
Abstract
The malignant phenotype of glioblastoma multiforme (GBM) is believed to be largely driven by glioma stem-like cells (GSCs), and targeting GSCs is now considered a promising new approach to treatment of this devastating disease. Here, we show that SN50, a cell-permeable peptide inhibitor of NFκB, induced robust differentiation of human GSCs, causing loss of their oncogenic potential. We observed that following treatment of GSCs with SN50, their differentiated progeny cells showed significant decreases in their capability to form neuro-spheres and to invade in vitro and a reduction in their tumorigenicity in mouse xenograft models, but had increased sensitivity to the chemotherapeutic drug temozolomide and to radiation treatment. These results suggest that blocking the NFκB pathway may be explored as a useful mean to induce differentiation of GSCs, and provide another supportive evidence for the promise of differentiation therapy in treatment of malignant brain tumors.
Introduction
Glioblastoma multiforme (GBM) is the most common form of brain malignancy in adults and children, and is a highly aggressive and fatal disease characterized by survival of glioma cells following initial treatment, invasion through the brain parenchyma and destruction of normal brain tissues, and ultimately resistance to conventional therapies. Despite apparently clean surgical margins, sophisticated radiation, and newer chemotherapy and targeted therapy, patients diagnosed with GBM typically have a median survival of less than 14.6 mo and fewer than 3% of glioblastoma patients are alive five years after diagnosis.Citation1-Citation3 Recent studies have revealed that malignant tumors including GBM are comprised of a heterogeneous mixture of both cancer stem cells (tumor-initiating cells) that possess the property of self-renewal, as well as more differentiated cancer cells that have limited proliferative potential and are not believed capable of self renewal.Citation4,Citation5 Cancer stem cells have been identified in blood, brain, and breast cancers and other types of malignancies, and are believed to be responsible for tumor development, progression, recurrence, and therapeutic resistance.Citation6-Citation8 It is generally believed that the discovery of cancer stem cells has provided an attractive target for cancer treatment, and various strategies to eradicating cancer stem cells are being investigated, include killing them or differentiating them into non-stem cancer cells.
Increasing evidences indicate that malignant gliomas originate from neural stem-cell populations, which are termed glioma stem-like cells (GSCs).Citation9 Recent studies have reported that targeting GSCs may effectively reduce tumor recurrence and significantly improve GBM treatment.Citation10-Citation12 Here we show that SN50, a cell-permeable peptide inhibitor of NFκB, can induce GSC differentiation, causing loss of their oncogenic phenotype. Our observations suggest that blocking the NFκB pathway may be an effective way to induce GSC differentiation and to ablate GSCs, and thus may be explored as a new approach to treatment of GBM.
Results
Induction of GSC differentiation by SN50
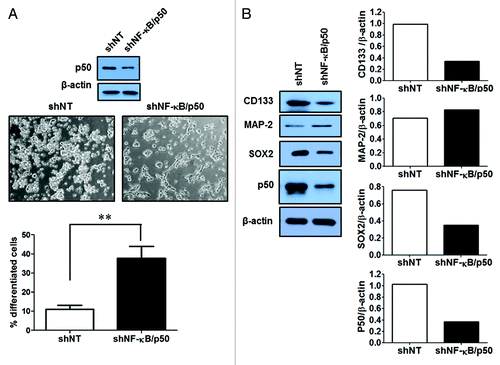
Two GSC cell lines established from human primary glioblastoma tissues,Citation13,Citation14 GSC11 and GSC23, were used in this study. These cell lines showed high expressions of the GSC markers, CD133 and SOX2 (), and acquired astrocytic (GFAP) and neuronal (MAP-2) phenotypes when cultured in the medium containing fetal calf serum (). We observed that following treatment with SN50, a cell-permeable peptide inhibitor of NFκB, GSC11 and GSC23 cells underwent robust differentiations (). The differentiation induced by SN50 was evidenced by a downregulation in the expressions of the GSC markers CD133, SOX2, and NAC1 () and an upregulation in the expressions of the differentiation markers, MAP-2 and GFAP (). The differentiated GSC progeny also showed remarkable alterations in their morphology, as compared with the GSC11 and GSC23 cells treated with vehicle (). To verify that the induction of GSC differentiation seen in the SN50-treated cells was a consequence of NFκB inhibition, we utilized an shRNA to silence the expression of NFκB/p50, and then examined the effect of this approach on GSCs. shows that silencing of NFκB/p50 expression also caused GSC differentiation, as evidenced by changes in the cellular morphology () and in the levels of the tumor stem cell markers ().
