Abstract
Glycogen synthase kinase-3 (GSK-3) plays a central role in cell survival and proliferation, in part by the regulation of transcription. Unlike most protein kinases, GSK-3 is active in quiescent cells in the absence of growth factor signaling. In a recent series of studies, we employed a systems-level approach to understanding the transcription network regulated by GSK-3 in a quiescent cell model. We identified a group of immediate early genes that were upregulated in quiescent cells solely by the inhibition of GSK-3 in the absence of growth factor stimulation. Computational analysis of the upstream sequences of these genes identified statistically over-represented binding sites for the transcription factors CREB, NFκB and AP-1, and the roles of these factors in regulating expression of GSK-3 target genes were verified by chromatin immunoprecipitation and RNA interference. In quiescent cells, GSK-3 inhibits CREB, NFκB and AP-1, thereby maintaining repression of their target genes and contributing to maintenance of cell cycle arrest.
Introduction
Glycogen synthase kinase-3 (GSK-3) was first identified as a metabolic regulatory enzyme that phosphorylated and inactivated glycogen synthase. However, it is now recognized that GSK-3 plays diverse roles in cell regulation and functions as a key element in signaling pathways that control cell proliferation, survival and differentiation.Citation1–Citation3 Consistent with its critical roles in normal cells, abnormalities of GSK-3 regulation have been implicated in cancer, diabetes, heart disease, Alzheimer disease and psychiatric disorders.
GSK-3 is a central element in both the Wnt and PI 3-kinase signaling pathways. In both cases, unlike most protein kinases, GSK-3 is active in the absence of growth factor stimulation. In the Wnt pathway, GSK-3 is associated with Axin and APC in a complex that phosphorylates β-catenin, leading to its ubiquitination and degradation. Wnt signaling disrupts this complex, preventing the phosphorylation and degradation of β-catenin and leading to the transcriptional activation of β-catenin/TCF target genes.Citation4 In the PI 3-kinase pathway, GSK-3 is likewise constitutively active in the absence of growth factor signaling and is inhibited as a result of phosphorylation by Akt following growth factor stimulation of PI 3-kinase. It appears that separate pools of GSK-3 function in Wnt and PI 3-kinase signaling, with Wnt signaling targeting a distinct pool of GSK-3 bound to Axin and dedicated to phosphorylation of β-catenin. In contrast, the pool of GSK-3 regulated by PI 3-kinase/Akt signaling phosphorylates a variety of substrates, including glycogen synthase and other metabolic enzymes, translation initiation factor eIF2B,Citation5,Citation6 multiple transcription factors,Citation1,Citation3 and regulators of cell cycle progression and apoptosis, such as cyclin D1,Citation7 and the Bcl-2 family member Mcl-1.Citation8
The PI 3-kinase pathway is a central regulator of mammalian cell proliferation and apoptosis and is one of the signaling pathways most commonly targeted by mutations in human cancers. Within this pathway, GSK-3 plays an important role as a pro-apoptotic protein kinase whose inhibition by Akt is critical to the survival of a wide range of cell types. In addition, the constitutive activity of GSK-3 in the absence of growth factor stimulation suggests a potential function of GSK-3 not only in apoptosis, but also in regulating gene expression in quiescent cells. It is interesting to note that mTOR, another downstream target of PI 3-kinase/Akt signaling, has been reported to play a role in determining the choice between quiescence and senescence in response to cell cycle arrest.Citation9,Citation10 In particular, inhibition of mTOR favors the entry of cells into the quiescent state, whereas mTOR activity favors senescence. Under conditions of growth factor deprivation, the opposing effects of PI 3-kinase signaling on mTOR and GSK-3 may serve to facilitate the entry of cells into quiescence via inhibition of mTOR and to maintain the quiescent state via activation of GSK-3.
We have addressed the function of GSK-3 in quiescent cells by studying its role in the control of gene expression downstream of PI 3-kinase signaling in cells that have been arrested in G0. As discussed below, these studies have revealed a significant role for GSK-3 in maintaining repression of growth factor-inducible genes during quiescence and have identified a network of three transcription factors (CREB, NFκB and AP-1) that mediate this regulation of gene expression by GSK-3.
