
Abstract
The present studies were to determine whether the multi-kinase inhibitor pazopanib interacted with histone deacetylase inhibitors (HDACI: valproate, vorinostat) to kill sarcoma cells. In multiple sarcoma cell lines, at clinically achievable doses, pazopanib and HDACI interacted in an additive to greater than additive fashion to cause tumor cell death. The drug combination increased the numbers of LC3-GFP and LC3-RFP vesicles. Knockdown of Beclin1 or ATG5 significantly suppressed drug combination lethality. Expression of c-FLIP-s, and to a lesser extent BCL-XL or dominant negative caspase 9 reduced drug combination toxicity; knock down of FADD or CD95 was protective. Expression of both activated AKT and activated MEK1 was required to strongly suppress drug combination lethality. The drug combination inactivated mTOR and expression of activated mTOR strongly suppressed drug combination lethality. Treatment of animals carrying sarcoma tumors with pazopanib and valproate resulted in a greater than additive reduction in tumor volume compared with either drug individually. As both pazopanib and HDACIs are FDA-approved agents, our data argue for further determination as to whether this drug combination is a useful sarcoma therapy in the clinic.
Introduction
Pazopanib (Votrient) is an inhibitor of multiple class III receptor tyrosine kinases including VEGFR1–3; PDGFRα/β, and c-Kit.Citation1,Citation2 These targets permit pazopanib to both slow tumor growth and to inhibit tumor angiogenesis.Citation3 In this respect, pazopanib has similar properties to the multi-kinase inhibitor sorafenib, except that pazopanib does not inhibit the RAF-1 ser/thr kinase.Citation4 Our prior in vitro and in vivo data have tended to argue using several (sorafenib + “drug”) combinations that PDGFRβ is a major target of sorafenib for its interactions with other agents e.g., with histone deacetylase (HDAC) inhibitors.Citation5 PDGFRβ inhibition has been linked to pazopanib biology.Citation6 In a recent study we also observed that the in vitro IC50 for sorafenib inhibition of P-VEGFR2 was ~2 μM.Citation7
Pazopanib is approved for the treatment of sarcoma. Sarcomas are a diverse group of tumors of mesenchymal origin and in the United States approximately 15 000 cases are diagnosed each year. Targeted therapy of oncogenic kinases within sarcomas was initially made in gastrointestinal stromal tumor (GIST) where the drug imatinib has activity against the c-Kit and PDGFRα driving mutations in this malignancy.Citation8 Subsequently other kinase inhibitor drugs including sunitinib and regorafenib have been used in this tumor type. Pazopanib is approved for use in soft tissue sarcomas as well as renal cell carcinoma, though the drug will probably have a wide range of effects on many tumors due to its actions on multiple receptor tyrosine kinases.Citation8,Citation9
A major biological effect of the class III RTK inhibitor sorafenib is the dose-dependent induction of an endoplasmic stress (ER)/unfolded protein response (UPR), with reduced expression of proteins that have short half-lives such as MCL-1 and BCL-XL.Citation5,Citation7 Reduced MCL-1 levels have been linked in many tumor types to increased levels of apoptosis. Studies by our group have also linked high dose single agent sorafenib exposure to an increase in the levels of autophagic markers including increased numbers of LC3-GFP vesicles.Citation5,Citation10 Much less is known about the biology of pazopanib, though high concentrations of pazopanib have also been shown to induce autophagy in bladder cancer cells, where the induction of autophagy was a toxic phenomenon.Citation11
Sodium valproate is an anti-convulsant used in the treatment of epilepsy, migraine, bipolar disorder, and prevention of seizures.Citation12 Side effects of the drug can include somnolence, tremors, and nausea. Valproate is a histone deacetylase (HDAC) inhibitor of class I, but weakly of class II, HDACs.Citation13 HDAC inhibitors modify the acetylation of proteins thereby altering the function of histones as well as multiple non-nuclear proteins, altering protein structure and function, and gene transcription. Genes whose expression is increased by HDAC inhibitors include the death receptor and ligand CD95/FAS-L and toxic BH3 domain proteins.Citation14,Citation15 HDAC inhibitors also increase ROS levels in cells and promote autophagy.Citation16 All of the above shifts in cell biology tend to predispose tumor cells toward cell death in response to HDAC inhibitors. HDAC inhibitors have also been claimed to reprogram tumor cells into a more stem-like biological state.Citation17
The present studies determined whether pazopanib interacted with HDAC inhibitors to kill a range of sarcoma cell lines in vitro and in vivo. Our data demonstrate an interaction between these drugs in multiple sarcoma cell types with killing that is due to both death receptor activation and a toxic form of autophagy.
