Sanchez CA, Dean PJ, et al. 17p allelic deletions and p53 protein overexpression in Barrett adenocarcinoma. Cancer Res 1991; 51:5482 - 6; PMID: 1680552
- Ramel S, Reid BJ, Sanchez CA, Blount PL, Levine DS, Neshat K, et al. Evaluation of p53 protein expression in Barrett esophagus by two-parameter flow cytometry. Gastroenterology 1992; 102:1220 - 8; PMID: 1551529
- Younes M, Lebovitz RM, Lechago LV, Lechago J. p53 protein accumulation in Barrett metaplasia, dysplasia, and carcinoma: a follow-up study. Gastroenterology 1993; 105:1637 - 42; PMID: 8253340
- Rice TW, Goldblum JR, Falk GW, Tubbs RR, Kirby TJ, Casey G. p53 immunoreactivity in Barrett metaplasia, dysplasia, and carcinoma. J Thorac Cardiovasc Surg 1994; 108:1132 - 7; PMID: 7983883
- Doak SH, Jenkins GJ, Parry EM, Griffiths AP, Shah V, Baxter JN, et al. Characterisation of p53 status at the gene, chromosomal and protein levels in oesophageal adenocarcinoma. Br J Cancer 2003; 89:1729 - 35; http://dx.doi.org/10.1038/sj.bjc.6601323; PMID: 14583777
- Hanazono K, Natsugoe S, Stein HJ, Aikou T, Hoefler H, Siewert JR. Distribution of p53 mutations in esophageal and gastric carcinomas and the relationship with p53 expression. Oncol Rep 2006; 15:821 - 4; PMID: 16525665
- Hritz I, Gyorffy H, Molnar B, Lakatos G, Sipos F, Pregun I, et al. Increased p53 expression in the malignant transformation of Barrett esophagus is accompanied by an upward shift of the proliferative compartment. Pathol Oncol Res 2009; 15:183 - 92; http://dx.doi.org/10.1007/s12253-008-9095-z; PMID: 18752044
- Binato M, Gurski RR, Fagundes RB, Meurer L, Edelweiss MI. P53 and Ki-67 overexpression in gastroesophageal reflux disease--Barrett esophagus and adenocarcinoma sequence. Dis Esophagus 2009; 22:588 - 95; http://dx.doi.org/10.1111/j.1442-2050.2009.00953.x; PMID: 19302208
- Kastelein F, Biermann K, Steyerberg EW, Verheij J, Kalisvaart M, Looijenga LH, et al, on behalf of the ProBar-study group. Aberrant p53 protein expression is associated with an increased risk of neoplastic progression in patients with Barrett oesophagus. Gut 2012; http://dx.doi.org/10.1136/gutjnl-2012-303594; PMID: 23256952
- Osterheld MC, Bian YS, Bosman FT, Benhattar J, Fontolliet C. Beta-catenin expression and its association with prognostic factors in adenocarcinoma developed in Barrett esophagus. Am J Clin Pathol 2002; 117:451 - 6; http://dx.doi.org/10.1309/1DB6-GFVH-RA6W-Q07Y; PMID: 11888085
- Wijnhoven BP, Nollet F, De Both NJ, Tilanus HW, Dinjens WN. Genetic alterations involving exon 3 of the beta-catenin gene do not play a role in adenocarcinomas of the esophagus. Int J Cancer 2000; 86:533 - 7; http://dx.doi.org/10.1002/(SICI)1097-0215(20000515)86:4<533::AID-IJC15>3.0.CO;2-O; PMID: 10797268
- Bian YS, Osterheld MC, Bosman FT, Fontolliet C, Benhattar J. Nuclear accumulation of beta-catenin is a common and early event during neoplastic progression of Barrett esophagus. Am J Clin Pathol 2000; 114:583 - 90; http://dx.doi.org/10.1309/3QLC-5MF1-JYXU-A5XX; PMID: 11026105
- Choi YW, Heath EI, Heitmiller R, Forastiere AA, Wu T-T. Mutations in beta-catenin and APC genes are uncommon in esophageal and esophagogastric junction adenocarcinomas. Mod Pathol 2000; 13:1055 - 9; http://dx.doi.org/10.1038/modpathol.3880194; PMID: 11048797