Transcriptional Regulation by PI 3-Kinase/Akt/GSK-3 Signaling
A variety of transcription factors are known to be regulated via phosphorylation by GSK-3, and many of these factors, including NFκB, c-Myc, p53, CREB and c-Jun, play important roles in cell proliferation and survival. However, studies of the regulation of individual transcription factors cannot yield an overall appreciation of the program of gene expression controlled by GSK-3 in specific physiological settings. We have approached this question by global expression analysis of genes regulated by PI 3-kinase/Akt/GSK-3 signaling in both quiescent cells and immediately following growth factor stimulation.
In order to identify genes that were regulated by PI 3-kinase signaling, microarray analysis was first used to characterize immediate early genes induced by platelet-derived growth factor (PDGF) stimulation of quiescent T98G glioblastoma cells. These cells have been widely used as a model for cell cycle analysis of human cells, because they undergo reversible cell cycle arrest upon growth factor deprivation.Citation11 Small-molecule inhibitors of either PI 3-kinase or MEK/ERK signaling were then used to identify genes whose induction was dependent upon each of these signaling pathways, allowing the induction of specific target genes to be attributed to individual signaling pathways activated downstream of the PDGF receptor tyrosine kinase. The results of these experiments indicated that approximately 20% of the immediate early genes induced by PDGF were principally dependent on PI 3-kinase signaling, 30% were principally dependent on MEK/ERK signaling, 35% were dependent on both the PI 3-kinase and the MEK/ERK pathways, and 15% were independent of both PI 3-kinase and MEK/ERK signaling ().Citation12 Subsequent studies using small-molecule inhibitors combined with global expression profiling have been used to identify genes specifically regulated by the MEK/ERK and PI 3-kinase signaling pathways in several other cell types.Citation13–Citation17
The identification of a set of genes whose induction in response to PDGF stimulation was dependent on PI 3-kinase signaling allowed us to address the role of GSK-3 in regulating gene expression in quiescent cells. Specifically, we asked whether inhibition of GSK-3 in response to growth factor stimulation played a role in the induction of immediate early genes via the PI 3-kinase pathway. If the high activity of GSK-3 was involved in maintaining repression of growth factor-inducible genes during quiescence, then inhibition of GSK-3 in the absence of growth factor stimulation might be sufficient to induce a subset of immediate early genes. This was tested by treating quiescent cells with a small-molecule inhibitor of GSK-3 in the absence of growth factor stimulation, mimicking the effect of PI 3-kinase signaling on GSK-3 without activating either Akt or other downstream targets of the PI 3-kinase pathway. These experiments demonstrated that approximately 40% of the PI 3-kinase-dependent genes could be induced solely by inhibition of GSK-3 ().Citation18 Moreover, for approximately half of these genes, the level of induction by inhibition of GSK-3 was comparable to their level of induction by PDGF, indicating that GSK-3 was the principal effector of their regulation. Thus, the constitutive activity of GSK-3 in quiescent cells plays a significant role in maintaining repression of growth factor-inducible genes. Like other immediate early genes, the genes induced by GSK-3 inhibition encoded transcription factors, growth factors and other signaling molecules with known roles in cell proliferation and motility ().
