
Abstract
Increasing health care costs in the US are due in a large part to the increasing prevalence of chronic diseases in an aging population. Current therapeutic strategies for treating chronic diseases alleviate symptoms allowing patients to live longer with these diseases, but they do little, however, to alter the underlying disease course. Recent advances in molecular biology are revealing new drug targets that may significantly alter the course of these diseases and, as a result, offer economic relief from burgeoning health care costs. Endoplasmic reticulum (ER) stress has been implicated as an underlying pathology in many chronic diseases, and, therefore, the development of therapies designed to ameliorate ER stress may yield novel, effective treatment strategies. Herein, we report that X-box binding protein 1 (XBP1) may be one of the earliest proteins engaged in response to ER stress. We show that a new signaling peptide derived from the ER-embedded transient receptor potential calcium channel protein 1 (TRPC1) engages XBP1 upstream of NLRP3 inflammasome-mediated maturation and secretion of IL-1β/IL-18. Moreover, we show that a synthetic homolog of this signaling peptide (Naclynamide™) administered intravenously twice weekly over a 4-week treatment course induced suppuration and evoked partial or complete resolution of lesions associated with a fibrotic granuloma, a lymphosarcoma, and a colo-rectal carcinoma in canine patients. The mode of action for Naclynamide™ as a first-in-class anti-cancer drug candidate is discussed.
Introduction
Uncontrolled endoplasmic reticulum (ER) stress has been implicated in the pathology that underlies a wide variety of acute and chronic diseases, including neurodegenerative diseases, prion diseases, ischemia/reperfusion injury, fibrosis, metabolic diseases, and cancers.Citation1 We have been investigating the molecular signaling that leads from cellular injury or infection to ER stress, inflammasome-mediated signaling, and the innate immune response as a therapeutically relevant target for the treatment of chronic diseases. Inflammasome biology in carcinogenesis and anticancer immune responses were recently reviewed, and as discussed therein, inflammasomes can have pleiotropic but sometimes contrasting anti-cancer effects.Citation2
The inflammasome comprises a family of cytosolic receptors called NOD-like receptors (NLR) that are involved in innate immune recognition of pathogen-associated molecular patterns as well as intracellular and extracellular damage-associated molecular patterns.Citation3 Thus far, more than 20 inflammasomes have been identified, and many are present in nearly every cell type. NLRP3 is the most extensively studied inflammasome and has been found to be activated by a diverse range of stimuli, including microbial derived products,Citation4-Citation7 environmental factors,Citation8-Citation10 and endogenous molecules.Citation11-Citation14 Ting et al. showed that NLRP3 signaling modulated colitis and colitis-associated cancer in a dextran sulfate induced colitis model in mice. In that study, mice that were deficient in NLRP3 inflammasome components had increased morbidity, polyp formation, and disease than the wild-type controls. Also shown was that an increased tumor burden correlated with decreased levels of two key markers of inflammasome activation, IL-1β and IL-18, at the tumor sites.Citation15
Cancer development has traditionally been thought to be the consequence of genetic mutations and chromosomal abnormalities that affect the function of genes involved in cell cycle regulation, differentiation, proliferation, and apoptosis. It has also been recognized, however, that the stromal environment in which the cancer arises plays an important role in carcinogenesis. Oncogenesis can arise as a result of chronic inflammation that is induced by many different factors, including persistent viral infection (human papilloma virus—cervical cancer) or bacteria (Helicobacter pylori—gastric cancer) and is often associated with fibrosis in the tissues of the affected organ. Artlett et al. recently reported that the NLRP3 inflammasome is driving fibrosisCitation16 and therefore may also play a contributing role in oncogenesis.
Thacker et al. reported the isolation, structure elucidation, and synthesis of 1-peptidyl-2,3-diacylglyclerol (“pDAG”) and the 18 amino acid sequence of the peptide moiety (“acALY18”).Citation17 Subsequently, Thacker et al. reported that acALY18 induces IL-1β, IL-18, and IL-33 secretion, immune cell recruitment and activation, and a potent innate immune response that is NLRP3 inflammasome-dependent.Citation18 It was postulated that, given the unique amino acid sequence of acALY18, it was likely to be a posttranslational modification of a peptide uniquely comprising the third extra-membrane loop on the lumenal side of the ER-embedded transient receptor potential calcium channel protein (TRPC1). Herein we show that, in response to pharmacologically induced ER calcium store depletion and ER stress, acALY18 covalently engages XBP1 via disulfide bond formation within minutes of ER stress.
It is well known that the transcription factor XBP1 is a regulator of the cellular response to ER stress known as the unfolded protein response (UPR).Citation19,Citation20 The molecular mechanisms, however, leading from ER stress to the UPR and either resolution of the cell stress or progression to disease are poorly understood. We show that acALY18, a newly discovered signaling peptide, abrogated fibrosis in diseased scleroderma cells in vitro and resolved a benign, chronic fibrotic tumor (acral granuloma) in a dog. We also provide clinical evidence that the synthetic homolog of this signaling peptide (“Naclynamide™”) is a viable drug candidate to induce a potent CTL response and tumor suppuration in a case of canine lymphoma and canine colorectal carcinoma. Finally, we postulate that XBP1 engagement of the signaling peptide modulates the UPR, NLRP3 inflammasome signaling, and the innate immune response, and is a new therapeutic target in the treatment of cancer.
Results
In vitro signaling experiments
TRPC1 is a store-operated calcium channel protein that is highly conserved in the animal kingdom and is present in most human cell phenotypes. Amino acids 2–18 in acALY18 are identical and unique to the third extra-membrane loop of ER embedded TRPC1 (corresponding to amino acids 558–574 in the parent protein) adjacent to a pore forming membrane spanning unit that is the putative store operated calcium channel. We had previously established that acALY18 induced inflammasome-dependent activation, maturation, and release of IL-1β, IL-18, and IL-33.Citation18 The specific deletion sequence in the Trpc1 gene to produce the TRPC1−/− mice left intact the region of the gene coding for acALY18. Constitutive mRNA containing the acALY18 sequence was confirmed to be present in the TRPC1−/− cells (Lutz Birnbaumer, NIEHS, Research Triangle Park, NC, personal communication). The TRPC1−/− cells are devoid of functional TRPC1 protein, but retain mRNA for TRPC1 protein comprising the specific region of the third extra-membrane loop. Even though there was a detectable level of this amino acid sequence, it was not inducible by thapsigargin treatment in TRPC1−/− cells.