Figure 1. Expression of the GSC markers in GSC11 and GSC23 cells. Immunofluorescence detection of CD133, SOX2, and MAP-2 (green)/GFAP (red) (with fetal calf serum for 3 d) in GSC11 and GSC23 cells.
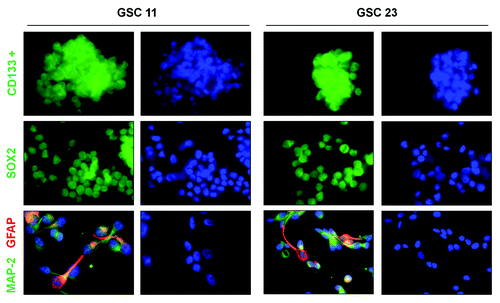
Figure 2. Effects of SN50 on differentiation of GSCs. GSC11 and GSC23 cells were cultured in the DMEM/F-12 medium containing 2% B-27, 20 ng/mL EGF, and 20 ng/mL β-FGF in the presence or absence of SN50 (25 μg/mL). Twenty-four hours later, (A) the cells were observed under a phase contrast microscopy (200× magnification). **P < 0.01, SN50 vs. Control; (B) cell lysates were prepared and subjected to western blot analysis of CD133, NAC1, Sox2, and MAP-2. β-actin was used as a loading control; (C) the cells were fixed on poly-l-lysine- coated coverslips and processed for immunofluorescence analysis of CD133; (D) the cells were fixed on poly-l-lysine-coated coverslips and processed for immunofluorescence analysis of MAP-2 and GFAP. Data shown are the representative of three identical experiments.
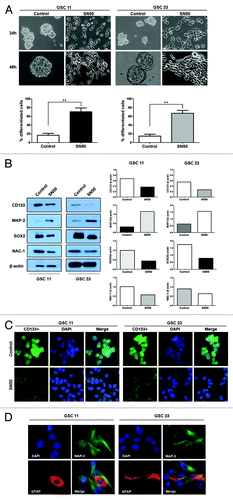
Figure 3. Effects of silencing of NFκB/p50 expression on differentiation of GSCs. (A) GSC11 cells were dissociated, suspended in serum-free DMEM medium in the absence of B-27, EGF, and β-FGF, and then were infected with an NFκB/p50-targeted shRNA lentiviral particles for 48 h in the presence of polybrene (5 μg/mL). Forty-eight hours later, (A) the cells were observed under a phase contrast microscopy (200× magnification). **P < 0.01, shNFκB/p50 vs. shNT control; (B) cell lysates were prepared and subjected to western blot analysis of CD133, Sox2, MAP-2, and p50. β-actin was used as a loading control.
SN50 inhibits the potential of GSCs to form neuro-spheres and to invade
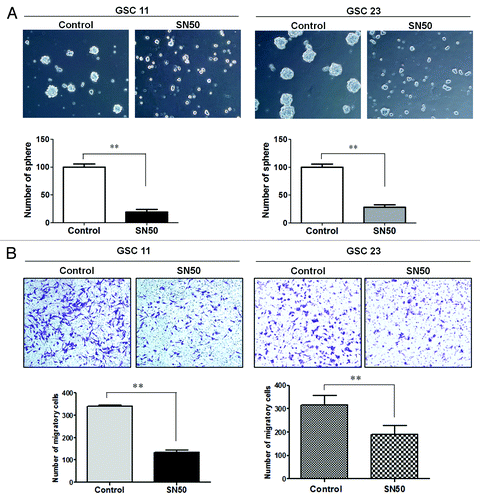
To determine whether the cellular differentiation induced by SN50 alters the malignant characteristics of GSCs, we determined and compared the GSCs’ ability to form neuro-spheres and to invade flowing treatment with SN50 or vehicle. shows that GS11 and GSC23 cells pre-treated with vehicle maintained their ability to form neuro-spheres; by contrast, the cells pre-treated with SN50 for 24 h formed significantly less neuro-spheres than the control cells. Similarly, GS11 and GSC23 cells subjected to pre-treatment with SN50 were significantly less invasive than the cells pre-treated with vehicle, as examined by the Matrigel Transwell invasion assay (). The lower rates of neuro-sphere formation and invasion observed in the SN50-treated samples () might partially result from those undifferentiated portion of the GSCs. These results suggest that the differentiated progeny cells resulting from SN50 treatment may have lost the malignant features of GSCs.