Results
Initial studies examined the response of sarcoma cells to pazopanib when combined with the FDA approved HDAC inhibitor sodium valproate. Cell lines chosen for these studies were: MES (uterine sarcoma), HT1080 (fibrosarcoma), Saos-2 (osteosarcoma), and SKES1 (Ewing osteosarcoma). In 3 of the 4 cell lines pazopanib and valproate interacted in an approximately additive manner to kill sarcoma cells (). In the fibrosarcoma HT1080 line pazopanib and valproate interacted in a greater than additive manner to kill (). Very similar cell killing data to that using sodium valproate were obtained using another FDA-approved chemically different HDAC inhibitor, vorinostat ().
Figure 1. Pazopanib and sodium valproate interact to kill multiple sarcoma cell lines. (A) Sarcoma cells were treated with vehicle (VEH, DMSO), pazopanib (PAZ, 5.0 μM) and/or sodium valproate (VAL, 500 μM) as indicated. Cells were isolated 24 h and 48 h after exposure and viability at each time point determined by trypan blue exclusion (n = 3, ± SEM) *P < greater than vehicle control. (B) HT1080 cells were treated with vehicle (VEH, DMSO), pazopanib (PAZ, 2.0 μM, 4.0 μM, 6 μM) and/or sodium valproate (VAL, 250 μM, 500 μM, 750 μM) as indicated. Cells were isolated 24 h after exposure and viability at each time point determined by trypan blue exclusion (n = 3, ± SEM) *P < greater than vehicle control.
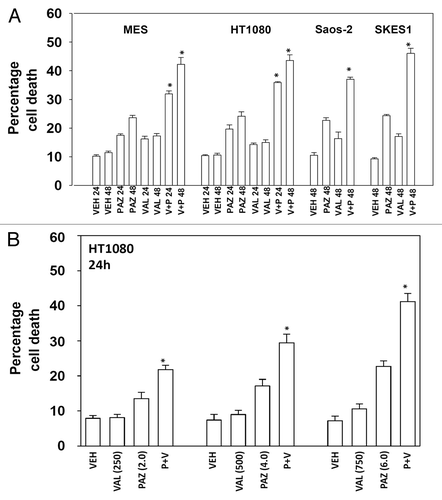
Figure 2. Pazopanib and vorinostat interact to kill multiple sarcoma cell lines. Sarcoma cells were treated with vehicle (VEH, DMSO), pazopanib (PAZ, 5.0 μM) and/or vorinostat (500 nM) as indicated. Cells were isolated 48h after exposure and viability determined by trypan blue exclusion (n = 3, ± SEM) *P < greater than vehicle control.
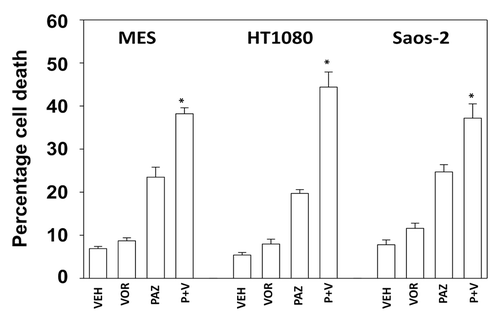
Pazopanib is an inhibitor of multiple class III receptor tyrosine kinases, including the PDGFRβ, and we next determined whether inhibition of PDGFRβ altered the viability of HT1080 cells exposed to sodium valproate. In a dose-dependent fashion increasing concentrations of valproate caused a modest elevation in tumor cell death, an effect that was significantly enhanced in cells in which PDGFRβ had been knocked down (). As knock down of PDGFRβ was not complete, our data tend to argue that this receptor represents a major target of pazopanib in HT1080 cells.
Figure 3. Knockdown of PDGFRβ enhances valproate toxicity in HT1080 cells. HT1080 cells were transfected with either a scrambled siRNA (siSCR) or an siRNA to knock down expression of PDGFRβ (siPDGFRβ). Thirty-six hours after transfection cells were treated with increasing concentrations of sodium valproate (VAL, 250 μM, 500 μM, 750 μM) as indicated. Cells were isolated 24 h after exposure and viability at each time point determined by trypan blue exclusion (n = 3, ± SEM) *P < greater than vehicle control.