We wanted to conclusively demonstrate that acALY18 is derived from TRPC1; therefore we examined TRPC1 knockout mouse embryonic fibroblasts (kind gift from Lutz Birnbaumer). The wild-type and TRPC1−/− cells were treated with thapsigargin and acALY18 levels were measured after 1, 5, 15, 30, and 60 min. shows the acALY18 peptide levels normalized to GAPDH over the time course in the wild-type and TRPC1−/− cells. Moreover, we identified a connection between TRPC1 and IL-1β secretion (). We had previously reported that the acALY18 effects (IL-1β, IL-18, and IL-33 maturation and secretion) were NLRP3 inflammasome-dependent.Citation18 Here, we show that acALY18 is derived from TRPC1 in response to ER stress induced by thapsigargin that is upstream of NLRP3 inflammasome activation and that, in the absence of functional TRPC1, the induced secretion of IL-1β is ablated. We then wanted to determine whether acALY18 itself was able to induce ER stress and we found that it did not as measured by GRP78 protein levels. Induction of ER stress with thapsigargin induced acALY18 synthesis at 60 min, but GRP78 was not induced until 24 h. The direct addition of acALY18 to cells was unable to induce either GRP78 or IRE1α at 24 h post-treatment or phosphorylation of eIF2α at 1 h post-treatment as compared with the thapsigargin positive control. The results are presented in .
Figure 1. acALY18 is derived from TRPC1. Western analysis of wild-type (WT) embryonic mouse fibroblasts (A) and TRPC1−/− embryonic mouse fibroblasts (B) treated with 25 nM thapsigargin and probed for acALY18. (C) The representative graph of acALY18 expression in WT (■) and TRPC1−/− (■) embryonic mouse fibroblasts normalized to GAPDH. (D) IL-1β secretion from WT (■) and TRPC1−/− (■) fibroblasts treated with 25 nM thapsigargin for 72 h.
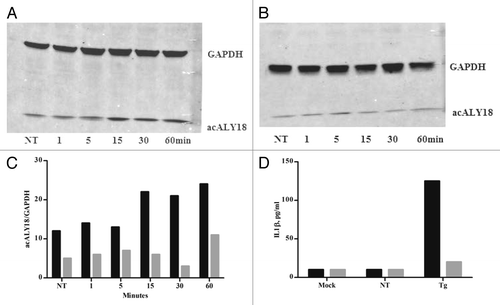
Figure 2. Administration of acALY18 to fibroblasts does not induce ER stress; however, ER stress induces acALY18. (A) A time course measuring the release of acALY18 from TRPC1 when human fibroblasts are treated with 25 nM thapsigargin. Results (top left) show that acALY18 expression precedes GRP78 expression (top right). Western analysis shows that acALY18 does not induce GRP78 in human fibroblasts treated with 25 nM thapsigargin (Tg), acALY18 (5 ng/ml), or mock treatment, for 24 h (bottom). NT, no treatment. (B) Normal human fibroblasts were treated with 25 nM thapsigargin, acALY18 (5 ng/ml), or mock treatment. Western analysis shows phosphorylation of eIF2α (p-eIF2α) after 1 h thapsigargin (Tg) treatment, but not acALY18 or mock treatment (left). Densitometry of the results is plotted (right) as the percentage p-eIF2α of total eIF2α. The expression of IRE1α after 24 h is only detectable in the thapsigargin treated cells (bottom).
We found that acALY18 localized to the cytosol of the cell () and that acALY18 forms a tetramer in solution (). Thapsigargin induced ER stress is known to activate IRE1α dimerization and unconventional mRNA processing of XBP1 mRNA followed by translation of the unspliced mRNA form to the spliced isoform of XBP1. The spliced isoform of XBP1 (XBP1s) is a transcription factor that localizes in the nucleus to induce the transcription of proteins as a part of the UPR. The unspliced isoform of XBP1 (XBP1u) is constitutively expressed, however XBP1s is not. Because the molecular mass of human XBP1u is 29 kDa and because acALY18 is detected on western blots at ~31 kDa, we thought that acALY18 may be targeting XBP1u. shows the immunoblots over the time course (1–60 min) after thapsigargin treatment and probed for either acALY18 or XBP1. Expression of acALY18 was increased >3-fold after 30 min and XBP1u protein expression increased almost 2-fold during the first hour.
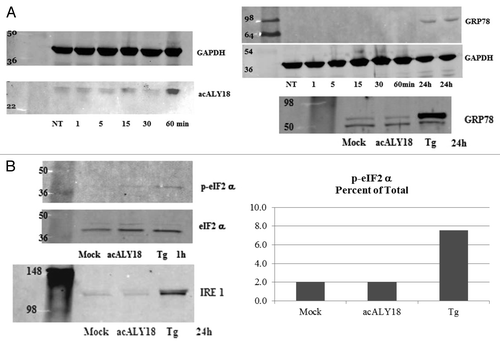
Figure 3. Administration of acALY18 directly to fibroblasts localizes to the cytosol of the cells and appears to form a tetramer. (A) Primary human fibroblasts were treated with 50 nM acALY18-AlexaFluor594 showing intracellular distribution (40× magnification). (B) Exogenously added acALY18 detected by western appears as a tetramer (~8 kDa).
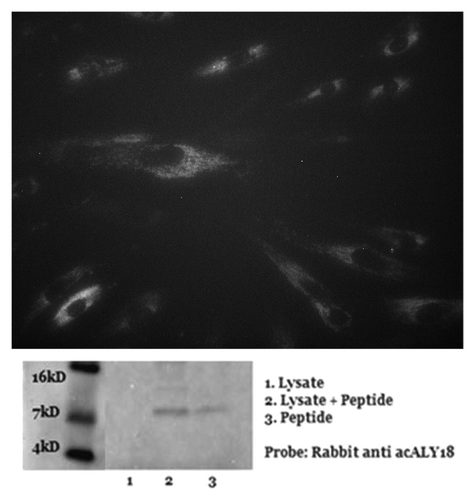
Figure 4. acALY18 forms a disulfide bond with its putative protein target XPB1. (A) Western analysis of human fibroblasts treated with 25 nM thapsigargin and probed for acALY18 (left) or XBP1 (right) at various time points post-treatment. The bar graph of the densitometry measurement shows increased XBP1 (■) expression preceding acALY18 (■) expression. (B) Normal human fibroblasts were treated for 30 min with 25 nM thapsigargin (Tg) and the whole cell lysates were reduced with DTT/BME for 15, 30, or 60 min and analyzed by western. Densitometry measurement of acALY18 levels were normalized to GAPDH (bar graph). NT, no treatment.