Identification of Transcription Factor Binding Sites Upstream of GSK-3-Regulated Genes
We have taken an integrated computational and experimental approach to identifying the transcription factors that control gene expression downstream of GSK-3. Computational prediction of transcription factor sites, particularly in mammalian cells, is challenging, because they consist of short, degenerate sequences that occur frequently by chance.Citation19 We have addressed this problem by using two approaches to predict transcription factor binding sites in the upstream promoter regions of the GSK-3-regulated genes. First, genes that are coordinately regulated might be expected to share common regulatory sequences, so we searched for transcription factor binding sites that are statistically over-represented in the upstream regions of the 12 genes that are induced following inhibition of GSK-3 in quiescent cells. In addition, predicted transcription factor binding sites are more likely to be functionally significant if they are phylogenetically conserved,Citation19,Citation20 so we focused on sites that are conserved between human and mouse genomes. This computational analysis predicted conserved binding sites for three families of transcription factors that are over-represented within the 5-kb upstream regions of the 12 genes induced by inhibition of GSK-3 in quiescent cells: CREB, NFκB and AP-1.Citation18,Citation21
CREB
Computational analysis identified binding sites for CREB in the upstream regions of 9 of the 12 GSK-3-regulated genes.Citation18 Chromatin immunoprecipitation (ChIP) assays confirmed the binding of CREB to predicted sites in the upstream regions of six of these genes. Since CREB is constitutively bound to its target sequences in both unstimulated and stimulated cells,Citation22 the physiological activity of these CREB binding sites was investigated by treatment of cells with forskolin, which directly activates cAMP signaling. Forskolin stimulated binding of the coactivator CBP to CREB sites upstream of all six genes, indicating that CREB bound to these sites was activated as a result of phosphorylation by protein kinase A. The binding of CBP was also stimulated by inhibition of GSK-3, indicating that inhibition of GSK-3 was sufficient to increase the transcriptional activity of CREB in quiescent cells. In addition, RNAi confirmed the role of CREB in the induction of five of these genes by forskolin and three genes by inhibition of GSK-3.
These results indicated that CREB is one target of GSK-3 that plays a role in repressing gene expression in quiescent cells. This is consistent with previous reports indicating that CREB is inhibited by GSK-3 phosphorylation.Citation23–Citation25 In addition, studies in monocytes and macrophages have shown that the regulation of CREB by GSK-3 plays a significant role in the Toll-like receptor inflammatory response.Citation26
NFκB
Phylogenetically conserved binding sites for NFκB were also predicted in the upstream regions of nine of the 12 GSK-3-regulated genes, and all of these sites were verified as physiologically functional binding sites for p65 in cells that were stimulated with TNFα to produce a robust activation of this NFκB family member.Citation21 The NFκB family is composed of five different members (p65, c-Rel, RelB, p50 and p52) that share a common DNA binding domain. We therefore performed ChIP assays for all five members of the NFκB family in quiescent cells as well as following inhibition of GSK-3. These studies demonstrated binding of p50 and p65 but not of the other family members to sites upstream of the GSK-3-regulated genes. p50 was bound to most of the predicted NFκB sites in quiescent cells as well those treated with SB-216763 to inhibit GSK-3. The co-repressor HDAC-1 was bound in association with p50, consistent with previous studies indicating that it is constitutively bound as a repressor to the promoters of most NFκB target genes.Citation27 In contrast, the binding of p65 (a strong activatorCitation28) to the upstream regions of four genes was stimulated by inhibition of GSK-3 as well as by growth factor stimulation. In addition, knockdown of p65 by siRNA inhibited the induction of three target genes following inhibition of GSK-3. These results indicate that p50 represses NFκB target genes in quiescent cells, and that GSK-3 serves to maintain repression of these genes by inhibiting p65.
In order to further elucidate the action of GSK-3, we investigated its effects on p65. In most cells, p65 is localized in the cytoplasm in an inhibitory complex with IκBα. Activated IKK phosphorylates IκBα, which targets it for ubiquitination and degradation in the proteasome. This liberates p65, which then translocates to the nucleus and activates transcription of its target genes.Citation28 Inhibition of GSK-3 in quiescent cells, like growth factor stimulation, resulted in activation of IKK, phosphorylation and degradation of IκBα and nuclear translocation of p65.Citation21 It therefore appears that the high activity of GSK-3 in quiescent cells inhibits IKK, thereby negatively regulating NFκB and maintaining repression of NFκB target genes.
In contrast to the inhibition of NFκB by GSK-3 that we observed in quiescent T98G cells, it has been reported that GSK-3 can stimulate NFκB in cells treated with TNFα and in some tumor cells with constitutively active NFκB signaling.Citation29–Citation32 The mechanism by which GSK-3 stimulates NFκB in these settings remains to be fully understood, but generally appears to affect the activity of nuclear p65 rather than the activation of IKK. On the other hand, consistent with our results, other studies have shown that high levels of GSK-3 activity associated with induction of apoptosis of neuronal cellsCitation33,Citation34 and serum deprivation of epithelial cellsCitation35 inhibits NFκB at the level of IκBα phosphorylation. It thus appears that the effects of GSK-3 on NFκB may vary with cell type and physiological circumstance. Under the common condition of activation of GSK-3 by growth factor deprivation, it appears that GSK-3 inhibits NFκB signaling, thereby contributing to either induction of apoptosis or cell cycle arrest.