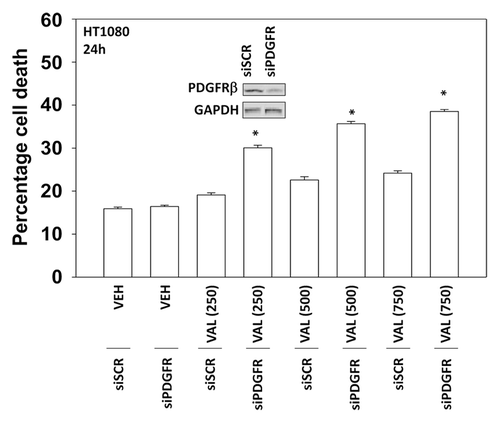
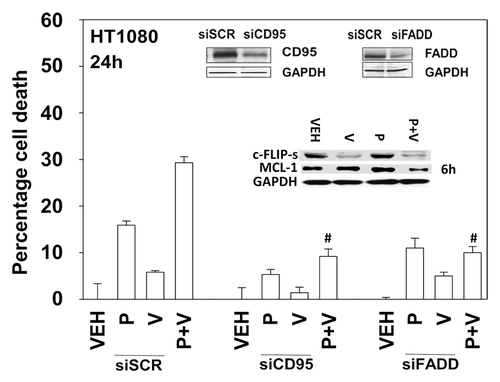
We next defined the molecular mechanisms by which pazopanib and sodium valproate killed HT1080 cells. Expression of the mitochondrial protective protein BCL-XL and to a greater extent the caspase 8 inhibitor c-FLIP-s protected cells from pazopanib + valproate toxicity (). As expression of the caspase 8 inhibitor c-FLIP-s was protective, we determined whether knock down of CD95 death receptor expression was protective. Knockdown of CD95 expression or that of the CD95/caspase 8 docking protein FADD reduced pazopanib + valproate toxicity (). Of note, treatment of HT1080 cells with valproate reduced expression of c-FLIP-s, and combined exposure of cells to valproate and pazopanib reduced expression of the mitochondrial protective protein MCL-1 (, inset panel). These alterations in protein expression would predispose HT1080 cells to killing through death receptor signaling and mitochondrial dysfunction.
Figure 4. Inhibition of caspase 8 protects cells from pazopanib and valproate treatment. HT1080 cells were infected (50 min.o.i.) with empty vector control virus (CMV) or with viruses to express dominant negative caspase 9, BCL-XL or the caspase 8 inhibitor c-FLIP-s. Twenty-four hours after infection cells were treated with vehicle (DMSO), pazopanib (P, 5.0 μM), and valproate (V, 500 μM). Cells were isolated 24 h after exposure and viability determined by trypan blue exclusion (n = 3, ± SEM) #P < less than corresponding value in CMV cells; ##P < less than corresponding value in BCL-XL cells.
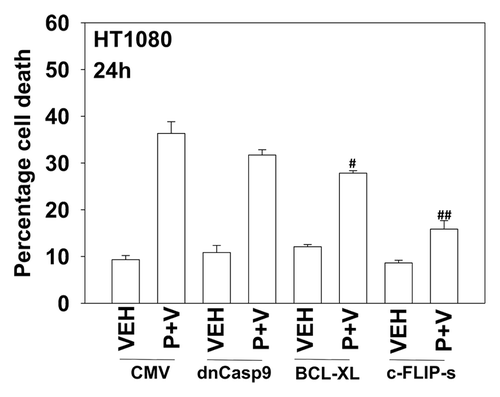
Figure 5. Knockdown of CD95 or FADD protects cells from pazopanib and valproate treatment. HT1080 cells were transfected with scrambled control siRNA (siSCR) or siRNA molecules to knockdown expression of CD95 (siCD95) or FADD (siFADD). Thirty-six hours after transfection cells were treated with vehicle (DMSO), pazopanib (P, 5.0 μM), and/or valproate (V, 500 μM). Cells were isolated 24 h after exposure and viability determined by trypan blue exclusion (n = 3, ± SEM). #P < 0.05 less than corresponding value in siSCR cells.
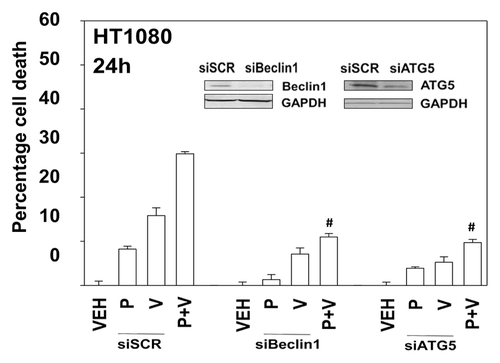
In prior studies using the multi-kinase inhibitor sorafenib we have shown that knock down of PDGFRβ can stimulate the number of autophagosome vesicles in cells.Citation5 As pazopanib is an inhibitor of PDGFRβ we next determined whether pazopanib induced autophagy, and if so, whether autophagy played a role in the survival/killing of drug treated cells. Treatment of HT1080 cells with pazopanib + valproate increased the numbers of LC3-GFP+ vesicles, indicative of early autophagosome formation as well as the numbers of LC3-RFP+ vesicles, indicative of increased levels of mature autolysosomes (). Chloroquine treatment, which inhibits autophagosome fusion with lysosomes decreased the numbers of LC3-RFP+ vesicles (data not shown). Collectively, this data argues that autophagy flux was being stimulated. We then defined the role of autophagy in the drug combination response. Knockdown of either Beclin1 or ATG5 expression suppressed autophagy (LC3-GFP+ vesicle numbers) and protected HT1080 cells from pazopanib + valproate exposure ( and data not shown).