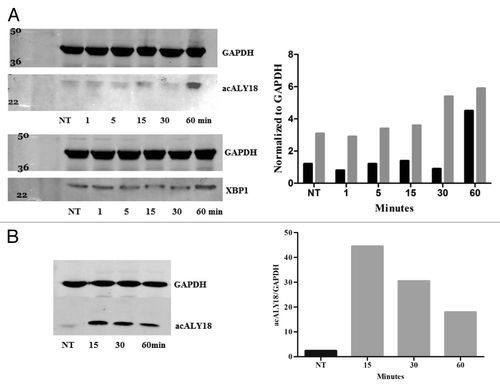
We were surprised to find that acALY18 was associated with a protein target under the denaturing conditions of western analysis that suggested covalent bond formation between acALY18 and XBP1. Because acALY18 contains a cysteine at position 15 and XBP1u contains 3 cysteines, we speculated that it was possible that a disulfide bond could covalently attach acALY18 to XBP1. Therefore, we induced acALY18 peptide in primary human fibroblasts with thapsigargin for 30 min. The whole cell lysates were treated with 50 mM DTT plus 2.5% β-mercaptoethanol for 15, 30, or 60 min to reduce disulfide bonds. Western analysis and quantitation of acALY18 normalized to GAPDH are presented in . The levels of acALY18 associated with the protein target were seen to linearly decline with increasing time of treatment with the reducing agents. This result establishes that acALY18 forms a disulfide bond with XBP1.
To confirm or refute the identity of the acALY18 protein target, we conducted a series of pulldown assays using immobilized anti-acALY18 antibody or anti-XBP1 antibody and thapsigargin-treated human fibroblasts or mouse fibroblasts in two independent laboratories. In the first experiment, primary human fibroblasts were treated for either 1 h or 24 h with thapsigargin. The cell lysates were incubated with anti-XBP1 immobilized on Protein-A coated magnetic beads. The protein was eluted with SDS and the captured protein was analyzed by western blotting. The immunoblots were cut in half, while one half was probed with anti-acALY18 antibody, the other half was probed with anti-XBP1 antibody. These results are presented in and show that acALY18 interacts with both the spliced and unspliced forms of XBP1.
Figure 5. Pulldown assay with XBP1 antibody after 1 and 24 h treatment with 25 nM thapsigargin. (A) Primary human fibroblasts were treated with 25 nM thapsigargin for 1 h or 24 h. Cell lysates were incubated with XBP1 antibody immobilized on Protein-A coated magnetic beads. The bead eluate was analyzed by western and probed for XBP1 (left) and acALY18 (right). Densitometry analysis indicates increasing expression of acALY18 (■) associated with either XBP1u (■) (left) or XBP1s (■) (right) consistent with the whole cell lysate western analysis shown in . (B) Immunoprecipitated protein isolated with immobilized acALY18 antibody and probed with XBP1 antibody (right) or isolated with immobilized XBP1 antibody and probed with acALY18 antibody (left). XBP1 and acALY18 co-localize and acALY18 appears to bind both the spliced and unspliced forms of XBP1.
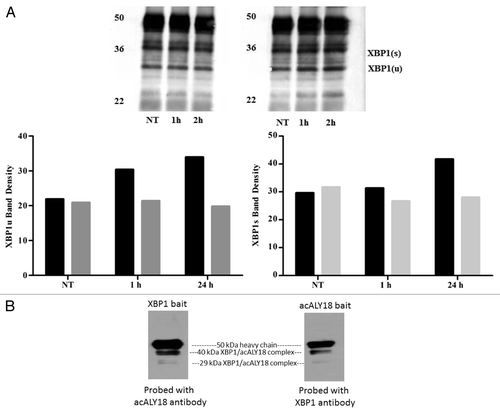
In the second experiment, C57BL/6 mouse fibroblasts were treated with thapsigargin for 1 h. Cell lysates were incubated with either Protein-A magnetic beads immobilized anti-acALY18 or anti-XBP1. The captured proteins were eluted and size fractionated on the same SDS PAGE gel and transferred to a PVDF membrane. The membrane was cut in half and probed with either anti-acALY18 or anti-XBP1 as appropriate. The results depicted in show that immobilized anti-XBP1 or anti-acALY18 pull out the same acALY18/XBP1 complexes, thus confirming that the intracellular protein target of acALY18 is, in fact, XBP1. In this study we also found that acALY18 associated with both XBP1u and XBP1s proteins.
In vitro fibrosis experiments
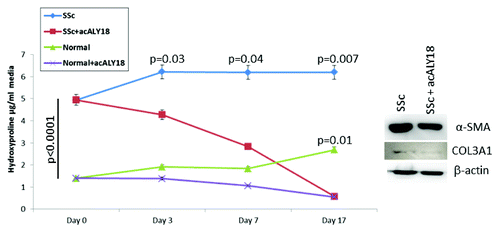
We found that acALY18 abrogated collagen synthesis in scleroderma fibroblasts over time. At the start of this experiment, human scleroderma fibroblasts synthesized 3.6-fold more collagen than normal fibroblasts (). Cyclical treatments with acALY18 (3 ng/ml) alternating every 3 or 4 d significantly lowered total collagen synthesis (as measured by hydroxyproline) in the scleroderma fibroblasts by 46% at day 3 (P = 0.03), by 69% at day 7 (P = 0.04), and by 91% at day 17 (P = 0.007). It was not until day 17 that we observed a significant reduction in collagen synthesis in normal fibroblasts (P = 0.01). Furthermore, when the study was terminated at day 17, we investigated protein levels of type III collagen and α-smooth muscle actin by western blotting and found these proteins to be lowered in the recovered cell lysates.
Figure 6. acALY18 abrogates fibrosis. Cells were cultured until confluent. At Day 0 media was removed and saved for hydroxyproline assay. acALY18 (3 ng/ml) was added to fresh media and the cells cultured for 3 or 4 d. At each addition of fresh media, the expired media was saved for hydroxyproline assessment and fresh acALY18 added. The study was terminated at Day 17. (Left) Hydroxyproline levels in all samples were determined at the same time. (Right) Cells were harvested on Day 17, lysed, and protein analyzed by western blotting for α-smooth muscle actin, collagen 3A1, and β-actin.
In vivo efficacy
A spayed, 7-y-old, female, black Labrador retriever weighing 45 kg presented with a 2 cm fibrotic lick granuloma on the right dorsal forepaw and a smaller lesion (1 cm) on the left dorsal forepaw that had been present for more than 1.5 y. The patient had successfully completed a treatment course of doxycycline and non-steroidal anti-inflammatory drugs (NSAIDs) for acute Lyme disease 3 mo prior, but this treatment had no effect on these lesions. The patient was administered a 0.6 ml dose of acALY18 (12 mg/ml in 0.1% Dextran-40/PBS, pH 7.2) intravenously into the left cephalic vein and was observed for any acute adverse reaction. A second dose was administered 3 d later. Two weeks later a second cycle was administered consisting of two 1 ml doses. Two weeks after this course, a third cycle was administered consisting of 1 ml and 0.8 ml doses. No concomitant treatments were given. No adverse effects were observed throughout and the blood chemistries and hematology were unremarkable. After the first course of treatment, the lesions on both paws became suppurative and by the third cycle, the lesions had completely disappeared. More than a year later there has been no recurrence of either of the fibrotic lesions.