- González MV, Artímez ML, Rodrigo L, López-Larrea C, Menéndez MJ, Alvarez V, et al. Mutation analysis of the p53, APC, and p16 genes in the Barrett oesophagus, dysplasia, and adenocarcinoma. J Clin Pathol 1997; 50:212 - 7; http://dx.doi.org/10.1136/jcp.50.3.212; PMID: 9155671
- Kawakami K, Brabender J, Lord RV, Groshen S, Greenwald BD, Krasna MJ, et al. Hypermethylated APC DNA in plasma and prognosis of patients with esophageal adenocarcinoma. J Natl Cancer Inst 2000; 92:1805 - 11; http://dx.doi.org/10.1093/jnci/92.22.1805; PMID: 11078757
- Eads CA, Lord RV, Wickramasinghe K, Long TI, Kurumboor SK, Bernstein L, et al. Epigenetic patterns in the progression of esophageal adenocarcinoma. Cancer Res 2001; 61:3410 - 8; PMID: 11309301
- Clément G, Braunschweig R, Pasquier N, Bosman FT, Benhattar J. Methylation of APC, TIMP3, and TERT: a new predictive marker to distinguish Barrett oesophagus patients at risk for malignant transformation. J Pathol 2006; 208:100 - 7; http://dx.doi.org/10.1002/path.1884; PMID: 16278815
- Clément G, Braunschweig R, Pasquier N, Bosman FT, Benhattar J. Alterations of the Wnt signaling pathway during the neoplastic progression of Barrett esophagus. Oncogene 2006; 25:3084 - 92; http://dx.doi.org/10.1038/sj.onc.1209338; PMID: 16407829
- Clément G, Guilleret I, He B, Yagui-Beltrán A, Lin Y-C, You L, et al. Epigenetic alteration of the Wnt inhibitory factor-1 promoter occurs early in the carcinogenesis of Barrett esophagus. Cancer Sci 2008; 99:46 - 53; PMID: 18005197
- Bailey T, Biddlestone L, Shepherd N, Barr H, Warner P, Jankowski J. Altered cadherin and catenin complexes in the Barrett esophagus-dysplasia-adenocarcinoma sequence: correlation with disease progression and dedifferentiation. Am J Pathol 1998; 152:135 - 44; PMID: 9422531
- Krishnadath KK, Tilanus HW, van Blankenstein M, Hop WC, Kremers ED, Dinjens WN, et al. Reduced expression of the cadherin-catenin complex in oesophageal adenocarcinoma correlates with poor prognosis. J Pathol 1997; 182:331 - 8; http://dx.doi.org/10.1002/(SICI)1096-9896(199707)182:3<331::AID-PATH860>3.0.CO;2-D; PMID: 9349237
- Corn PG, Heath EI, Heitmiller R, Fogt F, Forastiere AA, Herman JG, et al. Frequent hypermethylation of the 5′ CpG island of E-cadherin in esophageal adenocarcinoma. Clin Cancer Res 2001; 7:2765 - 9; PMID: 11555590
- Tselepis C, Perry I, Dawson C, Hardy R, Darnton SJ, McConkey C, et al. Tumour necrosis factor-alpha in Barrett oesophagus: a potential novel mechanism of action. Oncogene 2002; 21:6071 - 81; http://dx.doi.org/10.1038/sj.onc.1205731; PMID: 12203119
- Jethwa P, Naqvi M, Hardy RG, Hotchin NA, Roberts S, Spychal R, et al. Overexpression of Slug is associated with malignant progression of esophageal adenocarcinoma. World J Gastroenterol 2008; 14:1044 - 52; http://dx.doi.org/10.3748/wjg.14.1044; PMID: 18286686
- van Es JH, van Gijn ME, Riccio O, van den Born M, Vooijs M, Begthel H, et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature 2005; 435:959 - 63; http://dx.doi.org/10.1038/nature03659; PMID: 15959515
- Yang Q, Bermingham NA, Finegold MJ, Zoghbi HY. Requirement of Math1 for secretory cell lineage commitment in the mouse intestine. Science 2001; 294:2155 - 8; http://dx.doi.org/10.1126/science.1065718; PMID: 11739954