Figure 6. The regulation of autophagy by pazopanib and valproate. HT1080 cells were transfected with a plasmid to express LC3-GFP-RFP. Twenty-four hours after transfection cells were treated with vehicle (DMSO), pazopanib (PAZ, 5.0 μM), and/or valproate (VAL, 500 μM). Cells were microscopically examined 6 h and 12 h after exposure and the number of GFP and RFP punctae determined in at least 40 random cells per condition (n = 3, ± SEM). *P < 0.05 greater than value at 6 h.
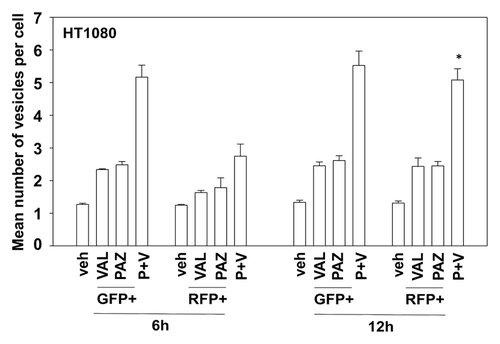
Figure 7. Knockdown of Beclin1 or ATG5 protects cells from pazopanib and valproate toxicity. HT1080 cells were transfected with scrambled control siRNA (siSCR) or siRNA molecules to knock down expression of Beclin1 (siB1) or ATG5 (siA5). Thirty-six hours after transfection cells were treated with vehicle (DMSO), pazopanib (P, 5.0 μM), and/or valproate (V, 500 μM). Cells were isolated 24 h after exposure and viability determined by trypan blue exclusion (n = 3, ± SEM). #P < less than corresponding value in siSCR cells.
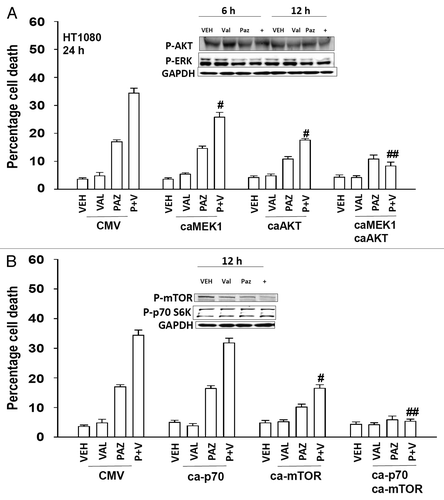
We next defined the roles of cytoprotective signaling pathways in the response to pazopanib + valproate treatment. We introduced activated forms of AKT and MEK1 into cells and determined the response to drug combination exposure. Activation of either the AKT or the MEK1 pathway resulted in a modest protection from drug combination toxicity (). Combined activation of the AKT and MEK1 pathways resulted in significantly enhanced cytoprotection. We introduced activated forms of p70 S6K and mTOR into cells and determined the response to pazopanib + valproate treatment. Activation of the mTOR pathway, but not the p70 S6K pathway, resulted in protection from drug combination toxicity (). Combined activation of the both pathways again resulted in enhanced cytoprotection.
Figure 8. Expression of activated AKT and MEK1 suppresses the toxicity of pazopanib and valproate. (A) HT1080 cells were infected (50 min.o.i.) with empty vector control virus (CMV) or with viruses to express activated MEK1 (caMEK1), activated AKT (caAKT), or caMEK1 and caAKT together. Twenty-four hours after infection cells were treated with vehicle (DMSO), pazopanib (PAZ, 5.0 μM), and valproate (VAL, 500 μM). Cells were isolated 24 h after exposure and viability determined by trypan blue exclusion (n = 3, ± SEM) #P < less than corresponding value in CMV infected cells; ##P < less than corresponding value in caAKT infected cells. (B) HT1080 cells were transfected with empty vector plasmid (CMV), with a plasmid to express activated p70 S6K (ca-p70), with a plasmid to express activated mTOR (ca-mTOR) or with both plasmids together. Twenty-four hours after infection cells were treated with vehicle (DMSO), pazopanib (PAZ, 5.0 μM), and valproate (VAL, 500 μM). Cells were isolated 24 h after exposure and viability determined by trypan blue exclusion (n = 3, ± SEM) #P < less than corresponding value in CMV transfected cells; ##P < less than corresponding value in ca-mTOR transfected cells.