Figure 4. Effects of SN50 on GSC neuro-sphere formation (A) and invasion (B). GSC11 and GSC23 cells were cultured in the DMEM/F-12 medium containing 2% B-27, 20 ng/mL EGF, and 20 ng/mL β-FGF in the presence or absence of SN50 (25 μg/mL). Twenty-four hours later, (A) the cells were dissociated and suspended in drug-free culture medium and seeded in 24-well tissue culture plates at a density of 5000 cells per well. Following 4 d incubation at 37 °C in a humidified incubator containing 5% CO2/95% air, sphere formations were observed and counted under a phase-contrast microscope; (B) the cells were dissociated and suspended in drug-free culture medium at 1 × 105 cells/mL, and then 300 µL of the cell suspension was added to the upper wells of the CytoSelect™ 24-Well Cell Migration/Invasion Assay system. The upper wells contained serum-free DMEM/F-12 medium and the lower wells contained DMEM/F-12 medium with 10% FBS. After 48 h of incubation, noninvading cells in the upper chambers were gently wiped away and the adherent cells on the lower side of each membrane were fixed and stained with 0.1% (w/v) crystal violet. Five fields were randomly selected and the stained cells were counted under a microscope. Data shown are the representative of three identical experiments. **P < 0.01, SN50 vs. control.
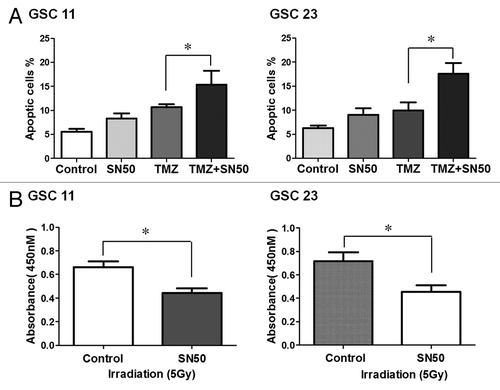
SN50 increases sensitivity of GSCs to temozolomide and radiation treatment
To assess whether SN50-induced cellular differentiation affects response of GSC11 and GSC23 cells to therapeutic intervention, we next tested the sensitivity of their differentiated progeny cells to the chemotherapeutic agent temozolomide (TMZ) and radiation treatment, which are commonly used in the treatment of GBM. shows that temozolomide caused a greater apoptosis in the differentiated progeny cells than in the undifferentiated GSC11 and GSC23 cells, as determined by flow cytometric analysis of Annexin V/7-aminoactinomycin D staining. Sensitizing effect of SN50 was also observed in the differentiated progeny of GSC11 and GSC23 cells subjected to irradiation treatment ().
Figure 5. Effects of SN50 on sensitivity of GSCs to temozolomide (A) and radiation treatment (B). (A) GSC11 and GSC23 cells were first treated with SN50 or vehicle for 24 h, and then were treated with 100 μM of TMZ for 3 d in the absence of SN50. At the end of treatment, apoptosis was determined by flow cytometric analysis of Annexin V/7-aminoactinomycin D staining. (B) GSC11 and GSC23 cells were first treated with SN50 or vehicle for 24 h, and then were irradiated with 5 Gy in the absence of SN50. Cell viability was measured using the CCK8 cell counting assay.