The drug candidate
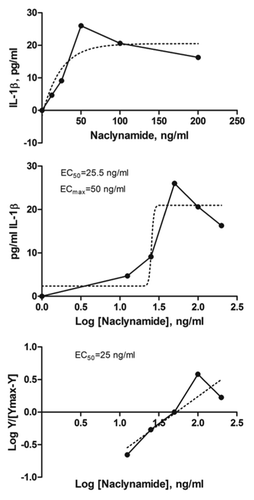
We had previously determined that membrane transport of acALY18 by passive diffusion was the rate limiting factor for bioavailability. Thus, we made the C-terminal amide homolog (Naclynamide) to be a neutral, amphipathic peptide to enhance the membrane permeability and retain the water solubility of the parent peptide, acALY18. A dose-response curve was established by measuring IL-1β secretion from primary human fibroblasts after exposure to increasing concentrations of Naclynamide (). The measured EC50 was 25 ng/ml and the EC99 was 50 ng/ml. The EC99 was chosen as the desired target blood concentration in the pilot clinical study. The EC99 for Naclynamide was approximately 10-fold lower than that measured for acALY18 and approximately 10-fold higher than the natural lipopeptide product. We determined that the target dose of Naclynamide would be 4 μg/kg.
Figure 7. Naclynamide dose response curve. (Top) Dose response curve obtained with primary human fibroblasts cultured for 48 h in the presence of increasing concentrations of Naclynamide. Secreted IL-1β was measured at the 48 h time point by ELISA. Each data point is the mean of 3 separate experiments. Dotted lines represent the regression curve and the calculated EC50 and EC99 from a semi-log plot (middle) and a Hill Plot (bottom) of the data.
Pilot clinical cancer study
MPI Research, Inc. conducted an escalating, single, intravenous dose, acute toxicity study in the mouse with the parent signaling peptide, acALY18. Up to a 2 mg dose (limit of solubility) for acALY18 was well tolerated with no adverse effects, and no significant alterations in blood chemistry, hematology, or cytokine/chemokine and acute phase protein blood levels over the 48 h study period. No gross abnormalities were noted on necropsy and histology of major organ tissues was negative.Citation21
Patient 1
A spayed, 9-y-old, female boxer had been cytologically diagnosed with stage IVb multicentric, high grade non-Hodgkin lymphoma, and grade II mast cell tumor. She had been diagnosed with lymphoma 5 mo prior to enrollment in the study and had failed various standard canine protocols (CHOP-based treatment, single agent Lomustine, prednisone, and MOPP). At the start of treatment, she had been on 0.5 mg/kg prednisone daily, and her tumor was rapidly progressive. She had generalized peripheral lymphadenopathy and suspected progression of lymphoma to the lung parenchyma based on clinical signs and thoracic radiographs. Her last treatment prior to Naclynamide had been her fourth dose of subcutaneous l-asparaginase, to which she had had a partial response that lasted 1.5 wk.
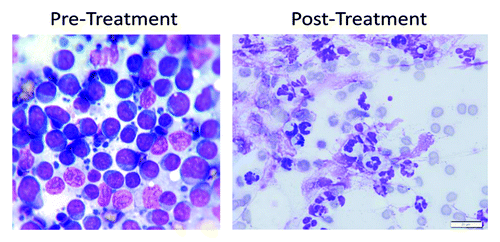
All peripheral lymph nodes measured approximately 2 cm in diameter except for the right prescapular lymph node, which measured 6.5 × 5 × 5 cm. We chose the prescapular lymph node to monitor the efficacy of Naclynamide treatment. During treatment all other peripheral lymph nodes either decreased in size or remained at 2 cm. On the third treatment visit (1.5 wk after the start of the treatment), the prescapular lymph node was reported to be painful, and was evaluated by cytology. Marked neutrophilic inflammation was noted without the obvious presence of bacteria (). A lymph node aspirate was evaluated cytologically by a board-certified clinical pathologist and it was reported to have no evidence of lymphoma and the cytological diagnosis was suppurative inflammation.
Figure 8. Cytology of the pre-scapular lymph node in the lymphoma patient. Pre-treatment, cytology shows a monomorphic population of lymphoblasts that have round to slightly irregular nuclei in a scant amount of circumferential basophilic cytoplasm. The nuclei also contain large nucleoli. Lymphoglandular bodies are present in the background. Post-treatment cytology shows many degenerate neutrophils, few macrophages, rare reactive fibroblasts, many ruptured cells, abundant streaming nuclear debris, and moderate numbers of erythrocytes. No atypical cells are identified.
Patient 2
A castrated, 11.5-y-old, male greyhound had been diagnosed with a metastatic colorectal carcinoma five years prior. He had been extensively treated with multiple surgeries and courses of chemotherapy (carboplatin, adriamycin, 5-FU, cytoxan, palladia, masitinib, piroxicam, deramax, and leukeran) and had also undergone 3 treatments for rectal stricture with an internal medicine specialist. The mass had been biopsied 4 times during the course of the previous treatments, and all biopsies confirmed the continued presence of rectal carcinoma. By early 2013, the patient’s tumor began to grow and protrude externally around the anus to an extent that compromised the anal region.
A board-certified surgeon evaluated the patient and determined that no further surgery should be performed. Given the lack of other treatment options, the patient was enrolled and received the first four doses of Naclynamide without incident. The tumor progressively decreased in size and began to suppurate. At this point the patient also began to lick at the tumor obscuring objective measurements of the tumor’s exterior appearance because of the consequent inflammation. Cytology was evaluated, and while there were still rafts of carcinoma cells present, the majority of the sample showed suppurative inflammation. Bacterial organisms were present, as would be expected based on tumor location. Internally, the patient’s rectal stricture improved markedly in diameter to approximately double that at the beginning of Naclynamide treatment. Initially on digital palpation, the rectal canal measured <5 mm in diameter but increased to approximately 1 cm during the course of treatment. The tumor itself shrank from 8 cm in its largest measurement to 4 cm.