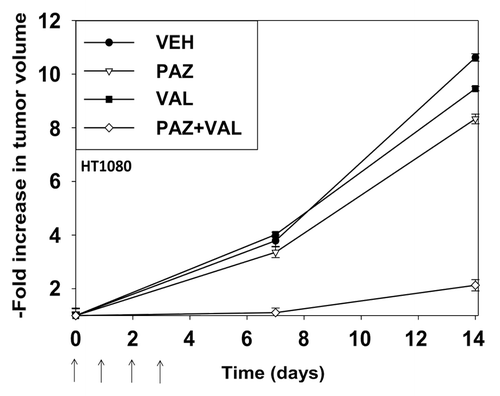
Pazopanib and valproate are both FDA-approved drugs. Based on our in vitro data we performed animal studies using pre-formed HT1080 tumors (~150 mm3) in athymic mice. In animals treated with vehicle control tumors increased their mass >10-fold within 14 d (). Treatment of animals with pazopanib or valproate resulted in modest decreases in tumor growth. Treatment of animals with pazopanib + valproate caused a significant decrease in growth beyond those observed with the individual drugs. This argues that our in vitro findings in HT1080 cells translate into an animal model system.
Figure 9. Pazopanib and valproate interact to suppress the growth of established HT1080 tumors. Mice were injected in the rear flank with 1.0 × 107 HT1080 cells in 100 μl of growth medium. Fourteen days after tumor cell implantation when tumors had grown to ~50 mm3 (defined as 1.00 in the graph) mice were mice were PO administered vehicle diluent; pazopanib (25 mg/kg, QD); valproate (50 mg/kg BID), or the drugs in combination for 4 d. Animals were monitored daily, tumor volumes taken every 7 d. (Values plotted are the fold change in mean tumor volume at each time point; ± SEM, 8 mice per treatment condition).
Discussion
The present studies were initiated to determine whether the multi-kinase inhibitor pazopanib interacted with HDAC inhibitors to kill sarcoma cells. Our data demonstrate that pazopanib and valproate/vorinostat interact in an additive to a greater than additive fashion to kill sarcoma cells in vitro. A recent study by Chan et al. in thyroid cancer cells using other HDAC inhibitors in combination with tyrosine kinase inhibitors, including pazopanib, supports our findings.Citation18
Prior studies by our group have shown that the multi-kinase inhibitor sorafenib increases the levels of LC3-GFP vesicles in tumor cells, suggestive of autophagy. The multi-kinase inhibitor pazopanib increased the levels of LC3-GFP+ vesicles and in a chloroquine-dependent and time-dependent manner to a lesser extent LC3-RFP+ vesicles, indicative that autophagic flux was occurring. Knock down of either Beclin1 or ATG5 blocked drug-induced LC3-GFP+/LC3-RFP+ vesicle formation and protected cells from the toxic effects of pazopanib + valproate treatment. Autophagy data using pazopanib similar to our own was recently reported in bladder cancer cells.Citation11 In addition to autophagy we also determined whether the intrinsic or extrinsic apoptosis pathways played a role in cell killing caused by the pazopanib/HDAC inhibitor drug combination. Overexpression of the mitochondrial protective protein BCL-XL or the caspase 8 inhibitor c-FLIP-s significantly reduce drug combination lethality. As caspase 8 appeared to be playing a role in cell death we determined whether death receptor signaling was involved. Knockdown of the death receptor CD95 or the death receptor/caspase 8 docking protein FADD also significantly reduced cell killing caused by the drug combination. Finally, we determined whether our in vitro findings translated into an animal model system. At the doses of drug used and for the treatment time, neither pazopanib nor valproate caused a large reduction in tumor mass. However, treatment of the animals with the drug combination resulted in a significantly reduced rate of tumor growth. Collectively our findings argue that pazopanib when combined with HDAC inhibitors may be a novel approach in the treatment of soft tissue sarcoma.
In our in vitro tumor cell studies, using clinically achievable doses of drugs, we noted relatively modest changes in the activities of signal transduction pathways that are normally associated with regulating cell survival. For example, after drug exposure the activity of the AKT pathway did not appreciably alter and the activity of ERK1/2 only modestly declined. However, expression of an activated form of AKT, or of MEK1 resulted in protection of cells from the drug combination with expression of both activated kinases almost abolishing cell killing. In contrast to our data for AKT and ERK1/2, the phosphorylation/activity of mTOR was almost abolished by drug combination treatment, which correlated with enhanced toxic autophagy. Of note, expression of activated mTOR abolished the induction of autophagy (unpublished data). Hence, we also performed studies in which activated forms of mTOR and p70 S6K were expressed and HT1080 cells treated with drugs; expression of activated mTOR significantly reduced drug combination lethality whereas that of p70 S6K had little to no effect.