Differentially-expressed genes in GSCs subjected to SN50 treatment
To explore the possible pathways involved in GSC differentiation caused by SN50, we performed a microarray analysis on the GSCs treated or untreated with SN50, using the Affymetrix GeneChip Human Genome U133 Plus 2.0 Array. We found that there were 10 genes showing a 1.5- to 1.8-fold upregulation and 10 genes showing a 2.2- to 2.8-fold downregulation in the SN50-treated cells as compared with the control cells treated with vehicle (). Whether the alterations of these gene expressions are the cause or consequence of the SN50-induced GSC differentiation are currently not clear.
Table 1. The differentially-expressed genes in GSCs treated with SN50
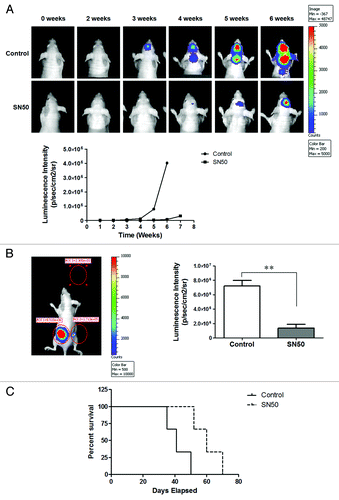
SN50-treated GSCs show a reduced tumorigenicity
As tumor-initiating capability is one of the identities of cancer stem cells, we next evaluated the tumorigenic potential of the differentiated progeny of GSCs following SN50 treatment. In these experiments, GSC11 cells expressing luciferase were treated with SN50 for 24 h to induce differentiation or with vehicle, and then those cells were inoculated into NCr-nu/nu mice either intra-cranially or subcutaneously. As shown in , in comparison to that of the control cells, the capability of the differentiated progeny cells to form brain tumors was remarkably decreased, as evidence by slower growth and smaller size of the tumors observed. The sub-cutaneous tumors of the differentiated GSC11 cells were also significantly smaller than that of the vehicle-treated control cells (). Furthermore, the mice inoculated intra-cranially with the SN50-treated GSC11 cells survived longer than the mice bearing control brain tumors (; median survival: 60 d vs. 41 d). These results clearly demonstrate that the differentiated GSC11 cells resulting from SN50 treatment lost their tumor-initiating capability.
Figure 6. Comparison of the tumorigenecity of GSC11 cells with their differentiated progeny cells. GSC11 cells expressing luciferase were treated with vehicle or SN50 for 24 h, and then the undifferentiated GSC11 cells or their differentiated progeny cells were transplanted into 4–5 wk old female nude mice either intra-cranially (5000 cells/mouse) or subcutaneously (1 × 106 cells per site). On days 14, 21, 28, 35, and 42 post tumor inoculation, bioluminescence was measured by non-invasive bioluminescence imaging. (A) Intra-cranial tumor; (B) subcutaneous tumors; (C) survival plot of mice bearing brain tumors.
Discussion
In this report, we demonstrate that SN50, a cell-permeable peptide inhibitor of NFκB, can induce robust differentiation of GSCs (), and that the differentiated progeny cells show loss of the characteristics of cancer stem cells, including neuro-sphere formation (), motility and invasion (), insensitivity to therapies (), and tumorigenicity (). As NFκB signaling pathway is activated in GSCs,Citation15 our results provide another supportive evidence for potential of targeting NFκB signaling as a therapeutic intervention for malignant gliomas.
The identification of cancer stem cells including GSCs, which constitute a reservoir of self-sustaining cells with potent tumorigenic potential, not only represents an important conceptual advance in cancer biology but also has therapeutic implications. When differentiated, cancer stem cells usually lose the capacity for long-term cellular repopulation, fail to propagate tumors, and show increased sensitivity to therapeutic intervention. Therefore, inducing differentiation of cancer stem cells has been explored as one of the strategies to eradicate those tumor-initiating cells and to suppress tumor development and progression.Citation16 For example, it was reported that silencing of the expression of YB-1, a transcription regulator that is highly expressed in the sub-ventricular zone of mouse fetal brain tissues, reduced growth of the neuro-spheres derived from GBM patients by triggering differentiation.Citation17 Rapamycin was shown to trigger CSCs differentiation in GBM through inhibiting mTOR, thereby sensitizing those cells to radiation therapy and reducing their ability to form brain tumors in a mouse model.Citation18 Combined treatment of rapamycin with a PI3K inhibitor was shown to promote GSC differentiation and have significant therapeutic effect.Citation19 Here, we report that the NFκB inhibitor, SN50, can promote GSC differentiation and inhibit gliomagenesis both in vitro and in vivo, presenting a possible new approach to differentiation therapy.