Patient 3
A castrated 6.5-y-old, male Yorkshire terrier presented 3-legged lame with a 6 cm, firm mass in the area of the right proximal tibia and stifle joint. Seventeen months prior, the same lesion was surgically biopsied and diagnosed as a sarcoma, most consistent with osteosarcoma. During that time, the lesion had grown and the patient had become progressively more lame, but 3-view thoracic radiographs showed no evidence of progressive disease. The owner declined other treatment, and was then offered Naclynamide. The patient was treated with intravenous Naclynamide for 6.5 wk with little to no change noted in the tumor size based on caliper measurement and repeat radiographs. Softening of the central tumor was noted, however, as evidenced by a progressively increasing range of motion starting in the second week of Naclynamide treatment. Eventually, the patient’s tumor and limb were amputated and the patient received a 2-wk course of Naclynamide to treat potential metastases. The mass was biopsied and again confirmed to be osteosarcoma.
Discussion
Thacker et al. had reported that an examination of the human TRPC1 gene had no stop codon corresponding to the C-terminal isoleucine in acALY18 and that it was unlikely that this peptide was transcribed from TRPC1 as a separate reading frame. The results presented here show that thapsigargin induces significant measurable expression of acALY18 within 60 min and detectable levels of GRP78 within 24 h. We have subsequently shown detectable levels of acALY18 within 15 min of thapsigargin treatment and GRP78 expression within 12 h. Therefore, it can be concluded that acALY18 expression precedes GRP78 expression after thapsigargin induced ER stress. Moreover, we show treatment with exogenous acALY18 does not induce an ER stress response as measured by GRP78, IRE1α, and eIF2α phosphorylation. We speculate that acALY18 may be enzymatically excised from the third extra-membrane loop of ER embedded TRPC1 during ER stress and that this process disrupts the calcium channel pore. We hypothesize that this limits calcium efflux from the ER store. Signal peptidases have been reviewed, and an ER membrane bound signal peptidase with pepsin-like specificity is a likely enzyme candidate to excise the third extra-membrane loop of ER-embedded TRPC1.Citation22
We have shown that XBP1 is the intracellular target for acALY18 and that, under pharmacologically induced ER calcium efflux and ER stress, acALY18 is excised from TRPC1 to engage XBP1 within minutes. The acALY18-induced IL-1β/IL-18/IL-33 secretion is NLRP3-dependent so it is tempting to suggest that acALY18/XBP1 engagement is somehow mediating NLRP3 inflammasome assembly and/or activation.
It is still unclear what cellular signaling events mediate inflammasome assembly and activation. The apoptotic speck-like protein containing a caspase recruitment domain (ASC) is an essential component of the fully assembled and active NLRP3 inflammasome. Stehlik et al. provided evidence that ASC is compartmentalized in the nucleus of resting monocytes and macrophages.Citation23 In that report it was shown that ASC redistributed to the cytosol within 30 min after exposure to E. coli total RNA and induced maturation and release of IL-1β. Moreover, when ASC relocation was inhibited, IL-1β maturation and release was ablated. Induction of reactive oxygen species within the mitochondria is another event that is known to precede NLRP3 inflammasome assembly and activation.Citation24
Herein, we show that XBP1 engagement of a new signaling peptide derived from ER embedded TRPC1 is a factor relating ER stress to inflammasome activation, maturation, and secretion of IL-1β/IL-18/IL-33, and downstream innate immune responses. XBP1 is an important transcription factor involved in the UPR in response to ER stress. As such, it has an important and significant role in many of the chronic diseases.
XBP1 has been suggested as a possible target for a novel anti-cancer strategy.Citation25 The role of XBP1 in cancer was elucidated in a study demonstrating that blockade of XBP1 splicing by inhibition of the UPR could be a promising therapeutic strategy for the treatment of multiple myelomaCitation26 and possibly other cancer types.Citation27,Citation28 Also, it has been shown that under hypoxic conditions, XBP1(s) promotes tumor growthCitation29,Citation30 lending credence to our hypothesis that engagement of XBP1 by Naclynamide may regulate XBP1 downstream gene transcription to abrogate tumorigenesis.
XBP1(s) was also reported to be elevated in scleroderma, an uncontrolled fibrotic disease of skin and internal organs.Citation31 Lenna et al. demonstrated increased UPR gene expression and altered XBP1 mRNA processing, which suggests that the UPR may be driving fibrosis in scleroderma. Interestingly, we have also found that there is a modest increased risk for cancer in these individuals.Citation32,Citation33
Inflammasome molecular biology is a rapidly expanding field of study, and the role the inflammasome plays, particularly in regards to cancer development and progression, is still not fully understood. Inflammasome biology is ripe for further study and exploitation as a druggable target in chronic disease. Our in vitro and laboratory animal studies suggest that Naclynamide engagement of XBP1 induces an innate immune response, tissue resident immune cell activation, recruitment and differentiation of monocytes and neutrophils, and a potent CTL response at the site of disease. The cytology of cancer tumors after Naclynamide administration and the observation of tumor suppuration with the absence or decline in the number of cancer cells in two cancer types (high grade non-Hodgkin lymphoma and rectal carcinoma), and the suppuration and resolution of benign fibrotic lesions support this mode of action in vivo. To the best of our knowledge, this is the first clinical evidence of a drug candidate specifically targeting XBP1 and inflammasome biology as a druggable target in cancer and fibrotic diseases.
Materials and Methods
Materials used
Unless otherwise specified general laboratory reagents and chemicals were provided by ThermoFisher Scientific, Inc.
The signaling peptide, acALY18, was synthesized as previously described.Citation9 The active pharmaceutical ingredient (Naclynamide) was synthesized in accordance with the US FDA cGMP standards by the contract manufacturer, Peptisyntha, Inc., a division of Solvay USA. The methodology was that as previously published with the modification as a C-terminal amide.
The acALY18 polyclonal antibody was prepared for TherimuneX Pharmaceuticals Inc., by Lampire Biological Laboratories. GRP78, GAPDH, and XBP1 antibodies were purchased from Santa Cruz Biotechnology. Primary human fibroblasts (accession number GM04190) were purchased from Coriell Institute for Medical Research and were maintained in DMEM supplemented with 10% FBS and penicillin/streptomycin.
IRE1α and eIF2α experiments
Primary human fibroblasts were treated with either 5 ng/mL of acALY18 or 25 nM thapsigargin for 1 h and 24 h. The cells were harvested by scraping into the media on ice. After centrifugation for 5 min at 2500 RPM and 4 °C, the resulting pellets were suspended in 500 uL of PBS supplemented with 50 mM sodium fluoride, 5 mM sodium pyrophosphate, 15 mM EDTA, 1× protease inhibitor cocktail (Sigma), 1× phosphates inhibitor cocktail II (Sigma), phosphatase inhibitor cocktail III (Sigma), and then collected by centrifugation for 5 min at 5000 RPM and 4 °C. The resulting pellet was lysed with 1× SDS sample buffer in PBS/0.5% EDTA. The cell lysates were analyzed by western and probed with antibodies to the phosphorylated forms of IRE1α (Santa Cruz) and eIF2α (Thermo Scientific). After stripping the western blots, they were probed again for total IRE1α (Santa Cruz) and eIF2α (Thermo Scientific). Band density was determined using Li-Cor’s Odyssey software. The phosphorylated band densities were compared with the total band densities and expressed as a percentage of the total band density.