We discovered that treatment of animals carrying pre-formed tumor for several days with pazopanib and valproate demonstrated that the drugs interacted to suppress the growth of established HT1080 tumors when compared with treatment with either drug individually. The body mass and behavior of the animals did not appreciably alter during the experiment (unpublished observations). This data, combined with our in vitro findings, argues that pazopanib and valproate combination therapy could have utility in soft tissue sarcoma treatment.
Pazopanib is presently being tested/investigated in a wide range of malignancies where its putative anti-cancer role as an anti-angiogenic agent is thought to act.Citation1-Citation4 Fewer studies have explored whether pazopanib can act as a direct anti-tumor cell agent. Our in vitro data using sarcoma tumor cell lines would however argue that pazopanib can directly act upon growth factor receptors in sarcoma cells, thereby enhancing their responsiveness to the pazopanib and valproate drug combination. Further studies outside the scope of the present manuscript will be required to determine in additional detail pazopanib and valproate interact to kill tumor cells.
Materials and Methods
Phospho-/total antibodies were purchased from Cell Signaling Technologies and Santa Cruz Biotech. All drugs were purchased from Selleckchem. Commercially available validated short hairpin RNA molecules to knockdown RNA/protein levels were from Qiagen. Antibody reagents, other kinase inhibitors, caspase inhibitors cell culture reagents, and non-commercial recombinant adenoviruses have been previously described.Citation5,Citation7,Citation10,Citation19
Cell culture and in vitro exposure of cells to drugs
All established cancer lines were cultured at 37 °C (5% [v/v] CO2) in vitro using RPMI supplemented with 10% (v/v) fetal calf serum and 10% (v/v) non-essential amino acids. For short-term cell killing assays and immunoblotting, cells were plated at a density of 3 × 103 per cm2 and 24 h after plating were treated with various drugs, as indicated. In vitro small molecule inhibitor treatments were from a 100 mM stock solution of each drug and the maximal concentration of vehicle (DMSO) in media was 0.02% (v/v). Cells were not cultured in reduced serum media during any study.
Cell treatments, SDS-PAGE, and western blot analysis
Cells were treated with various drug concentrations, as indicated in the figure legends. SDS PAGE and immunoblotting was performed as described in references Citation5, Citation7, Citation10, and Citation19.
Recombinant adenoviral vectors; infection in vitro
We generated and purchased previously noted recombinant adenoviruses as per refs. Cells were infected with these adenoviruses at an approximate m.o.i. as indicated in the figure legend (usually 50 min.o.i.). Cells were incubated for 24 h to ensure adequate expression of transduced gene products prior to drug exposures.
Detection of cell death by Trypan Blue, Hoechst, TUNEL, and flow cytometric assays
Cells were harvested by trypsinization with Trypsin/EDTA for ~10 min at 37 °C. Cell death assays were performed as described in references Citation5, Citation7, Citation10, and Citation19.
Assessment of autophagy
Cells were transfected with a plasmid to express a green fluorescent protein (GFP) and red fluorescent (RFP) doubly tagged form of LC3 (ATG8). For analysis of cells transfected with the RFP-GFP-LC3 construct, the RFP-GFP-LC3-positive vesicularized cells were examined under the 40× objective of a Zeiss Axiovert fluorescent microscope.
Plasmid transfection
Plasmids
Cells were plated as described above and 24 h after plating, transfected. Plasmids (0.5 μg) expressing a specific mRNA or appropriate vector control plasmid DNA was diluted in 50 μl serum-free and antibiotic-free medium (1 portion for each sample). Concurrently, 2 μl Lipofectamine 2000 (Invitrogen), was diluted into 50 μl of serum-free and antibiotic-free medium. Diluted DNA was added to the diluted Lipofectamine 2000 for each sample and incubated at room temperature for 30 min. This mixture was added to each well/dish of cells containing 200 μl serum-free and antibiotic-free medium for a total volume of 300 μl and the cells were incubated for 4 h at 37 °C. An equal volume of 2× medium was then added to each well. Cells were incubated for 48 h, then treated with drugs. To assess transfection efficiency of plasmids we used a plasmid to express GFP and defined the percentage of cells being infected as the percentage of GFP+ cells. For all cell lines the infection efficiency was >70%.