AP-1
AP-1 binding sites were likewise shown to be statistically overrepresented in the GSK-3-regulated genes, with phylogenetically conserved binding sites predicted upstream of the transcription start site of all 12 genes. AP-1 proteins are comprised of the Jun and Fos transcription factor families, which must dimerize to form functional transcription factors that bind to their target DNA sequences. The homo- and heterodimeric combinations of these two families, in turn, modulate AP-1-mediated gene transcription:Citation36 FosB, c-Fos and c-Jun are transcriptional activators, whereas JunB, JunD, Fra-1 and Fra-2 have weaker transactivation domains. JunB, JunD, Fra-1 and Fra-2 can therefore behave as repressors by competing for c-Fos, FosB and c-Jun binding.
As expected, Fra-1, Fra-2, c-Fos and FosB were not detected by immunoblots in quiescent T98G cells,Citation37 as their expression requires growth factor signaling.Citation38–Citation41 In contrast, c-Jun, JunD and JunB were all expressed in quiescent cells, and, therefore, were considered as candidate targets for regulation by GSK-3. Consistent with this, ChIP assays indicated that c-Jun and JunD but not JunB were bound to predicted AP-1 sites upstream of eight of the GSK-3-repressed genes.Citation37 The binding of c-Jun increased in response to both PDGF stimulation and direct GSK-3 inhibition, whereas the binding of JunD was unaffected, suggesting a direct effect of GSK-3 on c-Jun. In addition, targeted siRNA knockdown confirmed that c-Jun is required for the induction of three genes following GSK-3 inhibition.
These results indicated that c-Jun is a target of GSK-3 and plays a role in repressing gene expression in quiescent cells. This is consistent with the known physiological action of GSK-3 upon c-Jun. GSK-3 phosphorylates c-Jun on threonine 239,Citation42 thereby inhibiting its DNA binding activityCitation43 as well as targeting it for proteasomal degradation.Citation44 As expected, c-Jun was phosphorylated on threonine 239 in quiescent T98G cells.Citation33 This phosphorylation was rapidly reduced following inhibition of GSK-3, allowing AP-1 transcriptional activity to proceed.
Network of Transcriptional Regulation Downstream of GSK-3 in Quiescent Cells
As summarized above, the high activity of GSK-3 in quiescent cells maintains repression of growth factor-inducible genes by inhibiting at least three transcription factors: CREB, NFκB (p65) and AP-1 (c-Jun). These three transcription factors form a network regulating GSK-3-mediated gene expression (). Together, they bind to upstream regions of 10 of the 12 genes that are inducible by inhibition of GSK-3 (, gray and black lines). Notably, nine of the 10 genes are targeted by at least two of these transcription factors, and four of the genes (CYR61, NR4A2, FOSB, NR4A1) are targeted by all three factors.
The overlapping binding of these transcription factors to the promoters of most target genes suggests that they act coordinately to regulate gene expression. The relative contribution of each factor to the induction of specific target genes following inhibition of GSK-3 is indicated by the effects of siRNA against each factor on gene induction. Knockdown of CREB, p65 and c-Jun each significantly inhibited (more than 2-fold) the induction of three genes, with two genes (FOSB, NR4A1) inhibited by knockdown of both CREB and p65 (, black lines). Combined knockdown of either two or three transcription factors did not inhibit the induction of any additional target genes, suggesting that induction of each gene is dominated by one or two of the transcription factors targeted to its regulatory region. Of note, induction of NR4A1 was antagonized by AP-1 (c-Jun), suggesting that it is competing for CREB binding at a common binding site.