TRPC1 experiments
TRPC1−/− and the wild-type mouse embryonic fibroblasts were on a 129SvEv background and were the kind gift of Lutz Birnbaumer at the NIEHS. Cells were treated with 25 nM thapsigargin (Sigma-Aldrich) for 1, 5, 15, 30, and 60 min. Cells were harvested by scraping into 1× SDS (Boston BioProducts) sample buffer, analyzed by western, probed for acALY18, and acALY18 was normalized to GAPDH expression. IL-1β secretion was measured by ELISA (eBioScience) after 72 h thapsigargin (25 nM) treatment.
XBP1 experiments
Primary human fibroblasts were treated with 25 nM thapsigargin (1, 5, 15, 30, and 60 min), then harvested by scraping into 1× SDS sample buffer, analyzed by western that was probed for either acALY18, GAPDH, or XBP1 and protein levels were normalized to GAPDH.
Primary human fibroblasts were treated with 25 nM thapsigargin for 30 min and harvested by scraping into 1× SDS sample buffer. The cell lysate was then reduced with 50 mM DTT and 2.5% β-mercaptoethanol for 15, 30, or 60 min on ice and analyzed by western blotting and probed for acALY18 and GAPDH. The acALY18 levels were normalized to GAPDH.
Pulldown experiments
Primary human fibroblasts cultured in 6-well plates, were incubated with 25 nM thapsigargin for 60 min or for 24 h, then harvested by scraping with ice cold NET buffer (NaCl/EDTA/Tris) supplemented with 0.5% Triton X. Protein-A coated magnetic beads (Invitrogen) were prepared in accordance with the manufacturer’s directions and either polyclonal rabbit anti-human IL-1β, anti-acALY18, or anti-human XBP1 antibodies were adsorbed onto the beads. Three hundred microliters of whole cell lysate was centrifuged for 15 min to clear the lysate of any precipitate and then the supernatant was pre-cleared by incubating for 15 min with the non-target antibody-conjugated beads (anti-IL-1β). The pre-cleared lysate was collected after magnetic isolation of the beads and transferred to a tube containing anti-XBP1 antibody conjugated Protein-A beads and incubated at room temp for 15 min with gentle mixing. The beads were magnetically isolated from the lysate and washed 5 times with PBS. After the final wash the beads were suspended in 30 μL of SDS loading buffer and were stored frozen until used. For western analysis, the samples were thawed and heated to 95 °C for 5 min. The beads were magnetically isolated and the supernatant was loaded onto Tris/glycine PAGE gels and the proteins were separated by electrophoresis at 100 V for 120 min using a SDS/glycine running buffer. Proteins separated on the gel were transferred to PVDF (Invitrogen) using transfer buffer (Invitrogen) supplemented with 20% methanol at 230 mA for 120 min. The PVDF was cut in half and probed for either acALY18 or XBP1. The membranes were blocked for 60 min at room temp with Odyssey blocking buffer (LI-COR Biotechnology). XBP1 antibody was diluted 1:500 in Odyssey blocking buffer with 0.1% Tween-20 and applied to one half of the membrane and acALY18 antibody was diluted 1:500 in Odyssey blocking buffer with 0.1% Tween-20 and applied to the other half of the membrane. Membranes were incubated overnight at 4 °C with rocking and washed 3 times with PBS/0.05% Tween-20 for 5 min. Both membranes were then incubated with donkey anti-rabbit IR dye 800 (LI-COR) diluted 1:5000 in Odyssey blocking buffer with 0.1% Tween-20/ 0.1% SDS for 1 h, and washed 3 times in PBS/0.05% Tween-20 for 5 min and a final wash in PBS for 5 min.
C57BL/6 murine fibroblasts were cultured to confluence in a T75 flask. Cells were incubated with 25 nM thapsigargin for 60 min then harvested as described above and the cell pellet isolated by centrifugation. Cells were lysed in NET buffer (NaCl/EDTA/Tris) containing 0.5% Triton X. Two 50 μL aliquots of Protein-A coated magnetic beads were washed twice with PBS. One aliquot was incubated with acALY18 antibody and the other with XBP1 antibody for 30 min, then the beads were isolated by magnetic separation, washed 3 times with PBS, and stored in 100 μL PBS until needed. Two 50 μL aliquots of whole cell lysate were pre-cleared by incubation for 20 min with 50 μL of PBS washed unconjugated Protein A beads then the pre-cleared lysate was collected by magnetic separation of the beads and transferred to the tube containing anti-acALY18 or anti-XBP1 antibody conjugated Protein A beads. The cell lysate/bead mixture was incubated at room temp for 30 min with gentle mixing. The beads were isolated, and washed 3 times with PBS, then suspended in 30 μL of SDS loading buffer (Invitrogen) and boiled for 5 min. The beads were magnetically separated just prior to loading the supernatant on a precast gel (Invitrogen). The proteins were separated by electrophoresis at 140 V for 120 min using a SDS/glycine running buffer. Proteins separated on the gel were transferred to a PVDF using a transfer buffer (Invitrogen) supplemented with 10% methanol at 30 V for 60 min. The PVDF was cut in half to separate the protein pulled out by anti-acALY18 antibody from that pulled out by anti-XBP1 antibody, blocked for 60 min at room temperature with 5% skim milk powder/TBS. XBP1 antibody was diluted 1:500 in 3% BSA/TBS and applied to the membrane containing proteins pulled out with anti-acALY18 antibody. Anti-acALY18 antibody was diluted 1:1000 in 3% BSA/TBS and applied to the membrane containing proteins pulled out with anti-XBP1 antibody. Membranes were incubated overnight at 4 °C with rocking and washed 3 times for at least 20 min in TBS. Both membranes were then incubated with donkey anti-rabbit horseradish peroxidase (Jackson Immuno Research) diluted 1:2000 in 3% BSA/TBS for 3 h, then washed 3 times in TBS for at least 20 min and incubated with 2 mL of ECL Chemiluminescence Reagent (Pierce) for 10 min and exposed to X-ray film.