siRNA
Cells were plated in 60 mm dishes from a fresh culture growing in log phase as described above, and 24 h after plating transfected. Prior to transfection, the medium was aspirated and 1 ml serum-free medium was added to each plate. For transfection, 10 nM of the annealed siRNA, the positive sense control doubled stranded siRNA targeting GAPDH or the negative control (a “scrambled” sequence with no significant homology to any known gene sequences from mouse, rat or human cell lines) were used (predominantly Qiagen; occasional alternate siRNA molecules were purchased from Ambion, Inc.). Ten nanomolar siRNA (scrambled or experimental) was diluted in serum-free media. Four microliters Hiperfect (Qiagen) was added to this mixture and the solution was mixed by pipetting up and down several times. This solution was incubated at room temperature for 10 min, then added drop-wise to each dish. The medium in each dish was swirled gently to mix, then incubated at 37 °C for 2 h. One milliliter of 10% (v/v) serum-containing medium was added to each plate, and cells were incubated at 37 °C for 24–48 h before re-plating (50 × 103 cells each) onto 12-well plates. Cells were allowed to attach overnight, then treated with drugs (0–48 h). Trypan blue exclusion assays and SDS PAGE/immunoblotting analyses were then performed at the indicated time points.
Animal studies
Athymic female NCr-ν/ν mice (NCI-Fredrick) weighing ~20 g, were used for this study. Mice were maintained under pathogen-free conditions in facilities approved by the American Association for Accreditation of Laboratory Animal Care and in accordance with current regulations and standards of the US Department of Agriculture, the US Department of Health and Human Services, and the National Institutes of Health. Mice were injected with 1.0 × 107 HT1080 cells in 100 μl of medium. Fourteen days after tumor cell implantation when tumors had grown to ~150 mm3 mice were mice were PO administered vehicle diluent, pazopanib (25 mg/kg, QD), valproate (50 mg/kg BID), or the drugs in combination for 4 d. Animals were monitored daily, tumor volumes taken every 7 d, and moribund animals were killed as required based on IACUC rules.
Data analysis
Comparison of the effects of various treatments was performed using one way analysis of variance and a two tailed Student t test. Differences with a P value of <0.05 were considered statistically significant. Statistical examination of in vivo animal survival data utilized log rank statistical analyses between the different treatment groups. Experiments shown are the means of multiple individual points from multiple experiments (± SEM).
| Abbreviations: | ||
| ERK | = | extracellular regulated kinase |
| MEK | = | mitogen activated extracellular regulated kinase |
| PI3K | = | phosphatidyl inositol 3 kinase |
| MAPK | = | mitogen activated protein kinase |
| ca | = | constitutively active |
| dn | = | dominant negative |
| CMV | = | empty vector plasmid or virus |
| si | = | small interfering |
| SCR | = | scrambled |
| Ad | = | adenovirus |
| VEH | = | vehicle |
| P/PAZ | = | pazopanib |
| V/VAL | = | valproate |
| FADD | = | Fas-associated death domain |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Support for the present study was funded from PHS grants from the National Institutes of Health (R01-CA141704, R01-CA150214, R01-DK52825); the Department of Defense (W81XWH-10-1-0009). P.D. is the holder of the Universal Inc. Professorship in Signal Transduction Research. The authors have no conflicts of interest to report.
References
- Wilky BA, Meyer CF, Trent JC. Pazopanib in sarcomas: expanding the PALETTE. Curr Opin Oncol 2013; 25:373 - 8; http://dx.doi.org/10.1097/CCO.0b013e3283622d3a; PMID: 23666473
- Rajendra R, Jones RL, Pollack SM. Targeted treatment for advanced soft tissue sarcoma: profile of pazopanib. Onco Targets Ther 2013; 6:217 - 22; PMID: 23524973
- Petrillo M, Scambia G, Ferrandina G. Novel targets for VEGF-independent anti-angiogenic drugs. Expert Opin Investig Drugs 2012; 21:451 - 72; http://dx.doi.org/10.1517/13543784.2012.661715; PMID: 22339615
- Gotink KJ, Verheul HM. Anti-angiogenic tyrosine kinase inhibitors: what is their mechanism of action?. Angiogenesis 2010; 13:1 - 14; http://dx.doi.org/10.1007/s10456-009-9160-6; PMID: 20012482