NFκB is a transcription factor associated with cancer and many other diseases,Citation20,Citation21 and modulation of NFκB pathway is being explored as a new anti-cancer tactic.Citation22 SN50 is a 41-residue synthetic peptide containing a hydrophobic membrane-translocating region and the nuclear localization sequence of NFκB p50. SN50 can enter cells and compete with NFκB complexes for the machinery responsible for the nuclear translocation of this transcription factor.Citation23 SN50 has been shown to effectively inhibit the LPS- and TNFα-induced nuclear translocation of NFκB in different cell lines.Citation24 Here we show that this NFκB inhibitor can effectively reduce GSCs pool through inducing their differentiation and suppress the malignant features of GSCs, including their potential to form neuro-spheres and to invade, resistance to therapy, and tumor-initiating capability. In order to understand the mechanisms underlying the SN50-induced GSC differentiation, we performed a microarray analysis. Although our microarray analysis found that several genes are differentially expressed in the SN50-treated GSCs, we have not been able yet to determine whether those genes indeed play a role in driving GSC differentiation. Further studies would be needed to analyze the roles of those genes in inducing GSC differentiation. Additionally, it would be interesting and important to test the effects of other NFκB inhibitors, such as small molecule inhibitors, on differentiation of cancer stem cells to validate the role of NFκB in modulating stem cells differentiation.
In summary, we report here that the NFκB inhibitor SN50 has differentiation-inducing effect on GSCs, and that this action may account for its suppressive effects on oncogenic phenotypes of cancer stem cells. These functional and morphological evidence suggest that blocking the NFκB pathway may be further explored as a mean to induce differentiation of GSC, and support the potential of differentiation therapy in treatment of malignant brain tumors.
Materials and Methods
Cell cultures
The GSC lines, GSC11 and GSC23, were originally derived from human primary glioblastoma tissues,Citation13,Citation14 and are cultured in the Dulbecco’s modified Eagle’s medium/F-12 supplemented with B-27 (Invitrogen), 20 ng/mL of EGF, 20 ng/mL of β-FGF (GIBCO Invitrogen), 100 units/mL of penicillin, and 100 mg/mL of streptomycin. Cells were maintained at 37 °C in a humidified atmosphere containing 5% CO2/95% air. Every 4–5 d, neuro-spheres were dissociated using accutaseTM (Innovative Cell Technologies Inc.). All cell cultures were monitored routinely and found to be free of contamination by mycoplasma or fungi.
Reagents
AccutaseTM was purchased from Innovative Cell Technologies Inc.; fibroblast growth factor-basic (β-FGF) was purchased from GIBCO; epidermal growth factor (EGF) was purchased from PeproTech Inc.; mouse monoclonal antibodies (Alexa Fluor® 555 Conjugate) against glial fibrillary acidic protein, MAP-2, Sox2, and β-actin were purchased from Cell Signaling Technology Inc. Dulbecco’s modified Eagle’s medium/Ham’s F-12 (DMEM/F-12) was purchase from Lonza. SN50 was purchased from Enzo Life Sciences Inc.
Western blot analysis
Cells were lysed with Solulyse-M mammalian protein extraction reagent (Genlantis) supplemented with a cocktail of protease inhibitors (Roche). Following centrifugation at 14 000 × g for 10 min, cell lysates were collected and protein concentrations were measured. Protein (20 µg) were resolved by sodium dodecyl sulfate-PAGE (SDS-PAGE), and then transferred to PVDF membrane (Bio-Rad). The membranes were incubated with primary antibodies in 1% bovine serum albumin (BSA)/Tris-buffered saline/Tween-20 (TBST) at 4 °C for overnight, followed by incubation with secondary antibodies at room temperature for 2 h. The protein signals were detected by enhanced chemiluminescence (ECL) method.