Scleroderma experiments
Confluent primary scleroderma fibroblasts derived from skin explants were treated with 3 ng/ml acALY18 in Dulbecco’s modified Eagle’s medium supplemented with 10% FBS and 1× penicillin/streptomycin (Gibco). Media was changed alternating every 3 or 4 d at which time fresh acALY18 was added. The media was saved and assayed for hydroxyproline according to Artlett et al. (2011)Citation16 at the end of the study. The cells were also collected at the end of the study, lysed, and westerns were performed as previously published.Citation16
Pilot clinical study design
The trial was designed as an open-label, single arm, pilot study. Dogs were enrolled if they had been definitively diagnosed with cancer, the cancer had been refractory to prior chemotherapeutics, and either there were no other treatment options, or the owners had declined all other options. The patients also had not been treated with other chemotherapy agent for a minimum of 2 wk prior to starting the treatment with Naclynamide. Oral medications, such as non-steroidal anti-inflammatory drugs (NSAIDs) or steroids (prednisone) were allowed as long as the patients had been on these drugs continuously for greater than one month prior to the study. Staging included a serum chemistry analysis, complete blood count (CBC), and urinalysis. The patient’s tumors were measured, and where appropriate, pictures were taken to document changes that occurred during the study.
Vital signs, physical examination, and performance status were assessed at each visit. CBCs were assessed at least every other week. Serum chemistry analyses were performed at least one time per month. Tumor size was assessed by visual inspection, digital radiographic imaging, and caliper measurements. Anti-nausea or anti-diarrheal drugs were allowed to mitigate side effects if necessary.
Owners provided informed consent for treatment and signed an Owners Informed Consent and Waiver of Liability. Other than free treatment, financial benefits were not provided.
Drug dosing
Naclynamide (1000 μg) was dissolved in 4 ml of pre-sterilized vehicle (0.1% Dextran-40 in PBS, pH 7.2) and aseptically filled into sterile 5-ml vials for injection, stoppered, crimp sealed, labeled, and stored frozen (−20 °C) until the time of use (final Naclynamide concentration: 250 μg/ml). Naclynamide was then administered twice weekly by quick intravenous injection at 1 ml/60 lb of body weight. At this dosage, the blood concentration was expected to be 50 ng/ml, which is the EC99 for Naclynamide. Each dog received an initial loading dose (1 ml/30 lb of body weight) to overcome serum protein binding and a depot effect. This was then followed by the maintenance dosage (1 ml/60 lb of body weight) given twice weekly for a projected 8-wk treatment course.
Tumor cytology
A 22 gauge needle attached to a 10-ml syringe was used to collect cells from the cancerous lesions. Each lesion was sampled with a clean needle a minimum of 3 times. The collected cells were aspirated onto glass microscope slides and were stained with a standard Wright–Giemsa stain by personnel from Idexx Laboratories and evaluated by microscopy.
| Abbreviations: | ||
| BSA | = | bovine serum albumin |
| CBC | = | complete blood count |
| CTL | = | cytotoxic lymphocyte |
| GMP | = | current good manufacturing practices |
| CHOP | = | cyclophosphamide/hydroxydaunorubicin/oncovin/prednisone |
| DMEM | = | Dulbecco’s modified Eagle medium |
| FBS | = | fetal bovine serum |
| 5-FU | = | 5-fluorouracil |
| GAPDH | = | glyceraldehyde phosphate dehydrogenase |
| MOPP | = | mustagen/oncovin/procarbazine/prednisone |
| PBS | = | phosphate buffered saline |
| DTT | = | dithiothreitol |
| PVDF | = | polyvinylidene fluoride |
| SDS | = | sodium dodecyl sulfate |
| TBS | = | tris buffered saline |
| XBP1 | = | X box binding protein 1 |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by TherimuneX Pharmaceuticals, Inc. and Center for Animal Referral and Emergency Services (CARES).
The authors wish to acknowledge the support of Liz Little VMD, DACVP and Sami Fritz Waibel CVT at CARES for the technical support in the conduct of the pilot clinical study.
References
- Chakrabarti A, Chen AW, Varner JD. A review of the mammalian unfolded protein response. Biotechnol Bioeng 2011; 108:2777 - 93; http://dx.doi.org/10.1002/bit.23282; PMID: 21809331
- Zitvogel L, Kepp O, Galluzzi L, Kroemer G. Inflammasomes in carcinogenesis and anticancer immune responses. Nat Immunol 2012; 13:343 - 51; http://dx.doi.org/10.1038/ni.2224; PMID: 22430787
- Lamkanfi M. Emerging inflammasome effector mechanisms. Nat Rev Immunol 2011; 11:213 - 20; http://dx.doi.org/10.1038/nri2936; PMID: 21350580
- Chu J, Thomas LM, Watkins SC, Franchi L, Núñez G, Salter RD. Cholesterol-dependent cytolysins induce rapid release of mature IL-1beta from murine macrophages in a NLRP3 inflammasome and cathepsin B-dependent manner. J Leukoc Biol 2009; 86:1227 - 38; http://dx.doi.org/10.1189/jlb.0309164; PMID: 19675207
- Dostert C, Guarda G, Romero JF, Menu P, Gross O, Tardivel A, Suva M-L, Stehle J-C, Kopf M, Stamenkovic I, et al. Malarial hemozoin is a Nalp3 inflammasome activating danger signal. PLoS One 2009; 4:e6510; http://dx.doi.org/10.1371/journal.pone.0006510; PMID: 19652710
- Gurcel L, Abrami L, Girardin S, Tschopp J, van der Goot FG. Caspase-1 activation of lipid metabolic pathways in response to bacterial pore-forming toxins promotes cell survival. Cell 2006; 126:1135 - 45; http://dx.doi.org/10.1016/j.cell.2006.07.033; PMID: 16990137
- Thomas PG, Dash P, Aldridge JR Jr., Ellebedy AH, Reynolds C, Funk AJ, Martin WJ, Lamkanfi M, Webby RJ, Boyd KL, et al. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity 2009; 30:566 - 75; http://dx.doi.org/10.1016/j.immuni.2009.02.006; PMID: 19362023
- Cassel SL, Eisenbarth SC, Iyer SS, Sadler JJ, Colegio OR, Tephly LA, Carter AB, Rothman PB, Flavell RA, Sutterwala FS. The Nalp3 inflammasome is essential for the development of silicosis. Proc Natl Acad Sci U S A 2008; 105:9035 - 40; http://dx.doi.org/10.1073/pnas.0803933105; PMID: 18577586