- Menke V, van Es JH, de Lau W, van den Born M, Kuipers EJ, Siersema PD, et al. Conversion of metaplastic Barrett epithelium into post-mitotic goblet cells by gamma-secretase inhibition. Dis Model Mech 2010; 3:104 - 10; http://dx.doi.org/10.1242/dmm.003012; PMID: 20075383
- Tamagawa Y, Ishimura N, Uno G, Yuki T, Kazumori H, Ishihara S, et al. Notch signaling pathway and Cdx2 expression in the development of Barrett esophagus. Lab Invest 2012; 92:896 - 909; http://dx.doi.org/10.1038/labinvest.2012.56; PMID: 22449796
- Morrow DJ, Avissar NE, Toia L, Redmond EM, Watson TJ, Jones C, et al. Pathogenesis of Barrett esophagus: bile acids inhibit the Notch signaling pathway with induction of CDX2 gene expression in human esophageal cells. Surgery 2009; 146:714 - 21, discussion 721-2; http://dx.doi.org/10.1016/j.surg.2009.06.050; PMID: 19789031
- Mendelson J, Song S, Li Y, Maru DM, Mishra B, Davila M, et al. Dysfunctional transforming growth factor-β signaling with constitutively active Notch signaling in Barrett esophageal adenocarcinoma. Cancer 2011; 117:3691 - 702; http://dx.doi.org/10.1002/cncr.25861; PMID: 21305538
- Song S, Maru DM, Ajani JA, Chan CH, Honjo S, Lin HK, et al. Loss of TGF-β adaptor β2SP activates notch signaling and SOX9 expression in esophageal adenocarcinoma. Cancer Res 2013; 73:2159 - 69; http://dx.doi.org/10.1158/0008-5472.CAN-12-1962; PMID: 23536563
- van den Brink GR. Hedgehog signaling in development and homeostasis of the gastrointestinal tract. Physiol Rev 2007; 87:1343 - 75; http://dx.doi.org/10.1152/physrev.00054.2006; PMID: 17928586
- Litingtung Y, Lei L, Westphal H, Chiang C. Sonic hedgehog is essential to foregut development. Nat Genet 1998; 20:58 - 61; http://dx.doi.org/10.1038/1717; PMID: 9731532
- Wang DH, Clemons NJ, Miyashita T, Dupuy AJ, Zhang W, Szczepny A, et al. Aberrant epithelial-mesenchymal Hedgehog signaling characterizes Barrett metaplasia. Gastroenterology 2010; 138:1810 - 22; http://dx.doi.org/10.1053/j.gastro.2010.01.048; PMID: 20138038
- Yamanaka Y, Shiotani A, Fujimura Y, Ishii M, Fujita M, Matsumoto H, et al. Expression of Sonic hedgehog (SHH) and CDX2 in the columnar epithelium of the lower oesophagus. Dig Liver Dis 2011; 43:54 - 9; http://dx.doi.org/10.1016/j.dld.2010.04.014; PMID: 20619754
- Castillo D, Puig S, Iglesias M, Seoane A, de Bolós C, Munitiz V, et al. Activation of the BMP4 pathway and early expression of CDX2 characterize non-specialized columnar metaplasia in a human model of Barrett esophagus. J Gastrointest Surg 2012; 16:227 - 37, discussion 237; http://dx.doi.org/10.1007/s11605-011-1758-5; PMID: 22076569
- Milano F, van Baal JW, Buttar NS, Rygiel AM, de Kort F, DeMars CJ, et al. Bone morphogenetic protein 4 expressed in esophagitis induces a columnar phenotype in esophageal squamous cells. Gastroenterology 2007; 132:2412 - 21; http://dx.doi.org/10.1053/j.gastro.2007.03.026; PMID: 17570215
- Clemons NJ, Wang DH, Croagh D, Tikoo A, Fennell CM, Murone C, et al. Sox9 drives columnar differentiation of esophageal squamous epithelium: a possible role in the pathogenesis of Barrett esophagus. Am J Physiol Gastrointest Liver Physiol 2012; 303:G1335 - 46; http://dx.doi.org/10.1152/ajpgi.00291.2012; PMID: 23064761