- Park MA, Reinehr R, Häussinger D, Voelkel-Johnson C, Ogretmen B, Yacoub A, Grant S, Dent P. Sorafenib activates CD95 and promotes autophagy and cell death via Src family kinases in gastrointestinal tumor cells. Mol Cancer Ther 2010; 9:2220 - 31; http://dx.doi.org/10.1158/1535-7163.MCT-10-0274; PMID: 20682655
- Gril B, Palmieri D, Qian Y, Anwar T, Liewehr DJ, Steinberg SM, Andreu Z, Masana D, Fernández P, Steeg PS, et al. Pazopanib inhibits the activation of PDGFRβ-expressing astrocytes in the brain metastatic microenvironment of breast cancer cells. Am J Pathol 2013; 182:2368 - 79; http://dx.doi.org/10.1016/j.ajpath.2013.02.043; PMID: 23583652
- Sajithlal GB, Hamed HA, Cruickshanks N, Booth L, Tavallai S, Syed J, Grant S, Poklepovic A, Dent P. Sorafenib/regorafenib and phosphatidyl inositol 3 kinase/thymoma viral proto-oncogene inhibition interact to kill tumor cells. Mol Pharmacol 2013; 84:562 - 71; http://dx.doi.org/10.1124/mol.113.088005; PMID: 23877009
- Ranieri G, Mammì M, Donato Di Paola E, Russo E, Gallelli L, Citraro R, Gadaleta CD, Marech I, Ammendola M, De Sarro G. Pazopanib a tyrosine kinase inhibitor with strong anti-angiogenetic activity: A new treatment for metastatic soft tissue sarcoma. Crit Rev Oncol Hematol 2013; In press PMID: 24041629
- Iacovelli R, Verzoni E, De Braud F, Procopio G. First line treatment of metastatic renal cell carcinoma: Two standards with different toxicity profile. Cancer Biol Ther 2014; 15:19 - 21; http://dx.doi.org/10.4161/cbt.27150; PMID: 24253418
- Bareford MD, Park MA, Yacoub A, Hamed HA, Tang Y, Cruickshanks N, Eulitt P, Hubbard N, Tye G, Burow ME, et al. Sorafenib enhances pemetrexed cytotoxicity through an autophagy-dependent mechanism in cancer cells. Cancer Res 2011; 71:4955 - 67; http://dx.doi.org/10.1158/0008-5472.CAN-11-0898; PMID: 21622715
- Santoni M, Amantini C, Morelli MB, Liberati S, Farfariello V, Nabissi M, Bonfili L, Eleuteri AM, Mozzicafreddo M, Burattini L, et al. Pazopanib and sunitinib trigger autophagic and non-autophagic death of bladder tumour cells. Br J Cancer 2013; 109:1040 - 50; http://dx.doi.org/10.1038/bjc.2013.420; PMID: 23887605
- Michaelis M, Doerr HW, Cinatl J Jr.. Valproic acid as anti-cancer drug. Curr Pharm Des 2007; 13:3378 - 93; http://dx.doi.org/10.2174/138161207782360528; PMID: 18045192
- Atadja PW. HDAC inhibitors and cancer therapy. Prog Drug Res 2011; 67:175 - 95; PMID: 21141730
- Barbone D, Cheung P, Battula S, Busacca S, Gray SG, Longley DB, Bueno R, Sugarbaker DJ, Fennell DA, Broaddus VC. Vorinostat eliminates multicellular resistance of mesothelioma 3D spheroids via restoration of Noxa expression. PLoS One 2012; 7:e52753; http://dx.doi.org/10.1371/journal.pone.0052753; PMID: 23300762
- Premkumar DR, Jane EP, Agostino NR, DiDomenico JD, Pollack IF. Bortezomib-induced sensitization of malignant human glioma cells to vorinostat-induced apoptosis depends on reactive oxygen species production, mitochondrial dysfunction, Noxa upregulation, Mcl-1 cleavage, and DNA damage. Mol Carcinog 2013; 52:118 - 33; http://dx.doi.org/10.1002/mc.21835; PMID: 22086447
- Shubassi G, Robert T, Vanoli F, Minucci S, Foiani M. Acetylation: a novel link between double-strand break repair and autophagy. Cancer Res 2012; 72:1332 - 5; http://dx.doi.org/10.1158/0008-5472.CAN-11-3172; PMID: 22422989
- Jeong SG, Ohn T, Kim SH, Cho GW. Valproic acid promotes neuronal differentiation by induction of neuroprogenitors in human bone-marrow mesenchymal stromal cells. Neurosci Lett 2013; 554:22 - 7; http://dx.doi.org/10.1016/j.neulet.2013.08.059; PMID: 24021810
- Chan D, Zheng Y, Tyner JW, Chng WJ, Chien WW, Gery S, Leong G, Braunstein GD, Koeffler HP. Belinostat and panobinostat (HDACI): in vitro and in vivo studies in thyroid cancer. J Cancer Res Clin Oncol 2013; 139:1507 - 14; http://dx.doi.org/10.1007/s00432-013-1465-6; PMID: 23824064
- Cruickshanks N, Hamed HA, Booth L, Tavallai S, Syed J, Sajithlal GB, Grant S, Poklepovic A, Dent P. Histone deacetylase inhibitors restore toxic BH3 domain protein expression in anoikis-resistant mammary and brain cancer stem cells, thereby enhancing the response to anti-ERBB1/ERBB2 therapy. Cancer Biol Ther 2013; 14:982 - 96; http://dx.doi.org/10.4161/cbt.26234; PMID: 24025251