AP-1,Citation45,Citation46 NFκBCitation28,Citation47 and CREBCitation48,Citation49 all play key roles in the induction of immediate early genes in response to growth factor stimulation of quiescent cells. Furthermore, previous studies have shown that AP-1,Citation43,Citation50 NFκBCitation33–Citation35 and CREBCitation18,Citation23–Citation26 can all be inhibited by GSK-3. Our results indicate that these three factors comprise a transcriptional network whose inhibition by GSK-3 plays an important role in maintaining repression of growth factor-inducible genes during quiescence. By regulating this network of transcription factors, GSK-3 acts as a check to inhibit the expression of growth factor-inducible genes in the absence of appropriate stimulatory signals.
Figures and Tables
Figure 1 Identification of genes repressed by GSK-3. Microarray analysis was used to identify immediate early genes that were induced by 30 min of PDGF stimulation of quiescent T98G cells.Citation12 Small-molecule inhibitors were used to identify genes that were dependent on PI 3-kinase and MEK/ERK for their induction. The genes showing PI 3-kinase dependence were subsequently tested for their ability to be induced by direct inhibition of GSK-3 (using the small-molecule inhibitor SB-216763) in the absence of growth factor stimulation.Citation18
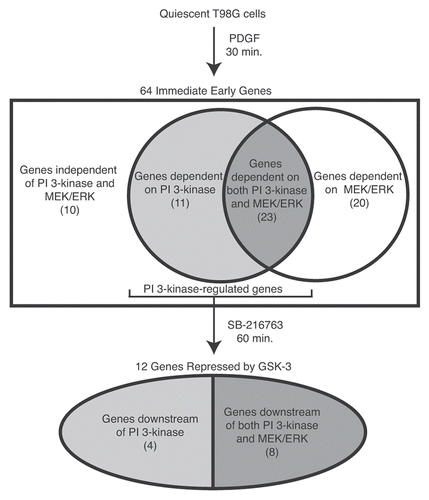
Figure 2 GSK-3 transcriptional network in quiescent cells. Grey and black arrows indicate positive ChIP binding by the indicated transcription factor. Black arrows indicate that siRNA against the transcription factor blocked induction of the gene upon direct inhibition of GSK-3 using the small-molecule inhibitor SB-216763. The blunt-ended line between AP-1 and NR4A1 indicates an inhibition of induction in the presence of AP-1. RND3 and CCL8 are excluded, as no functional connections could be established with AP-1, NFκB or CREB.
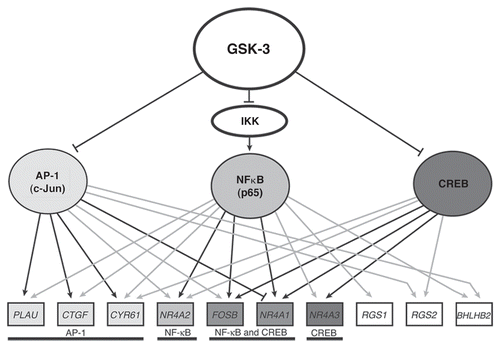
Table 1 Functional categories of the GSK-3-repressed genes
Acknowledgments
This work was supported by the National Institutes of Health Grant R01 CA18689 (G.M.C.) and by American Cancer Society Grant IRG-72-001-33-IRG (J.W.T.).