Immunofluorescence staining
Cells were fixed on poly-l-lysine-coated coverslips, permeabilized with 0.3% triton 100/ phosphate-buffered saline (PBS), blocked with 1% BSA (bovine serum albumin), and then incubated with a primary antibody for overnight at 4 °C. At the end of incubation, the cells were washed in PBS and incubated with a second antibody for 2 h at room temperature. For counterstaining of the nuclei, the cells were washed in PBS and incubated with DAPI (4’,6-diamidino-2-phenylindole, dihydrochloride) for 10 min at room temperature. Negative controls were established by omitting the incubation with primary antibodies.
Viral infection
GSC11 cells were dissociated, plated into 6-well plates, and then infected with an NFκB/p50-targeted shRNA lentiviral particles (Santa Cruz Biotechnology, Inc.) in the presence of 5 μg/mL of polybrene (Sigma-Aldrich) for 24 h. The cells were then selected with puromycin.
Neuro-sphere formation assay
GSC11 or GSC23 cells were cultured in the presence or absence of SN50 (25 µg/mL) for 24 h, and then dissociated, suspended in the drug-free culture medium, and seeded in 24-well tissue culture plates at a density of 5000 cells per well. Following 4 d incubation at 37 °C in a humidified incubator containing 5% CO2/95% air, the number of neuro-spheres was counted under a phase-contrast microscope (Olympus).
In vitro invasion assay
The invasiveness of GSC11 and GSC23 cells and their differentiated progeny cells was measured using CytoSelect™ 24-Well Cell Migration and Invasion Assay, 8 μm, Colorimetric Format (Cell Biolabs, Inc.). Briefly, GSC11 or GSC23 cells were trypsinized into single-cell suspensions, and 300 µL of the cell suspension (1 × 105 cells/mL) were added to each of the upper wells. The upper wells contained serum-free DMEM/F-12 medium and the lower wells contained DMEM/F-12 medium with 10% FBS. Neither of the media contained growth factors or supplements. After 48 h of incubation, noninvading cells in the upper chambers were gently wiped away, and the adherent cells on the lower side of each membrane were fixed and stained with 0.1% (w/v) crystal violet (Cell Biolabs, Inc.). Five fields were randomly selected and the stained cells were counted under a microscope.
Apoptosis assay
Cellular apoptosis were measured using flow cytometric analysis of cells double-stained with Annexin V and 7-aminoactinomycin D. Briefly, samples were collected, dissociated, and washed with cold phosphate buffered saline (PBS); then, 100 µL of Guava Nexin reagent was added to 1 × 105 cells in 100 µL, and the cells were incubated with the reagent for 20 min at room temperature in the dark. At the end of incubation, the cells were analyzed by Guava EasyCyteTM plus Flow Cytometry System (Millipore).
Cell viability assay
Cellular viability was determined using the CCK8 cell counting kit (Dojindo Molecular Technologies, Inc.). Optical density (OD) was measured at 450 nm on a microplate reader (Bio Rad).
RNA preparation and microarray analysis
GSCs were cultured in DMEM/ F-12 medium with or without SN50 for 12 h, and then RNA samples were prepared using the Trizol isolation reagent (Invitrogen). Using GeneChip® One-Cycle cDNA Synthesis Kit (Affymetrix, P/N 900431), 1 μg of total RNAs were subjected to reverse transcription to synthesize first-strand cDNA by use of a T7 Oligo (dT) promoter. Following RNase H-mediated second-strand cDNA synthesis, the double-stranded cDNAs were purified and served as a template in the subsequent in vitro transcription reaction, which synthesizes cRNA and incorporates a biotin-conjugated nucleotide. The cRNA was then purified using GeneChip® Sample Cleanup Module (Affymetrix), fragmented and then hybridized to the probe array (Affymetrix GeneChip Human Genome U133 Plus 2.0 Array) for 16 h at 45 °C in a GeneChip hybridization oven. Following washing and staining, the probe array was scanned using the GeneChip® Scanner 3000 7G.