- Berman DM, Karhadkar SS, Maitra A, Montes De Oca R, Gerstenblith MR, Briggs K, et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 2003; 425:846 - 51; http://dx.doi.org/10.1038/nature01972; PMID: 14520411
- Rizvi S, Demars CJ, Comba A, Gainullin VG, Rizvi Z, Almada LL, et al. Combinatorial chemoprevention reveals a novel smoothened-independent role of GLI1 in esophageal carcinogenesis. Cancer Res 2010; 70:6787 - 96; http://dx.doi.org/10.1158/0008-5472.CAN-10-0197; PMID: 20647328
- Wang Y, Ding Q, Yen CJ, Xia W, Izzo JG, Lang JY, et al. The crosstalk of mTOR/S6K1 and Hedgehog pathways. Cancer Cell 2012; 21:374 - 87; http://dx.doi.org/10.1016/j.ccr.2011.12.028; PMID: 22439934
- Yen CJ, Izzo JG, Lee DF, Guha S, Wei Y, Wu TT, et al. Bile acid exposure up-regulates tuberous sclerosis complex 1/mammalian target of rapamycin pathway in Barrett-associated esophageal adenocarcinoma. Cancer Res 2008; 68:2632 - 40; http://dx.doi.org/10.1158/0008-5472.CAN-07-5460; PMID: 18413730
- Schmidt MK, Meurer L, Volkweis BS, Edelweiss MI, Schirmer CC, Kruel CDP, et al. c-Myc overexpression is strongly associated with metaplasia-dysplasia-adenocarcinoma sequence in the esophagus. Dis Esophagus 2007; 20:212 - 6; http://dx.doi.org/10.1111/j.1442-2050.2007.00673.x; PMID: 17509117
- Tselepis C, Morris CD, Wakelin D, Hardy R, Perry I, Luong QT, et al. Upregulation of the oncogene c-myc in Barrett adenocarcinoma: induction of c-myc by acidified bile acid in vitro. Gut 2003; 52:174 - 80; http://dx.doi.org/10.1136/gut.52.2.174; PMID: 12524396
- von Rahden BH, Stein HJ, Pühringer-Oppermann F, Sarbia M. c-myc amplification is frequent in esophageal adenocarcinoma and correlated with the upregulation of VEGF-A expression. Neoplasia 2006; 8:702 - 7; http://dx.doi.org/10.1593/neo.06277; PMID: 16984727
- Nancarrow DJ, Handoko HY, Smithers BM, Gotley DC, Drew PA, Watson DI, et al. Genome-wide copy number analysis in esophageal adenocarcinoma using high-density single-nucleotide polymorphism arrays. Cancer Res 2008; 68:4163 - 72; http://dx.doi.org/10.1158/0008-5472.CAN-07-6710; PMID: 18519675
- Stairs DB, Nakagawa H, Klein-Szanto A, Mitchell SD, Silberg DG, Tobias JW, et al. Cdx1 and c-Myc foster the initiation of transdifferentiation of the normal esophageal squamous epithelium toward Barrett esophagus. PLoS One 2008; 3:e3534; http://dx.doi.org/10.1371/journal.pone.0003534; PMID: 18953412
- Hoyo C, Cook MB, Kamangar F, Freedman ND, Whiteman DC, Bernstein L, et al. Body mass index in relation to oesophageal and oesophagogastric junction adenocarcinomas: a pooled analysis from the International BEACON Consortium. Int J Epidemiol 2012; 41:1706 - 18; http://dx.doi.org/10.1093/ije/dys176; PMID: 23148106
- Kubo A, Cook MB, Shaheen NJ, Vaughan TL, Whiteman DC, Murray L, et al. Sex-specific associations between body mass index, waist circumference and the risk of Barrett oesophagus: a pooled analysis from the international BEACON consortium. Gut 2013; http://dx.doi.org/10.1136/gutjnl-2012-303753; PMID: 23355549
- Su Z, Gay LJ, Strange A, Palles C, Band G, Whiteman DC, et al, Esophageal Adenocarcinoma Genetics Consortium, Wellcome Trust Case Control Consortium 2. Common variants at the MHC locus and at chromosome 16q24.1 predispose to Barrett esophagus. Nat Genet 2012; 44:1131 - 6; http://dx.doi.org/10.1038/ng.2408; PMID: 22961001