References
- Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci 2003; 116:1175 - 1186; PMID: 12615961; http://dx.doi.org/10.1242/jcs.00384
- Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochem J 2001; 359:1 - 16; PMID: 11563964; http://dx.doi.org/10.1042/0264-6021:3590001
- Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci 2004; 29:95 - 102; PMID: 15102436; http://dx.doi.org/10.1016/j.tibs.2003.12.004
- Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol 2004; 20:781 - 810; PMID: 15473860; http://dx.doi.org/10.1146/annurev.cellbio.20.010403.113126
- Hardt SE, Tomita H, Katus HA, Sadoshima J. Phosphorylation of eukaryotic translation initiation factor 2B-ε by glycogen synthase kinase-3β regulates β-adrenergic cardiac myocyte hypertrophy. Circ Res 2004; 94:926 - 935; PMID: 15001529; http://dx.doi.org/10.1161/01.RES.0000124977.59827.80
- Pap M, Cooper GM. Role of translation initiation factor 2B in control of cell survival by the phosphatidylinositol-3-kinase/Akt/glycogen synthase kinase3β signaling pathway. Mol Cell Biol 2002; 22:578 - 586; PMID: 11756553; http://dx.doi.org/10.1128/MCB.22.2.578-86.2002
- Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3β regulates cyclin D1 proteolysis and subcellular localization. Genes Dev 1998; 12:3499 - 3511; PMID: 9832503; http://dx.doi.org/10.1101/gad.12.22.3499
- Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell 2006; 21:749 - 760; PMID: 16543145; http://dx.doi.org/10.1016/j.molcel.2006.02.009
- Demidenko ZN, Zubova SG, Bukreeva EI, Pospelov VA, Pospelova TV, Blagosklonny MV. Rapamycin decelerates cellular senescence. Cell Cycle 2009; 8:1888 - 1895; PMID: 19471117; http://dx.doi.org/10.4161/cc.8.12.8606
- Korotchkina LG, Leontieva OV, Bukreeva EI, Demidenko ZN, Gudkov AV, Blagosklonny MV. The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway. Aging (Albany NY) 2010; 2:344 - 352; PMID: 20606252
- Stein GH. T98G: an anchorage-independent human tumor cell line that exhibits stationary phase G1 arrest in vitro. J Cell Physiol 1979; 99:43 - 54; PMID: 222778; http://dx.doi.org/10.1002/jcp.1040990107
- Tullai JW, Schaffer ME, Mullenbrock S, Kasif S, Cooper GM. Identification of transcription factor binding sites upstream of human genes regulated by the phosphatidylinositol-3-kinase and MEK/ERK signaling pathways. J Biol Chem 2004; 279:20167 - 20177; PMID: 14769801; http://dx.doi.org/10.1074/jbc.M309260200
- Bromann PA, Korkaya H, Webb CP, Miller J, Calvin TL, Courtneidge SA. Platelet-derived growth factor stimulates Src-dependent mRNA stabilization of specific early genes in fibroblasts. J Biol Chem 2005; 280:10253 - 10263; PMID: 15637050; http://dx.doi.org/10.1074/jbc.M413806200
- Gu J, Iyer VR. PI3K signaling and miRNA expression during the response of quiescent human fibroblasts to distinct proliferative stimuli. Genome Biol 2006; 7:42; PMID: 16737555; http://dx.doi.org/10.1186/gb-2006-7-5-r42
- Hjortsberg L, Lindvall C, Corcoran M, Arulampalam V, Chan D, Thyrell L, et al. Phosphoinositide-3-kinase regulates a subset of interferon-α-stimulated genes. Exp Cell Res 2007; 313:404 - 414; PMID: 17141757; http://dx.doi.org/10.1016/j.yexcr.2006.10.022
- Jürchott K, Kuban RJ, Krech T, Bluthgen N, Stein U, Walther W, et al. Identification of Y-box binding protein 1 as a core regulator of MEK/ERK pathway-dependent gene signatures in colorectal cancer cells. PLoS Genet 2010; 6:1001231; PMID: 21170361; http://dx.doi.org/10.1371/journal.pgen.1001231
- Ulici V, James CG, Hoenselaar KD, Beier F. Regulation of gene expression by PI3K in mouse growth plate chondrocytes. PLoS ONE 2010; 5:8866; PMID: 20111593; http://dx.doi.org/10.1371/journal.pone.0008866