In vivo tumorigenicity assay
GSC11 cells were first transduced with a luciferase retrovirus vector, and the cells expressing the luciferase were then treated with vehicle or SN50 for 24 h. GSC11 cells or their differentiated progeny cells were transplanted to 4–5 wk old female athymic (NCr-nu/nu mice (National Cancer Institute) either intra-cranially (5000 cells/mouse) or subcutaneously (1 × 106 cells per site), respectively. A non-invasive bioluminescence imaging was performed to monitor the tumor growth using the IVIS® animal imaging system with LivingImageTM software (Xenogen). In these experiments, the mice were first given D-Luciferin potassium salt (Caliper Life Sciences), the substrate for firefly luciferase, at a dose of 150 mg/kg body weight. During image acquisition, the mice were anesthetized with i.p. administration of ketamine (80 mg/kg) and xylazine (10 mg/kg). The animals were observed daily, and bioluminescence was measured on day 14, 21, 28, 35, and 42 post inoculation. The animal studies were approved by the Institutional Animal Care and Use Committee of the Penn State College of Medicine.
Statistical analysis
The Student t test was performed to analyze differences between treatments. Differences were considered significant when P < 0.05.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Supported by grants from the US Public Health Service CA135038, the National Natural Sciences Foundation of China (81101913), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
References
- Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, et al, European Organisation for Research and Treatment of Cancer Brain Tumour and Radiation Oncology Groups, National Cancer Institute of Canada Clinical Trials Group. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol 2009; 10:459 - 66; http://dx.doi.org/10.1016/S1470-2045(09)70025-7; PMID: 19269895
- Lee J, Son MJ, Woolard K, Donin NM, Li A, Cheng CH, Kotliarova S, Kotliarov Y, Walling J, Ahn S, et al. Epigenetic-mediated dysfunction of the bone morphogenetic protein pathway inhibits differentiation of glioblastoma-initiating cells. Cancer Cell 2008; 13:69 - 80; http://dx.doi.org/10.1016/j.ccr.2007.12.005; PMID: 18167341
- Grossman SA, Ye X, Piantadosi S, Desideri S, Nabors LB, Rosenfeld M, Fisher J, NABTT CNS Consortium. Survival of patients with newly diagnosed glioblastoma treated with radiation and temozolomide in research studies in the United States. Clin Cancer Res 2010; 16:2443 - 9; http://dx.doi.org/10.1158/1078-0432.CCR-09-3106; PMID: 20371685
- Visvader JE, Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell Stem Cell 2012; 10:717 - 28; http://dx.doi.org/10.1016/j.stem.2012.05.007; PMID: 22704512
- Akhtar K, Bussen W, Scott SP. Cancer stem cells - from initiation to elimination, how far have we reached? (Review). [Review] Int J Oncol 2009; 34:1491 - 503; PMID: 19424566
- Becker MW, Jordan CT. Leukemia stem cells in 2010: current understanding and future directions. Blood Rev 2011; 25:75 - 81; http://dx.doi.org/10.1016/j.blre.2010.11.001; PMID: 21216511
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006; 444:756 - 60; http://dx.doi.org/10.1038/nature05236; PMID: 17051156
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A 2003; 100:3983 - 8; http://dx.doi.org/10.1073/pnas.0530291100; PMID: 12629218
- Sanai N, Alvarez-Buylla A, Berger MS. Neural stem cells and the origin of gliomas. N Engl J Med 2005; 353:811 - 22; http://dx.doi.org/10.1056/NEJMra043666; PMID: 16120861
- Cheng L, Bao S, Rich JN. Potential therapeutic implications of cancer stem cells in glioblastoma. Biochem Pharmacol 2010; 80:654 - 65; http://dx.doi.org/10.1016/j.bcp.2010.04.035; PMID: 20457135