- Dostert C, Pétrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 2008; 320:674 - 7; http://dx.doi.org/10.1126/science.1156995; PMID: 18403674
- Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 2008; 9:847 - 56; http://dx.doi.org/10.1038/ni.1631; PMID: 18604214
- Gasse P, Riteau N, Charron S, Girre S, Fick L, Pétrilli V, Tschopp J, Lagente V, Quesniaux VFJ, Ryffel B, et al. Uric acid is a danger signal activating NALP3 inflammasome in lung injury inflammation and fibrosis. Am J Respir Crit Care Med 2009; 179:903 - 13; http://dx.doi.org/10.1164/rccm.200808-1274OC; PMID: 19218193
- Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006; 440:228 - 32; http://dx.doi.org/10.1038/nature04515; PMID: 16407890
- Salminen A, Ojala J, Suuronen T, Kaarniranta K, Kauppinen A. Amyloid-beta oligomers set fire to inflammasomes and induce Alzheimer’s pathology. J Cell Mol Med 2008; 12:6A 2255 - 62; http://dx.doi.org/10.1111/j.1582-4934.2008.00496.x; PMID: 18793350
- Yamasaki K, Muto J, Taylor KR, Cogen AL, Audish D, Bertin J, Grant EP, Coyle AJ, Misaghi A, Hoffman HM, et al. NLRP3/cryopyrin is necessary for interleukin-1beta (IL-1beta) release in response to hyaluronan, an endogenous trigger of inflammation in response to injury. J Biol Chem 2009; 284:12762 - 71; http://dx.doi.org/10.1074/jbc.M806084200; PMID: 19258328
- Allen IC, TeKippe EM, Woodford RM, Uronis JM, Holl EK, Rogers AB, Herfarth HH, Jobin C, Ting JP. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J Exp Med 2010; 207:1045 - 56; http://dx.doi.org/10.1084/jem.20100050; PMID: 20385749
- Artlett CM, Sassi-Gaha S, Rieger JL, Boesteanu AC, Feghali-Bostwick CA, Katsikis PD. The inflammasome activating caspase 1 mediates fibrosis and myofibroblast differentiation in systemic sclerosis. Arthritis Rheum 2011; 63:3563 - 74; http://dx.doi.org/10.1002/art.30568; PMID: 21792841
- Thacker JD, Brown MA, Rest RF, Purohit M, Sassi-Gaha S, Artlett CM. 1-Peptidyl-2-arachidonoyl-3-stearoyl-sn-glyceride: an immunologically active lipopeptide from goat serum (Capra hircus) is an endogenous damage-associated molecular pattern. J Nat Prod 2009; 72:1993 - 9; http://dx.doi.org/10.1021/np900360m; PMID: 19835390
- Thacker JD, Balin BJ, Appelt DM, Sassi-Gaha S, Purohit M, Rest RF, Artlett CM. NLRP3 inflammasome is a target for development of broad-spectrum anti-infective drugs. Antimicrob Agents Chemother 2012; 56:1921 - 30; http://dx.doi.org/10.1128/AAC.06372-11; PMID: 22290938
- Majumder M, Huang C, Snider MD, Komar AA, Tanaka J, Kaufman RJ, Krokowski D, Hatzoglou M. A novel feedback loop regulates the response to endoplasmic reticulum stress via the cooperation of cytoplasmic splicing and mRNA translation. Mol Cell Biol 2012; 32:992 - 1003; http://dx.doi.org/10.1128/MCB.06665-11; PMID: 22215619
- Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature 2008; 454:455 - 62; http://dx.doi.org/10.1038/nature07203; PMID: 18650916
- Luckett EM. THX314: Intravenous toxicity study in female C57Bl/6 mice. MPI Research, Inc. Mattawan, MI 49071 February 25, 2010.
- Paetzel M, Karla A, Strynadka NCJ, Dalbey RE. Signal peptidases. Chem Rev 2002; 102:4549 - 80; http://dx.doi.org/10.1021/cr010166y; PMID: 12475201
- Bryan NB, Dorfleutner A, Rojanasakul Y, Stehlik C. Activation of inflammasomes requires intracellular redistribution of the apoptotic speck-like protein containing a caspase recruitment domain. J Immunol 2009; 182:3173 - 82; http://dx.doi.org/10.4049/jimmunol.0802367; PMID: 19234215
- Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011; 469:221 - 5; http://dx.doi.org/10.1038/nature09663; PMID: 21124315
- Koong AC, Chauhan V, Romero-Ramirez L. Targeting XBP-1 as a novel anti-cancer strategy. Cancer Biol Ther 2006; 5:756 - 9; http://dx.doi.org/10.4161/cbt.5.7.2973; PMID: 16861911
- Mimura N, Fulciniti M, Gorgun G, Tai Y-T, Cirstea D, Santo L, Hu Y, Fabre C, Minami J, Ohguchi H, et al. Blockade of XBP1 splicing by inhibition of IRE1α is a promising therapeutic option in multiple myeloma. Blood 2012; 119:5772 - 81; http://dx.doi.org/10.1182/blood-2011-07-366633; PMID: 22538852
- Liu Y, Zhang X, Liang Y, Yu H, Chen X, Zheng T, Zheng B, Wang L, Zhao L, Shi C, et al. Targeting X box-binding protein-1 (XBP1) enhances sensitivity of glioma cells to oxidative stress. Neuropathol Appl Neurobiol 2011; 37:395 - 405; http://dx.doi.org/10.1111/j.1365-2990.2010.01155.x; PMID: 21138464
- Andres SA, Wittliff JL. Relationships of ESR1 and XBP1 expression in human breast carcinoma and stromal cells isolated by laser capture microdissection compared to intact breast cancer tissue. Endocrine 2011; 40:212 - 21; http://dx.doi.org/10.1007/s12020-011-9522-x; PMID: 21858728
- Romero-Ramirez L, Cao H, Nelson D, Hammond E, Lee AH, Yoshida H, Mori K, Glimcher LH, Denko NC, Giaccia AJ, et al. XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res 2004; 64:5943 - 7; http://dx.doi.org/10.1158/0008-5472.CAN-04-1606; PMID: 15342372
- Iwakoshi NN, Pypaert M, Glimcher LH. The transcription factor XBP-1 is essential for the development and survival of dendritic cells. J Exp Med 2007; 204:2267 - 75; http://dx.doi.org/10.1084/jem.20070525; PMID: 17875675
- Lenna S, Farina AG, Martyanov V, Christmann RB, Wood TA, Farber HW, Scorza R, Whitfield ML, Lafyatis R, Trojanowska M. Increased expression of endoplasmic reticulum stress and unfolded protein response genes in peripheral blood mononuclear cells from patients with limited cutaneous systemic sclerosis and pulmonary arterial hypertension. Arthritis Rheum 2013; 65:1357 - 66; http://dx.doi.org/10.1002/art.37891; PMID: 23400395
- Derk CT, Artlett CM, Jimenez SA. Morbidity and mortality of patients diagnosed with systemic sclerosis after the age of 75: a nested case-control study. Clin Rheumatol 2006; 25:831 - 4; http://dx.doi.org/10.1007/s10067-005-0177-y; PMID: 16435160
- Derk CT, Rasheed M, Artlett CM, Jimenez SA. A cohort study of cancer incidence in systemic sclerosis. J Rheumatol 2006; 33:1113 - 6; PMID: 16622904