- Tullai JW, Chen J, Schaffer ME, Kamenetsky E, Kasif S, Cooper GM. Glycogen synthase kinase-3 represses cyclic AMP response element-binding protein (CREB)-targeted immediate early genes in quiescent cells. J Biol Chem 2007; 282:9482 - 9491; PMID: 17277356; http://dx.doi.org/10.1074/jbc.M700067200
- Wasserman WW, Sandelin A. Applied bioinformatics for the identification of regulatory elements. Nat Rev Genet 2004; 5:276 - 287; PMID: 15131651; http://dx.doi.org/10.1038/nrg1315
- Sauer T, Shelest E, Wingender E. Evaluating phylogenetic footprinting for human-rodent comparisons. Bioinformatics 2006; 22:430 - 437; PMID: 16332706; http://dx.doi.org/10.1093/bioinformatics/bti819
- Graham JR, Tullai JW, Cooper GM. GSK-3 represses growth factor-inducible genes by inhibiting NFκB in quiescent cells. J Biol Chem 2010; 285:4472 - 4480; PMID: 20018891; http://dx.doi.org/10.1074/jbc.M109.053785
- Conkright MD, Guzman E, Flechner L, Su AI, Hogenesch JB, Montminy M. Genome-wide analysis of CREB target genes reveals A core promoter requirement for cAMP responsiveness. Mol Cell 2003; 11:1101 - 1108; PMID: 12718894; http://dx.doi.org/10.1016/S1097-2765(03)00134-5
- Bullock BP, Habener JF. Phosphorylation of the cAMP response element binding protein CREB by cAMP-dependent protein kinase A and glycogen synthase kinase-3 alters DNA-binding affinity, conformation and increases net charge. Biochemistry 1998; 37:3795 - 3809; PMID: 9521699; http://dx.doi.org/10.1021/bi970982t
- Grimes CA, Jope RS. CREB DNA binding activity is inhibited by glycogen synthase kinase-3β and facilitated by lithium. J Neurochem 2001; 78:1219 - 1232; PMID: 11579131; http://dx.doi.org/10.1046/j.14714159.2001.00495.x
- Liang MH, Chuang DM. Differential roles of glycogen synthase kinase-3 isoforms in the regulation of transcriptional activation. J Biol Chem 2006; 281:30479 - 30484; PMID: 16912034; http://dx.doi.org/10.1074/jbc.M607468200
- Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol 2005; 6:777 - 784; PMID: 16007092; http://dx.doi.org/10.1038/ni1221
- Schreiber J, Jenner RG, Murray HL, Gerber GK, Gifford DK, Young RA. Coordinated binding of NFκB family members in the response of human cells to lipopolysaccharide. Proc Natl Acad Sci USA 2006; 103:5899 - 5904; PMID: 16595631; http://dx.doi.org/10.1073/pnas.0510996103
- Hayden MS, Ghosh S. Shared principles in NFκB signaling. Cell 2008; 132:344 - 362; PMID: 18267068; http://dx.doi.org/10.1016/j.cell.2008.01.020
- Gong R, Rifai A, Ge Y, Chen S, Dworkin LD. Hepatocyte growth factor suppresses proinflammatory NFκB activation through GSK3β inactivation in renal tubular epithelial cells. J Biol Chem 2008; 283:7401 - 7410; PMID: 18201972; http://dx.doi.org/10.1074/jbc.M710396200
- Götschel F, Kern C, Lang S, Sparna T, Markmann C, Schwager J, et al. Inhibition of GSK3 differentially modulates NFκB, CREB, AP-1 and β-catenin signaling in hepatocytes, but fails to promote TNFα-induced apoptosis. Exp Cell Res 2008; 314:1351 - 1366; PMID: 18261723; http://dx.doi.org/10.1016/j.yexcr.2007.12.015
- Schwabe RF, Brenner DA. Role of glycogen synthase kinase-3 in TNFα-induced NFκB activation and apoptosis in hepatocytes. Am J Physiol Gastrointest Liver Physiol 2002; 283:204 - 211; PMID: 12065308
- Steinbrecher KA, Wilson W 3rd, Cogswell PC, Baldwin AS. Glycogen synthase kinase 3β functions to specify gene-specific, NFκB-dependent transcription. Mol Cell Biol 2005; 25:8444 - 8455; PMID: 16166627; http://dx.doi.org/10.1128/MCB.25.19.844455.2005
- Bournat JC, Brown AM, Soler AP. Wnt-1 dependent activation of the survival factor NFκB in PC12 cells. J Neurosci Res 2000; 61:21 - 32; PMID: 10861796; http://dx.doi.org/10.1002/1097-4547(20000701)61:1<21::AIDJNR3>3.0.CO;2-7