- Piccirillo SG, Reynolds BA, Zanetti N, Lamorte G, Binda E, Broggi G, Brem H, Olivi A, Dimeco F, Vescovi AL. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature 2006; 444:761 - 5; http://dx.doi.org/10.1038/nature05349; PMID: 17151667
- Chan XH, Nama S, Gopal F, Rizk P, Ramasamy S, Sundaram G, Ow GS, Ivshina AV, Tanavde V, Haybaeck J, et al. Targeting glioma stem cells by functional inhibition of a prosurvival oncomiR-138 in malignant gliomas. Cell Rep 2012; 2:591 - 602; http://dx.doi.org/10.1016/j.celrep.2012.07.012; PMID: 22921398
- Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, Fiocco R, Foroni C, Dimeco F, Vescovi A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res 2004; 64:7011 - 21; http://dx.doi.org/10.1158/0008-5472.CAN-04-1364; PMID: 15466194
- McCord AM, Jamal M, Shankavaram UT, Lang FF, Camphausen K, Tofilon PJ. Physiologic oxygen concentration enhances the stem-like properties of CD133+ human glioblastoma cells in vitro. Mol Cancer Res 2009; 7:489 - 97; http://dx.doi.org/10.1158/1541-7786.MCR-08-0360; PMID: 19372578
- Garner JM, Fan M, Yang CH, Du Z, Sims M, Davidoff AM, Pfeffer LM. Constitutive activation of signal transducer and activator of transcription 3 (STAT3) and nuclear factor κB signaling in glioblastoma cancer stem cells regulates the Notch pathway. J Biol Chem 2013; 288:26167 - 76; http://dx.doi.org/10.1074/jbc.M113.477950; PMID: 23902772
- Frank NY, Schatton T, Frank MH. The therapeutic promise of the cancer stem cell concept. J Clin Invest 2010; 120:41 - 50; http://dx.doi.org/10.1172/JCI41004; PMID: 20051635
- Fotovati A, Abu-Ali S, Wang PS, Deleyrolle LP, Lee C, Triscott J, Chen JY, Franciosi S, Nakamura Y, Sugita Y, et al. YB-1 bridges neural stem cells and brain tumor-initiating cells via its roles in differentiation and cell growth. Cancer Res 2011; 71:5569 - 78; http://dx.doi.org/10.1158/0008-5472.CAN-10-2805; PMID: 21730024
- Zhuang W, Li B, Long L, Chen L, Huang Q, Liang Z. Induction of autophagy promotes differentiation of glioma-initiating cells and their radiosensitivity. Int J Cancer 2011; 129:2720 - 31; http://dx.doi.org/10.1002/ijc.25975; PMID: 21384342
- Sunayama J, Sato A, Matsuda K, Tachibana K, Suzuki K, Narita Y, Shibui S, Sakurada K, Kayama T, Tomiyama A, et al. Dual blocking of mTor and PI3K elicits a prodifferentiation effect on glioblastoma stem-like cells. Neuro Oncol 2010; 12:1205 - 19; PMID: 20861085
- Naugler WE, Karin M. NF-kappaB and cancer-identifying targets and mechanisms. Curr Opin Genet Dev 2008; 18:19 - 26; http://dx.doi.org/10.1016/j.gde.2008.01.020; PMID: 18440219
- Hoesel B, Schmid JA. The complexity of NF-κB signaling in inflammation and cancer. Mol Cancer 2013; 12:86; http://dx.doi.org/10.1186/1476-4598-12-86; PMID: 23915189
- Nogueira L, Ruiz-Ontañon P, Vazquez-Barquero A, Moris F, Fernandez-Luna JL. The NFκB pathway: a therapeutic target in glioblastoma. Oncotarget 2011; 2:646 - 53; PMID: 21896960
- Lin YZ, Yao SY, Veach RA, Torgerson TR, Hawiger J. Inhibition of nuclear translocation of transcription factor NF-kappa B by a synthetic peptide containing a cell membrane-permeable motif and nuclear localization sequence. J Biol Chem 1995; 270:14255 - 8; http://dx.doi.org/10.1074/jbc.270.24.14255; PMID: 7782278
- Abate A, Oberle S, Schröder H. Lipopolysaccharide-induced expression of cyclooxygenase-2 in mouse macrophages is inhibited by chloromethylketones and a direct inhibitor of NF-kappa B translocation. Prostaglandins Other Lipid Mediat 1998; 56:277 - 90; http://dx.doi.org/10.1016/S0090-6980(98)00061-6; PMID: 9990673