- Sanchez JF, Sniderhan LF, Williamson AL, Fan S, Chakraborty-Sett S, Maggirwar SB. Glycogen synthase kinase 3β-mediated apoptosis of primary cortical astrocytes involves inhibition of nuclear factor κB signaling. Mol Cell Biol 2003; 23:4649 - 4662; PMID: 12808104; http://dx.doi.org/10.1128/MCB.23.13.4649-62.2003
- Bachelder RE, Yoon SO, Franci C, de Herreros AG, Mercurio AM. Glycogen synthase kinase-3 is an endogenous inhibitor of Snail transcription: implications for the epithelial-mesenchymal transition. J Cell Biol 2005; 168:29 - 33; PMID: 15631989; http://dx.doi.org/10.1083/jcb.200409067
- Angel P, Karin M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim Biophys Acta 1991; 1072:129 - 157; PMID: 1751545
- Tullai JW, Tacheva S, Owens LJ, Graham JR, Cooper GM. AP-1 Is a component of the transcriptional network regulated by GSK-3 in quiescent cells. PLoS ONE 2011; 6:20150; PMID: 21647439; http://dx.doi.org/10.1371/journal.pone.0020150
- Cohen DR, Curran T. fra-1: a serum-inducible, cellular immediate early gene that encodes a fosrelated antigen. Mol Cell Biol 1988; 8:2063 - 2069; PMID: 3133553
- Kovary K, Bravo R. Expression of different Jun and Fos proteins during the G0-to-G1 transition in mouse fibroblasts: in vitro and in vivo associations. Mol Cell Biol 1991; 11:2451 - 2459; PMID: 1901942
- Müller R, Bravo R, Burckhardt J, Curran T. Induction of c-fos gene and protein by growth factors precedes activation of c-myc. Nature 1984; 312:716 - 720; PMID: 6334806; http://dx.doi.org/10.1038/312716a0
- Nishina H, Sato H, Suzuki T, Sato M, Iba H. Isolation and characterization of fra-2, an additional member of the fos gene family. Proc Natl Acad Sci USA 1990; 87:3619 - 3623; PMID: 2110368; http://dx.doi.org/10.1073/pnas.87.9.3619
- Morton S, Davis RJ, McLaren A, Cohen P. A reinvestigation of the multisite phosphorylation of the transcription factor c-Jun. EMBO J 2003; 22:3876 - 3886; PMID: 12881422; http://dx.doi.org/10.1093/emboj/cdg388
- Boyle WJ, Smeal T, Defize LH, Angel P, Woodgett JR, Karin M, et al. Activation of protein kinase C decreases phosphorylation of c-Jun at sites that negatively regulate its DNA-binding activity. Cell 1991; 64:573 - 584; PMID: 1846781; http://dx.doi.org/10.1016/0092-8674(91)90241-P
- Wei W, Jin J, Schlisio S, Harper JW, Kaelin WG Jr. The v-Jun point mutation allows c-Jun to escape GSK3-dependent recognition and destruction by the Fbw7 ubiquitin ligase. Cancer Cell 2005; 8:25 - 33; PMID: 16023596; http://dx.doi.org/10.1016/j.ccr.2005.06.005
- Chinenov Y, Kerppola TK. Close encounters of many kinds: Fos-Jun interactions that mediate transcription regulatory specificity. Oncogene 2001; 20:2438 - 2452; PMID: 11402339; http://dx.doi.org/10.1038/sj.onc.1204385
- Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer 2003; 3:859 - 868; PMID: 14668816; http://dx.doi.org/10.1038/nrc1209
- Ghosh S, Karin M. Missing pieces in the NFκB puzzle. Cell 2002; 109:81 - 96; PMID: 11983155; http://dx.doi.org/10.1016/S0092-8674(02)00703-1
- Carlezon WA Jr, Duman RS, Nestler EJ. The many faces of CREB. Trends Neurosci 2005; 28:436 - 445; PMID: 15982754; http://dx.doi.org/10.1016/j.tins.2005.06.005
- Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol 2001; 2:599 - 609; PMID: 11483993; http://dx.doi.org/10.1038/35085068
- Nikolakaki E, Coffer PJ, Hemelsoet R, Woodgett JR, Defize LH. Glycogen synthase kinase-3 phosphorylates Jun family members in vitro and negatively regulates their transactivating potential in intact cells. Oncogene 1993; 8:833 - 840; PMID: 8384354